

 , Xinlin Fu 2,†, Lixin Xie 1,*
, Xinlin Fu 2,†, Lixin Xie 1,* , Shoulong Deng 3,*
, Shoulong Deng 3,*1 College of Pulmonary and Critical Care Medicine, Chinese PLA General Hospital, 100853 Beijing, China
2 College of Animal Science and Technology, China Agricultural University, 100193 Beijing, China
3 NHC Key Laboratory of Human Disease Comparative Medicine, Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences and Comparative Medicine Center, Peking Union Medical College, 100021 Beijing, China
†These authors contributed equally.
Abstract
Acute lung injury (ALI) is a severe condition characterized by an inflammatory response and increased vascular permeability, with its pathological mechanisms closely associated with the dysfunction of vascular smooth muscle cells (VSMCs). The present study investigates the molecular mechanisms through which lactate dehydrogenase A (LDHA) influences the inflammatory response in ALI by modulating VSMC metabolic reprogramming. It was observed that under pathological conditions, hypoxia and the inflammatory microenvironment significantly upregulate LDHA expression in lung VSMCs via the activation of the hypoxia-inducible factor (HIF) signaling pathway. The LDHA-mediated Warburg effect not only provides energetic support to VSMCs but also exacerbates inflammatory responses through both direct and indirect mechanisms. This review highlights the critical role of LDHA as a metabolic-inflammatory nexus in ALI and offers a theoretical foundation for targeting LDHA to regulate metabolic reprogramming as a means to mitigate the progression of ALI. Future research will further investigate the specific mechanisms by which LDHA regulates VSMC metabolic reprogramming and will seek to identify effective intervention strategies.
Keywords
- ALI
- LDHA
- smooth muscle cell
- inflammation
Acute lung injury (ALI) is characterized as an acute, diffuse inflammatory
condition of the lungs, precipitated by a variety of intra- and extra-pulmonary
pathogenic factors of non-cardiogenic origin. This condition can lead to severe
hypoxemia and acute respiratory failure, potentially progressing to the critical
illness, known as acute respiratory distress syndrome (ARDS). The reprogramming
of glucose metabolism is intricately associated with immune responses and
inflammatory processes. Study indicates that sepsis link to hyperlactatemia;
during septic ALI, macrophages recruited and activated by lipopolysaccharides
(LPS) exhibit enhanced glycolysis, which further exacerbates the inflammatory
response and worsens oxidative stress, leading to progression of ALI and
potentially ARDS or even death, suggesting that glycolytic inhibition is a potent
anti-inflammatory strategy for the treatment of ALI [1]. ARDS is a critical
respiratory condition characterized by an unclear pathogenesis, a lack of
effective pharmacological treatments, and a low quality of life and survival rate
for affected individuals. Currently, the incidence of new ALI cases worldwide
exceeds three million annually, with clinical meta-analyses revealing a mortality
rate of 43%. Furthermore, up to 50% of survivors experience significant
pulmonary dysfunction. Persistent inflammation, coupled with the loss of vascular
barrier integrity, results in pulmonary edema and multiorgan dysfunction, which
are significant contributors to the elevated mortality associated with ALI and
ARDS. The pulmonary vascular system, due to its extensive surface area, is
particularly susceptible to the activation of inflammatory responses. In the
context of ALI, the infiltration of inflammatory cells and the release of
inflammatory mediators, along with increased tissue metabolic activity, lead to a
heightened demand for oxygen. Additionally, inflammation-induced vasoconstriction
further diminishes oxygen supply to the affected regions of the lung, resulting
in hypoxemia. This hypoxic state subsequently triggers the overexpression of HIF,
which directly promotes the upregulation of LDHA and the degradation of Inhibitor
of kappa B zeta (I
Vascular endothelial cells (VECs) constitute a layer of mononuclear cells
