











 , Cheng-Yang Huang 2,3
, Cheng-Yang Huang 2,31 Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, NE 68198-6025, USA
2 Department of Biomedical Sciences, Chung Shan Medical University, 40201 Taichung, Taiwan
3 Department of Medical Research, Chung Shan Medical University Hospital, 40202 Taichung, Taiwan
Abstract
To maintain genome stability, the coordinated actions of multiple proteins and protein complexes, which are collectively known as genome guardians, are required. In prokaryotes, one such 20-member genome guardian family known as the single-stranded DNA binding protein (SSB) interactome exists. Proteins within this essential family contain oligonucleotide/oligosaccharide-binding folds (OB-fold). These structurally conserved OB-folds bind to the intrinsically disordered linkers characteristic of SSB protein C-termini, resulting in partner regulation. The mechanism of binding employed is similar to that utilized by Src homology 3 domain (SH3) proteins in eukaryotes. Binding requires the interaction of conserved PXXP motifs in the SSB linker with the OB-fold in the partner. A second region of SSB C-termini, an 8–10 stretch of predominantly acidic amino acids functions to maintain the linker domain in a biologically active conformation, while simultaneously preventing it from adhering to the OB-folds of the SSB tetramer from which it emanates. In addition, this acidic domain also functions as a secondary binding site docking with a distal site in the partner, stabilizing the linker/OB-fold interactions. The interaction of an SSB with its partner proteins is genus-specific and results in the loading of partners onto the genome at various stages of the cell cycle thereby maintaining genome stability.
Keywords
- single-strand DNA binding protein
- SSB
- genome stability
- SSB interactome
- OB-fold
- SH3 domain
- prokaryotic
- protein-protein interactions