situated between the bloodstream and the vascular wall tissues. These cells are
capable of secreting a variety of vasoactive substances through autocrine,
endocrine, and paracrine mechanisms, thereby playing crucial roles in regulating
vascular tone, inhibiting the proliferation of smooth muscle cells, and
modulating inflammatory responses within the vascular wall. The structural and
functional integrity of VECs is essential for the maintenance of vascular wall
function and lung homeostasis. Under homeostatic conditions, VECs remain
quiescent and exhibit antioxidant and anti-inflammatory properties. However,
during inflammatory events, VECs participate in the interplay between local and
systemic immune responses and serve as significant targets for inflammatory
processes. This involvement can enhance innate immune responses through the
production and release of cytokines and chemokines, including the activation of
the NF-
VECs are influenced by various factors that prompt them to lose their original characteristics and undergo significant alterations in polarity, morphology, and function. This process leads to their transformation into mesenchymal cells, such as fibroblasts and smooth muscle cells, a phenomenon known as endothelial-to-mesenchymal transition (EndMT) [3]. Pyruvate dehydrogenase kinase (PDK) phosphorylates pyruvate dehydrogenase complex (PDH) during EndMT, which inhibits the mitochondrial catabolism of pyruvate in endothelial cells, resulting in the reduction of endothelial pyruvate by lactate dehydrogenase (LDH) reduces pyruvate from endothelial cells to lactate, resulting in a paradigm shift in the energy metabolism of VECs.
Abnormal communication between VECs and VSMCs is a critical factor in the initiation and progression of vasculopathic alterations. In ALI, lung epithelial cells and vascular endothelial cells leads to diffuse interstitial and alveolar edema, progressive hypoxemia, and respiratory distress, which subsequently triggers hypoxic pulmonary vasoconstriction (HPV) [4]. Although early HPV is a protective immune response, persistent inflammation and hypoxia can result in the hyperproliferation of VSMCs, thickening of the vascular wall, and ultimately the development of ARDS. Nitric oxide and carbon monoxide in VECs indirectly induce vasodilation and modulate the inflammatory response through crosstalk between VECs and VSMCs, while also promoting the proliferation and migration of VSMCs. The knockdown of vesicle-associated membrane protein 3 (VAMP3) and synaptosomal-associated protein 23 (SNAP23) in endothelial cells has been shown to inhibit the proliferation, migration, and phenotypic transformation of smooth muscle cells co-cultured with endothelial cells. Furthermore, microRNA-126-3p secreted by endothelial cells enhances the proliferation and dedifferentiated phenotype of smooth muscle cells, contributing to the thickening of the neointima in arterial vessels in murine models [5].
VSMCs are a highly differentiated cell type characterized by the expression of
various proteins associated with vascular contraction, including smooth muscle
myosin heavy chain (SMMHC) and alpha-smooth muscle actin (
During vascular injury, VSMCs migrate from the mesentery to the intima in response to growth factors secreted by endothelial cells, resulting in intimal hyperplasia and restenosis. To mitigate excessive proliferation, nitric oxide (NO) and prostacyclin (PGI2) released by endothelial cells inhibit the over-proliferation and migration of VSMCs, thereby maintaining the stability of the vessel wall. Furthermore, VSMCs may undergo phenotypic transformation, adopting a myofibroblast-like phenotype that is more conducive to tissue repair, characterized by enhanced proliferation and increased secretion of extracellular matrix (ECM). Consequently, investigating the interactions between vascular smooth muscle cells and vascular endothelial cells in the context of inflammatory responses and cell proliferation during the development of ALI is an effective way to ameliorate the adverse prognosis associated with ALI and to prevent vessel wall thickening.