The SSB interactome is essential to maintaining genome integrity in bacteria [1, 2, 3, 4]. The interactome consists of at least 20, oligonucleotide/oligosaccharide binding fold (OB-fold) containing, genome guardian proteins and includes exonucleases, DNA helicases, DNA polymerases, DNA primases, recombination mediators, DNA repair enzymes, and topoisomerases (Fig. 1, Ref. [5, 6, 7, 8, 9, 10, 11, 12, 13] and Table 1, Ref. [14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36]). The central player controlling interactome function is also an interactome member and is the product of the essential ssb gene, the SSB protein [37, 38, 39, 40, 41].
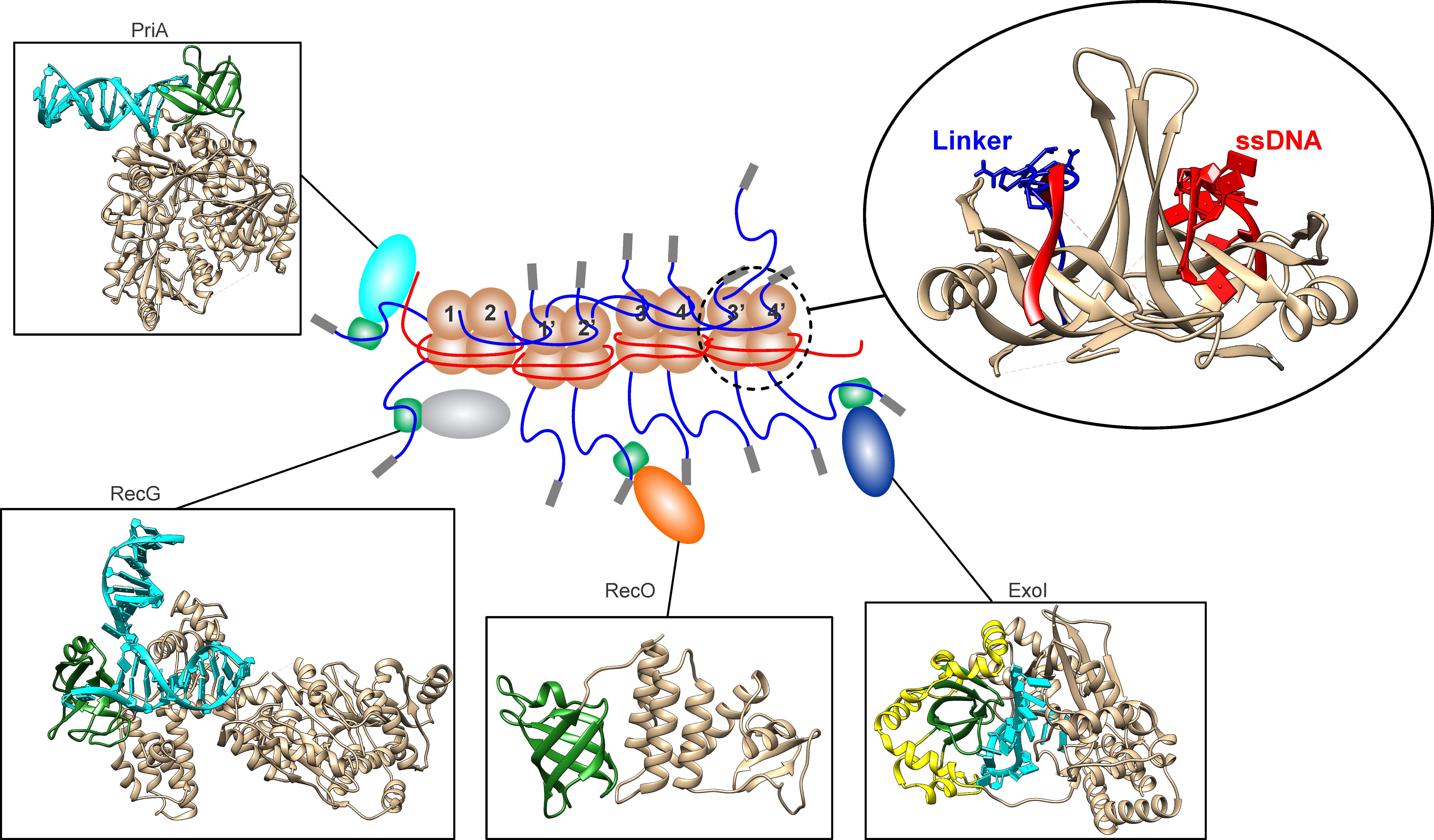 Fig. 1.
Fig. 1.
The SSB interactome is regulated by the linker/OB-fold mechanism. SSB proteins are bound contiguously to ssDNA (red) forming a stable complex. Within this complex, the OB-folds (light brown) of some subunits are bound to ssDNA (red) while others bind to the linkers (blue) of adjacent SSB monomers (inset, upper right). When bound to ssDNA, the linkers of monomers 1 and 2, bind to the OB-folds of monomers 1’ and 2’, respectively. Concurrently, the linkers of monomers 1’ and 2’ bind to the OB-folds of monomers 3 and 4, and their linkers bind to monomers 3’ and 4’, respectively. In the SSB-ssDNA complex, linker/OB-fold binding is responsible for the cooperative interactions between SSB tetramers. In addition, some SSB linkers are available for partner binding, docking with the OB-folds (green) in PriA, RecG, RecO or Exonuclease I. In this image the following PDB files were used: Exonuclease I (4JS5), the extended SH3 domain is colored yellow; RecO (3Q8D); PriA (6DGD) and RecG (1GM5) [6, 7, 8, 9, 10]. For EcSSB, PDB file 4MZ9 was used and TM-align utilized to model the linker from SH3 domain 2XKC and ssDNA from 1EYG [5, 11, 12, 13]. The figure was made using Corel Designer v2018 (Alludo; Ottawa, Canada). OB-fold, oligonucleotide/oligosaccharide binding fold.
| Protein (E.coli name)b | Function | OB-fold present | Bacterial species identified inc |
| SSB | Binds ssDNA and partner proteins | Yes | 68,567d |
| AlkB [21] | Reversal of alkylation damage | Yes | 11,338 |
| DinG [24] | Structure-specific DNA repair DNA helicase | Yes | 13,855 |
| DNA Polymerase II [14] | DNA polymerase involved in DNA repair | Yes | 9372 |
| DNA Polymerase III, |
DNA replication | Yes | 10,410 |
| DNA Polymerase III, |
DNA replication | No | 770 |
| DNA Polymerase III, |
DNA replication | No | 3332 |
| DNA Polymerase III, |
DNA replication | No | 1981 |
| DnaG [29] | DNA primase | Yes | 29,943 |
| DNA Polymerase IV [20] | DNA polymerase involved in translesion synthesis | Yes | 29,968 |
| Exonuclease I [17, 30] | Exonuclease involved in chromosome replication | Yes (extended SH3 domain) | 2030 |
| PriA [22] | Replication restart | Yes | 31,953 |
| DNA helicase | |||
| PriB [18] | Primosome component; replication restart | Yes | 6341 |
| PriC [31] | primosome component involved in replication restart | No | 5329 |
| RadD [32] | DNA repair (putative DNA Helicase) | Yes | 251 |
| RecG [19] | Unwinds fork structures | Yes | 32,397 |
| RecJ [27] | Exonuclease in RecF recombination and other DNA repair pathways | Yes | 30,763 |
| RecO [33] | Recombination mediator | Yes | 30,154 |
| RecQ [34] | DNA helicase involved in the RecF recombination pathway and inhibition of illegitimate recombination | Unclear | 38,045 |
| RnaseHI [25] | Endonuclease that removes RNA in RNA:DNA hybrids | Unclear | 1363 |
| Topoisomerase III [27] | Chromosome decatenation | Yes | 18,259 |
| Uracil DNA glycosylase [23] | Base excision repair | Yes | 53,102 |
a. This is not an exhaustive list and not all SSB interactome partners are present in all organisms. One example is the recombination mediator RecO.
b. For simplicity, only the E.coli names for proteins are listed. In organisms such as B. subtilis, protein names are different [15, 26].
c. Identified by an initial BLAST search and confirmed with Ensembl Genomes [35, 36].
d. The value shown is the number of genes present in the Ensembl Genomes database with the search limited to all bacterial species.
SH3, Src homology 3 domain; SSB, single-stranded DNA binding protein.
The activities of the interactome proteins must be temporally and spatially regulated as their binding to the DNA at the right time is essential to cell viability. This regulation involves the interaction of SSB with the inner membrane, itself, and separately, other interactome partners. Binding to the acidic phospholipids of the inner membrane sequesters excess SSB protein thereby minimizing spurious duplex DNA unwinding that could be deleterious to the cell [42, 43, 44, 45]. Localization is comparable to what is seen for DnaA, RecA, LacI, and the RNA degradosome [46, 47, 48, 49, 50, 51]. The interaction of SSB with itself is required for rapid and cooperative single-stranded DNA (ssDNA) binding which protects the exposed single strand from nucleolytic damage while simultaneously maintaining the nucleic acid as an optimal substrate for downstream processing. Finally, partner binding is necessary for dynamically mediating partner-DNA interactions essential to maintaining genome integrity (Fig. 1) [1, 2, 3, 52, 53, 54, 55, 56]. How a single protein can facilitate such a variety of interactions thereby enabling the maintenance of genome integrity is the focus of this review.
The prototypical Escherichia coli single-stranded DNA binding protein (SSB), like its homologs in other bacteria, is essential to all aspects of DNA metabolism, including the SOS response to DNA damaging agents [1, 37, 38, 57, 58]. SSB was originally discovered as the E.coli DNA unwinding protein by Sigal et al. [14] as a protein that resisted displacement from ssDNA cellulose by dextran sulfate but was displaced by 2M NaCl from the resin and formed complexes on ssDNA similar to those of bacteriophage T4 gene 32 protein. Seven years later, using a temperature-sensitive mutant defective for DNA replication at 42 °C, the Kornberg group identified the gene and renamed the protein, the single strand binding protein [59]. Sequencing of the gene subsequently revealed the now well-established 3 “regions or domains” common to most, if not all, bacterial SSB proteins (Fig. 2A, Ref. [19]) [15, 60, 61, 62, 63, 64, 65, 66, 67, 68]. These 3 regions known as the core, intrinsically disordered linker, and acidic tip or C-peptide, differ in their sequence conservation and pI, with the acidic tip having a conservation score of 9.33 out of 10 and a pI of 3.3; the N-terminal domain a score of 6 and pI of 8.01 with the linker having a conservation score of 2.44 and a pI of 9.9 [69, 70, 71].