The human LDHA gene is localized on chromosome 11, 11p15.4, and encodes a protein containing 332 amino acids, including 24 splice variants and several unknown pseudogenes [7]. LDHA is expressed predominantly in skeletal muscle and has a high affinity for pyruvate, preferentially converting pyruvate to lactate while converting nicotinamide adenine dinucleotide (NADH) to NAD+ [8]. During conditions of hypoxia or high-intensity exercise, skeletal muscle generates elevated levels of lactic acid through the action of LDHA and subsequently releases it into the bloodstream. This lactic acid is then transported throughout the body, including to the lungs, via the circulatory system. In the lungs, smooth muscle cells uptake lactic acid from the blood, utilizing it as an energy source. The clearance of lactate produced by skeletal muscle occurs in the liver through the Cori cycle. Conversely, lactate generated by VSMCs in the lungs exerts direct effects on local immune cells, such as macrophages, and endothelial cells, thereby establishing a positive feedback loop that exacerbates the progression of ALI [9], which is important for us to explore the relationship of lactate and LDHA, and the inflammatory response, to the inflammatory response in the lungs. Lactate and LDHA are intricately associated with the inflammatory response in the lungs. Recent studies have demonstrated that the regulatory effects of LDHA primarily manifest through transcriptional regulation, post-transcriptional regulation, and post-translational modifications [10]. LDHA is mainly found in the cytoplasm, but it is also present in the nucleus of some tumor cells. It has been reported that tyrosine residues of LDHA appear in the nucleus after phosphorylation, suggesting that LDHA may have a unique function in gene transcription and RNA stability.
LDH is classified within the 2-hydroxy oxidoreductase family. In humans, LDH is encoded by three genes: LDHA, LDHB, and LDHC. Notably, LDHC is specifically expressed in testicular tissues, while LDHA and LDHB are ubiquitously expressed across various tissues and are recognized as some of the most abundant proteins found in the cytoplasm [11]. LDH functions as a homo- or heterotetramer composed of two subunits, LDHA (M) and LDHB (H), which are encoded by the respective LDHA and LDHB genes. Among the different isoforms, LDH-5 (4M) exhibits the highest catalytic efficiency in the conversion of pyruvate to lactate [12].
The promoter region of the LDHA gene encompasses multiple transcription factor binding sites and is subject to regulation by various transcription factors. Hypoxia-inducible factor-1 (HIF-1) interacts with the hypoxia-responsive element (HRE) to activate LDHA expression. Additionally, the proto-oncogene c-Myc forms a heterodimer with myc-associated factor X (MAX), which subsequently binds to the E-box within the LDHA promoter, thereby enhancing LDHA expression [12]. Furthermore, forkhead box protein M1 (FOXM1) directly binds to the LDHA promoter region, modulating the transcriptional regulation of the LDHA gene, a process that is crucial for aerobic glycolysis and tumorigenesis in patients with pancreatic cancer [13]. Moreover, KLF4 is involved in the regulation of epithelial and mesenchymal gene expression, which is significant for cell proliferation and differentiation. Research indicates that KLF4 exhibits a negative correlation with LDHA expression level [14].
LDHA was directly regulated by factors such as HIF-1, c-Myc, FOXM1, and KLF4, suggesting that LDHA plays a significant role in cellular phenotypic transformation, cell proliferation, and apoptosis [15]. Additionally, the pro-inflammatory effects exhibited by VSMCs in inflammatory contexts, mediated by elevated lactate production, further underscore its critical involvement in cellular energy metabolism. Nevertheless, research concerning the role of LDHA in cell proliferation remains insufficient, revealing numerous gaps in the current understanding. Investigating how LDHA regulates inflammatory responses, cell proliferation, and vascularization in ALI is a novel and significant area for future research.
Recombinant LDHA is a pivotal enzyme in the glycolytic pathway, significantly influencing the proliferation, migration, and metabolic processes of VSMCs. The lactate microenvironment generated by LDHA activity facilitates the transition of VSMCs from a contractile phenotype to a secretory phenotype, thereby enhancing their proliferation and migration, as well as promoting the secretion of ECM. This process contributes to the thickening of the vascular wall and an increase in pulmonary vascular resistance [16]. The modifications of LDHA, specifically crotonylation and monoubiquitination, were found to be elevated in proliferating VSMCs and neovascular endothelium. The crotonylation occurred at lysine 5 (K5), while monoubiquitination was observed at lysine 76 (K76). The crotonylation at K5 enhanced LDHA activity and increased intracellular lactic acid levels by facilitating its tetramer formation, thereby promoting cellular growth. Conversely, the monoubiquitination at K76 contributed to cell proliferation by mediating mitochondrial division and enhancing VSMC migration [16, 17].
In a mouse ALI model, the knockdown of LDHA in VSMCs significantly decreased
lung edema and neutrophil infiltration while enhancing the oxygenation index
[18]. Conversely, LDHA in lung VSMCs exacerbated alveolar injury by promoting
lactate accumulation, directly activating the NF-
Metabolic reprogramming of vascular smooth muscle cells is a crucial component of the vascular remodeling process. LDHA plays a significant role in this context by facilitating glycolysis and lactate production, thereby supplying the necessary energy and metabolic intermediates. The restoration of mitochondrial function has been shown to normalize hypoxia-induced oxidative stress and mitigate the inflammatory response in murine models by decreasing excessive lactate accumulation and correcting abnormal glycolytic activity [20]. Furthermore, the overexpression of LDHA has been confirmed in human vascular smooth muscle cell lines, promoting a transition to a synthetic phenotype. Conversely, the knockdown of LDHA results in reduced proliferation and lactate production in human coronary artery smooth muscle cells (HACSMCs) [19].
Energy metabolism serves as a crucial regulator of cellular function and is significantly altered when tissues and cells experience pathological changes. Under normal physiological conditions, cells are efficiently supplied with adenosine triphosphate (ATP) through glycolysis and oxidative phosphorylation. One molecule of glucose can yield a net production of 36 ATP molecules, which provide the necessary energy for various cellular activities. In hypoxic conditions, cells are unable to utilize oxidative phosphorylation effectively for ATP production; instead, they resort to aerobic glycolysis facilitated by M2-type pyruvate kinase (PKM2) and LDHA, resulting in a net production of only 2 ATP molecules. Concurrently, LDHA sustains aerobic glycolysis by regenerating NAD+ from NADH. Although aerobic glycolysis is less efficient, it occurs at a rate that is approximately 100 times faster than oxidative phosphorylation, thereby meeting the short-term energy demands of the cell in the absence of adequate oxygen, albeit at the cost of increased glucose consumption and lactate production. Notably, certain cell types, including activated immune cells, tumor cells, and infected cells, adopt an aerobic glycolytic pathway to convert glucose to lactate even under normoxic conditions. This metabolic shift signifies a change in cellular function and signaling pathways and is commonly referred to as the Warburg effect [21].
Acute injury leads to an insufficient supply of oxygen to lung tissue, prompting
VSMCs to depend primarily on LDHA-mediated glycolysis for energy production,
rather than on mitochondrial oxidative phosphorylation. This metabolic
reprogramming represents a crucial adaptive mechanism for VSMCs to manage
oxidative stress and inflammation. In conditions of hypoxia, hypoxia-inducible
factor 1 alpha (HIF1
Research has demonstrated that the supplementation of lactate in growth medium enhances the synthetic phenotype of VSMCs derived from human induced pluripotent stem cells [25]. Furthermore, lactate promotes the polarization of alternatively activated macrophages exhibiting an anti-inflammatory (M2-like) phenotype. These macrophages play a crucial role in angiogenesis and tissue remodeling, as well as stimulating the release of vascular endothelial growth factor from endothelial cells, thereby facilitating wound healing and tumor-associated angiogenesis [26, 27, 28, 29].
The etiology of ALI/ARDS has not been fully defined, and its pathogenesis
encompassing a range of direct and indirect risk factors. It is widely
acknowledged that uncontrolled inflammation, either localized within the lungs or
systemic, constitutes the primary mechanism underlying ALI/ARDS. The principal
contributors to this inflammatory response include biological, chemical, and
physical factors. ① Biological stimuli, such as bacteria or viruses that
invade the lungs, induce inflammation through their proliferation within the
host, the production of toxins, and their antigenic properties.