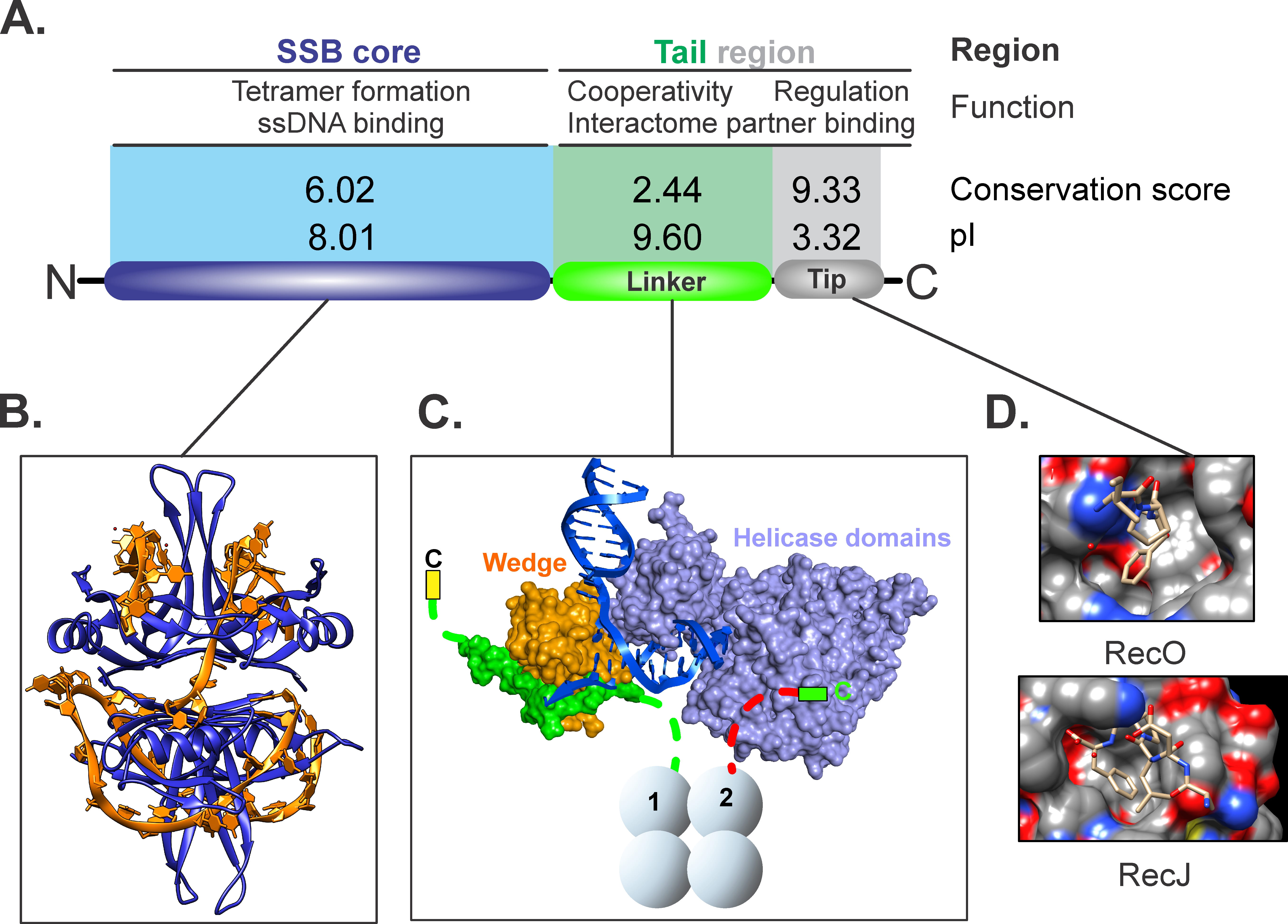 Fig. 2.
Fig. 2.
The three domains of SSB are critical for protein function. (A) The 3-domain structure of an SSB monomer is shared among prokaryotic homologs. (B) The OB-folds of the SSB core forms an intimate complex with ssDNA wrapping the nucleic acid around the tetramer. (C) The linker binds to the OB-fold present in the partner, here RecG. For this DNA helicase, the stoichiometry of binding is 2 RecG molecules per SSB tetramer [19]. In this complex, primary binding is mediated by the linker of one SSB monomer binding to the OB-fold of RecG leaving its acidic tip exposed. The linker of a second monomer directs its acidic tip to bind to a second, distant site, possibly stabilizing the complex (Bianco, unpublished). (D) Acidic tips can bind to pockets in partners. Here two representative binding pockets are shown. The figure was made using Corel Designer v2018.