Pathogen-associated molecular patterns (PAMPs), including LPS, unmethylated CpG
DNA, and RNA derived from pathogens, interact with various surface and
intracellular pattern recognition receptors (PRRs) present on immune cells. These
receptors encompass Toll-like receptors, nucleotide oligomerization domain-like
receptors (NLRs), and C-type lectins (which recognize end products of late
glycosylation). The engagement of these PRRs activates the expression and
secretion of inflammatory mediators, such as interleukin-1 (IL-1), tumor necrosis
factor (TNF), and reactive oxygen species. ② Chemical or physical
stimuli, such as lung tissue damage resulting from the inhalation of noxious
gases, radiation, and contusion, lead to the production of damage-associated
molecular patterns (DAMPs). These include heat shock proteins (HSP), high
mobility group protein B1 (HMGB-1), uric acid, and extracellular ATP. In ALI, the
hypoxic and inflammatory microenvironment of the lung significantly upregulates
the expression in lung VSMC through the activation of signaling pathways,
including HIF-1
During glycolysis, NADH engages with the Rossman folding domain of LDHA, and
through the catalytic activity of LDHA and the thermodynamic stabilization of
free radical intermediates, electrons are transferred from NADH to
oxygen-containing compounds at an accelerated rate. This process leads to the
production of increased levels of oxygen radicals, which may contribute to
oxidative damage and cytotoxicity, as well as to the promotion of inflammatory
responses [10], It also promotes T-cell effector function by increasing
acetylation and transcription of IFNG, thus highlighting the key role of LDH in
inflammation [17]. The application of
(S)-N-Hydroxy-3-(4-methoxybenzylsulfonyl)-2,2-dimethylchroman-4-carboxamide
(FX11), an inhibitor of LDHA, in chondrocytes resulted in a significant reduction
in the expression of the inflammatory factors IL-6 and matrix
metalloproteinase-13 (MMP13). Although I
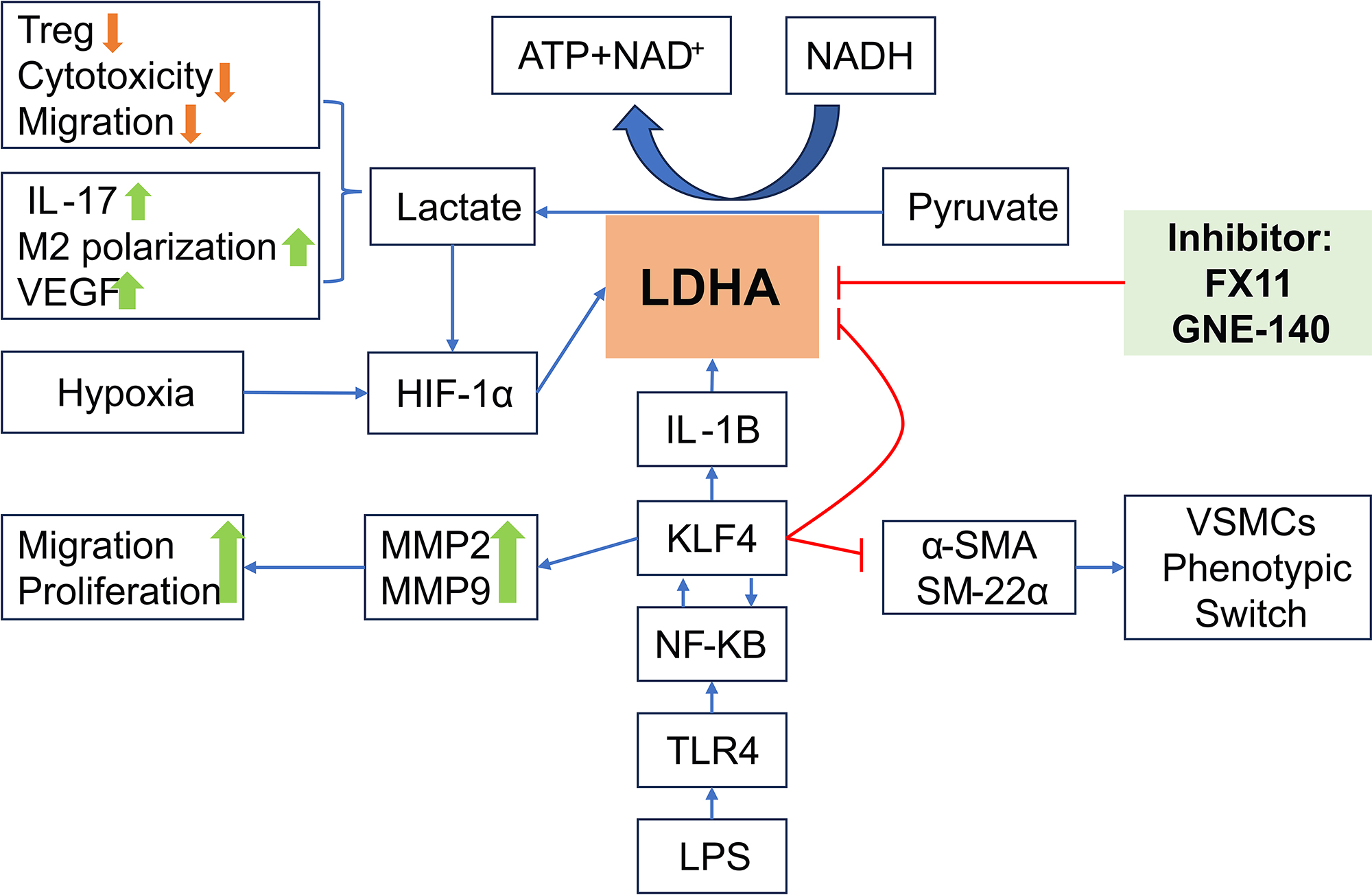 Fig. 1.
Fig. 1.
LDHA affects the inflammatory response in acute lung injury by