The positioning of the three domains of SSB is: (I), the N-terminally positioned
core that contains the OB-fold and binds competitively to ssDNA, the linker
region of the other SSB tetramers or, the acidic phospholipids of the inner
membrane, and, it also contains the sequence elements required for tetramer
formation (Fig. 2B); (II), the almost centrally placed linker that is essential
for cooperative ssDNA binding, partner OB-fold binding, and, for the release of
tetramers from ssDNA (Fig. 2C); and (III), the C-peptide, often referred to as
the acidic tip that is predominantly
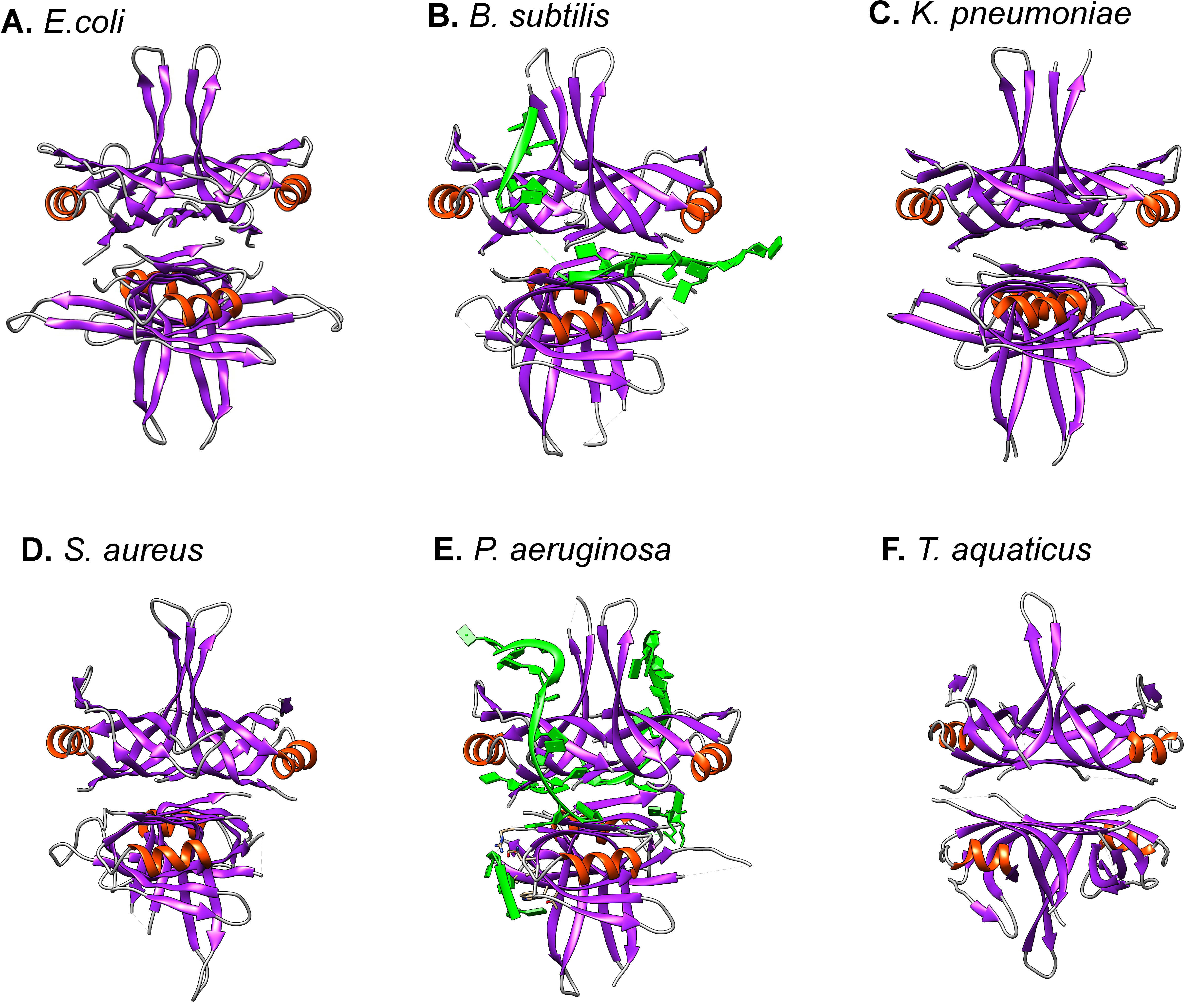 Fig. 3.
Fig. 3.
The overall structure of SSB proteins is conserved. The
structures of six proteins are shown. For structures in panels (A–E) the SSB is
a homo-tetramer. (F). For T. aquaticus, the protein is a homodimer,
containing 4 OB-folds. Images were generated using UCSF Chimera with
Although SSB was known to bind ssDNA, the first detailed characterizations showed it was a tetramer that bound to both RNA and DNA with negligible binding to duplex DNA [92, 93, 94]. An apparent equilibrium dissociation constant of 2 nM for ssDNA was obtained and the protein was also shown to bind cooperatively to the nucleic acid lattice. In addition, Molineux and Gefter demonstrated the first complex formation of SSB with a partner protein, DNA polymerase II [95]. Binding occurred in the absence of DNA, facilitated the association of DNA polymerase II with the nucleic acid lattice, and enhanced the DNA synthetic ability of the enzyme. Over the years the number of binding partners for E.coli SSB has increased to twenty and this family of proteins is now known as the SSB interactome [1, 3, 53, 54]. There are homologs for each interactome partner in other bacteria, although the way the cognate SSB binds to the partners varies [87, 96, 97]. Since the initial publication by Molineux and Gefter in 1974, elucidating the mechanism of both ssDNA and partner binding has required an additional almost 50 years of research.
SSB proteins wrap ssDNA around themselves, resulting in a decrease in the nucleic acid length [93, 98, 99, 100, 101]. The mechanism for binding was revealed from crystal structures (Fig. 1 and Fig. 2B, Fig. 3B,E) [5, 56, 76, 102]. Here the four OB-folds form an intimate association with the nucleic acid, which binds within the “mouth” of the fold. DNA binding by this domain is non-specific and the wrapping of ssDNA around the SSB tetramer utilizes an extensive network of electrostatic and base-stacking interactions with the phosphodiester backbone and nucleotide bases, respectively. This non-specific binding mechanism appears to be generally conserved being observed for at least the SSB proteins from Bacillus, Coxiella, Deinococcus, Helicobacter, Herbaspirillum, Klebsiella, Mycobacteria, Pseudomonas, Salmonella, Staphylococcus, Streptomyces, Streptococcus and Thermus [64, 66, 88, 91, 101, 103, 104, 105, 106, 107, 108, 109, 110]. However, the details of the interaction with the DNA show intriguing differences. For PaSSB the ssDNA binding contributions from aromatic residues are significantly different from that of EcSSB and surprisingly, even though the protein is a homotetramer, only two OB-folds are required to form a stable DNA protein complex (Fig. 3E) [101].
The discovery of the interactome was made in Bacillus subtilis [2, 53].
SSB was shown to be responsible for directing multiple repair proteins to damaged
DNA including RecA, PriA, RecG, and RecQ. This activity is dependent on the
presence of the C-terminal 35 residues of BsSSB as when these amino acids are
deleted (ssb
While the mechanism of ssDNA binding by prokaryotic SSB proteins is well
established, the mechanism of partner binding and their regulation has been more
controversial. Included in this list of partners are DNA polymerase II, the Chi
subunit of DNA polymerase III, Exonuclease I, PriA, PriB, RecG, RecJ, RecO,
Red
The earliest proposal for the tip model came from the work of Curth [111]. The
first biochemical evidence came from the work of the O’Donnell group which showed
in vitro that the SSB mutant SSB-113 (P176S) had a reduced affinity for
the psi subunit of the gamma complex clamp loader [16]. They also showed that a
peptide consisting of the 15 C-terminal residues of SSB bound to psi. Similarly,
work from Curth’s group showed that the C-terminal 26 amino acids of SSB were
required to bind the chi subunit of the DNA polymerase III complex [119].