modulating metabolic reprogramming in vascular smooth muscle. ATP, adenosine
triphosphate; NAD+, Nicotinamide Adenine Dinucleotide (oxidized form);
HIF-1
Under normoxia, LDHA is a key enzyme associated with aerobic glycolysis that contributes to lactate production in cancer cells [31, 32], tumor cells convert 69–80% of glucose into lactate; while under hypoxia, the conversion efficiency reaches 90%. Lactate, the end product of glycolysis, was once considered a metabolic waste product, but current research suggests that lactate has a variety of important biological functions. Lactate serves a crucial physiological function by facilitating the exchange between lactate-producing and lactate-consuming cells within the body. It has been identified as an amplifier of inflammation, with its accumulation exacerbating the inflammatory response. Elevated levels of LDH activity can impair immune cell functionality, while high concentrations of lactate may diminish the efficacy of effector T cells and promote the production of interleukin-17 (IL-17) by Cluster of Differentiation 4 (CD4+) T cells through the PKM2/STAT3 signaling pathway [33]; Inhibition of LDHA activity in macrophages downregulates the expression of pro-inflammatory cytokines, inducible nitric oxide synthase (iNOS), and other anti-inflammatory effects [34]. During ALI, hyperlactatemia arises as a consequence of the upregulation of anaerobic glycolysis, which is activated by an inadequate supply of oxygen. Anaerobic glycolysis refers to the metabolic conversion of glucose to lactate, a process regulated by various rate-limiting glycolytic enzymes, including LDHA. A recent study has demonstrated that the inhibition of the glycolytic pathway using the inhibitor PFKFB3, specifically 3-(3-pyridyl)-1-(4-pyridyl)-2-propen-1-one (3PO), results in reduced glucose uptake and inhibits the flux of glycolysis to lactate. This intervention effectively obstructs the energy source for VSMCs and has been identified as a promising strategy for preventing sepsis-associated ALI [9] (Table 1, Ref. [10, 17, 18, 31, 32, 33, 34, 35, 36, 37, 38, 39]).