Subsequent work from several groups has obtained structures of portions of the
C-peptide bound to a partner protein (Fig. 2D) (for review see [54]). In
addition, multiple groups have performed extensive biochemical and biophysical
studies of the binding of a peptide comprised of the C-terminal 8–10 residues of
SSB and a partner (reviewed in [54]). Separately, other groups have utilized
intact, mutant SSB tetramers and analyzed their binding to partners both
in vivo and in vitro [19, 117, 120]. These mutants include
SSB-113 (P117S) and SSB
Several groups have assessed the ability of acidic tip peptides to compete with full-length EcSSB for partner binding in activity assays. For DNA polymerase IV, 10- to 15,076 molecules of peptide per polymerase were required for inhibition of DNA synthesis [20]. For Exonuclease I, 106 molecules of peptide per enzyme molecule, were required to reduce nuclease activity to levels observed in the absence of SSB [121]. Thus while there is competition for binding, extremely high concentrations of peptide are essential for inhibition to be observed. Under these conditions, the 1300 Da peptide, which has a pI of 3.32, can in principle compete for binding at the tip docking site on the partner as well as at the SSB and partner OB-folds where ssDNA binding occurs. The combination of the binding of a large excess of peptide to multiple targets would then result in the observed inhibition.
Although the acidic tip of SSB is the most highly conserved part of the protein, the docking sites in partners are not well conserved (Fig. 2D). They do have in common a basic lip element, a basic ridge, and a hydrophobic pocket [54]. As the C-peptide is the most well-conserved sequence among prokaryotic SSB proteins, further analysis of these pockets is critical to understanding how this unique sequence in SSB can interact with so many different proteins.
To date, 10 different partner proteins complexed with the tip are available in the PDB, and this information will help reveal the binding environment and the relative contributions to complex formation. For the 10 available structures, hydrogen bonding (Fig. 4A, Ref. [122, 123]) and hydrophobic interactions (Fig. 4B) were examined. Surprisingly, the results show that the number of interactions observed in the crystal structures is limited, as indicated by the large number of “NI” (no interaction) entries. This is consistent with the weak binding affinity observed in assays. For example, only one tip residue is found to interact with PriA, and this interaction is subunit-dependent: chain A shows no interaction, while R699 of chain B interacts with D of DDDIPF, and R697 of chain E interacts with F of DDDIPF. Surprisingly, the structure of tri-D in the DDDIPF (the main reason for naming the acidic tip) complexed with a partner has not been solved, which is consistent with a dynamic and unstable interaction. In addition, partner interactions with the tip are not conserved among partners, even within the same protein from different species. For example, in uracil DNA glycosylase, the terminal F in EcSSB is critical for hydrogen bonding for binding, whereas the same residue in DrSSB does not contribute to hydrogen bonding. Collectively, these data suggest that the C-peptide binding alone may be insufficient to mediate stable SSB-partner complex formation while simultaneously regulating the activity of the bound partner.