| Modes of action | Machanisms | Concrete content | Reference |
| Direct effect | • NADH-LDHA interaction | • During glycolysis, it promotes electron transfer, increases oxygen radical production, and enhances oxidative killing and inflammatory responses. | [10, 35] |
| • T-cell effector function promotion | • Increased acetylation and transcription of interferon- |
[17, 36] | |
| • Inflammatory factor regulation | • The LDHA inhibitor FX11 significantly inhibited IL-6 and MMP13 expression, increased I |
[18, 37] | |
| • Immunocytosis | • Direct activation of immune cells promotes their inflammatory response; macrophages produce LDHA-containing exocysts that promote glycolysis and inhibit macrophage infiltration, reducing inflammation. | [38] | |
| Indirect effect | • Lactic acid production and effects | • Promotes massive conversion of glucose to lactate, and lactate accumulation amplifies the inflammatory response. | [31, 32] |
| • Suppression of immune cell function | • Increased LDH activity suppresses immune cell function, and high concentrations of lactate decrease effector T cell activity and promote IL-17 production by CD4+ T cells. | [33, 39] | |
| • Anti-inflammatory effect | • Inhibition of LDHA activity down-regulates pro-inflammatory factors and iNOS expression and exerts anti-inflammatory effects. | [34, 37] |
NADH, nicotinamide adenine dinucleotide; LDHA, lactate dehydrogenase A; FX11,
(S)-N-Hydroxy-3-(4-methoxybenzylsulfonyl)-2,2-dimethylchroman-4-carboxamide;
IL-6, interleukin-6; MMP13, matrix metalloproteinase-13; I
The etiology of ALI is multifaceted, resulting in damage to alveolar epithelial cells and vascular endothelial cells. This condition triggers an inflammatory response and induces persistent hypoxemia, ultimately leading to the development of ARDS, and the high mortality associated with ALI has garnered significant attention within the medical community. LDHA is a crucial metabolic enzyme that regulates hypoxemia during the progression of ALI. It induces alterations in the energy metabolism of VSMCs and provides energy for cellular proliferation and differentiation through aerobic glycolysis. Additionally, LDHA promotes the repair of damaged tissues, vasoconstriction, endothelial-mesenchymal transition, cell proliferation, and migration, while also generating reactive oxygen species that further exacerbate inflammatory responses. LDHA serves not only as an enzyme involved in energy metabolism but also as a protein factor that regulates various biological processes. It is recognized as an important biomarker and potential therapeutic target for ALI, ARDS, tumor development, and poor prognosis. Consequently, comprehensive research into the pro-inflammatory and metabolic functions of LDHA, along with the identification of novel genes that synergistically interact with LDHA, will significantly enhance the foundational theoretical understanding of ALI pathogenesis.
Numerous studies have substantiated the efficacy of various LDHA inhibitors in
models of lung injury and tumors. FX11, a competitive inhibitor of NADH, has been
shown to reduce bacterial load and inhibit necrotic lesions in the lungs of
tuberculosis-infected mice by impeding glycolysis [40]. Furthermore, its
nanocarrier formulation (FX11@TPEG-WS2) targets mitochondria and enhances
antitumor efficacy when used in conjunction with acoustic kinetic therapy [41].
In combination with metformin, FX11 activates the AMPK
The comprehensive investigation of the LDHA regulatory network is crucial for
addressing the challenges associated with clinical translation. Clarifying the
role of LDHA in modulating endothelial inflammation and permeability via the
lactate-NF-
ALI represents a critical inflammatory disorder characterized by elevated
mortality rates, primarily resulting from dysregulated metabolism in VSMCs and
sustained inflammatory responses. This review aims to clarify the significant
function of LDHA in the metabolic reprogramming of VSMCs and the enhancement of
inflammatory processes during ALI. In the context of hypoxia and inflammation,
LDHA expression is increased through the HIF-1
DL and XLF contributed equally to the study design, data analysis, literature search, and preparation of the initial draft of the manuscript. LXX was responsible for the study conception and supervision, while LXX and SLD were involved in the design, editing, and revision of the manuscript. All authors have approved the final version of the manuscript and have participated sufficiently in the work, thereby agreeing to be accountable for all aspects of the research.
Not applicable.
Not applicable.
This research received funding from China NSFC Key Grant (82341119).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.