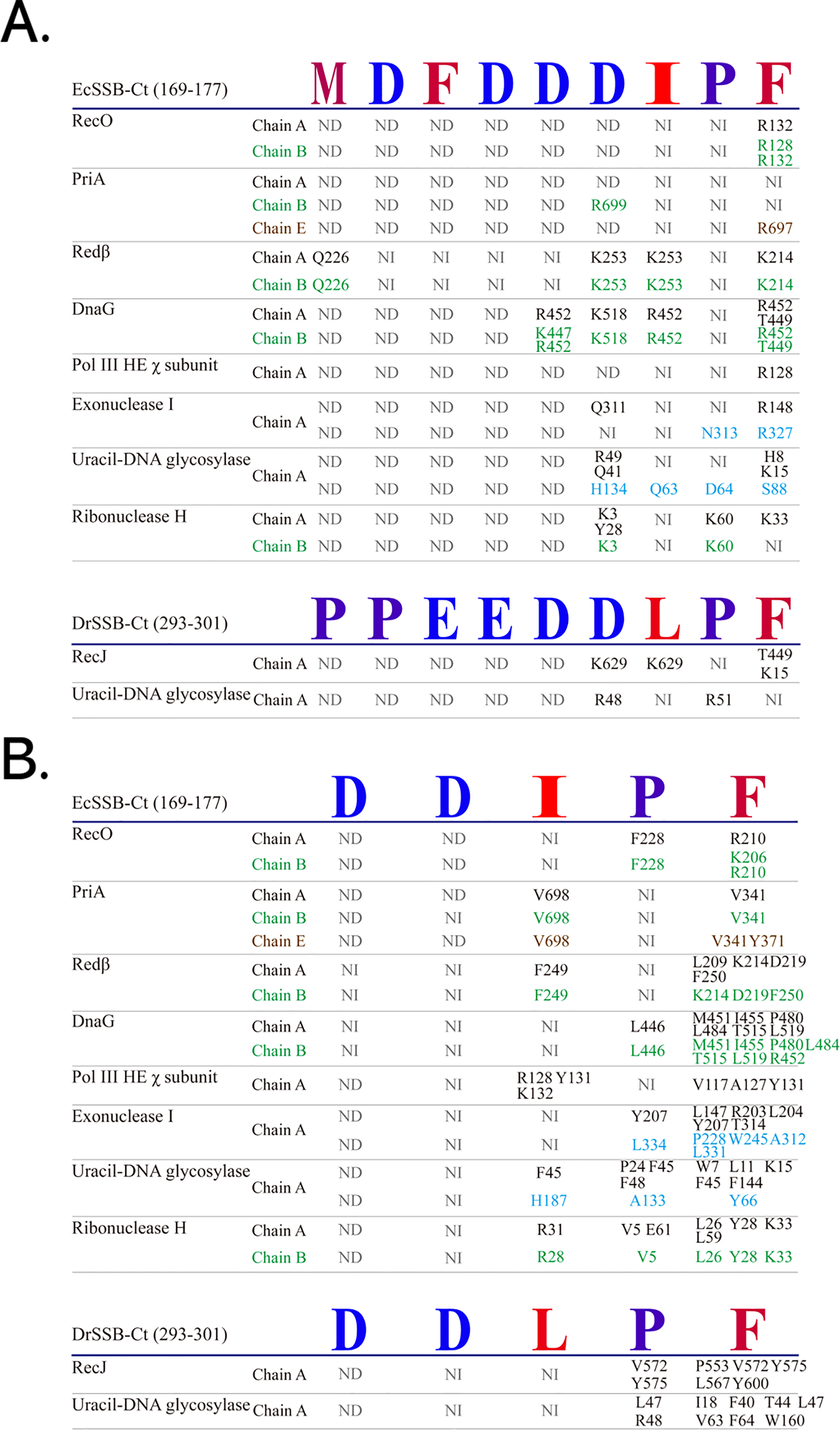 Fig. 4.
Fig. 4.
Analysis of tip binding sites. Ten structures of proteins complexed with the C-peptide (EcSSB-Ct or DrSSB-Ct) in the PDB were analyzed. (A) Hydrogen bonding between the protein and the C-peptide in these structures was evaluated using the PISA (Protein Interfaces, Surfaces, and Assemblies; https://www.ebi.ac.uk/pdbe/pisa/) software [122]. (B) Hydrophobic interactions were assessed using the PLIP (the protein–ligand interaction profiler; https://plip-tool.biotec.tu-dresden.de) software [123]. Interactions in different subunits are colored differently. The most hydrophobic residues are colored red, and the most hydrophilic ones are colored blue. NI, no interaction. ND, structure not determined. The figure was made using Corel Designer v2018.
One unique protein, when considering the EcSSB tip structure, is worth mentioning here: myosin-2 (PDB ID 2XO8) [124]. The C-terminal domain of EcSSB (S164-F177), including the tip, was fused to this myosin-2 protein at the C-terminus and used as a purification tag. The C-terminal domain of SSB proteins, including EcSSB, is generally flexible and unobserved in X-ray structures; however, it is resolved in this myosin-2-EcSSB-S164-F177 structure (Fig. 5). Despite this, the EcSSB-S164-F177 region in this structure does not interact with myosin-2. Whether the flexibility of this C-terminal region of EcSSB is specific to SSBs when present in the tail (e.g., interactions that drive its movement in SSBs or other OB-fold proteins) should be further investigated biochemically and structurally.
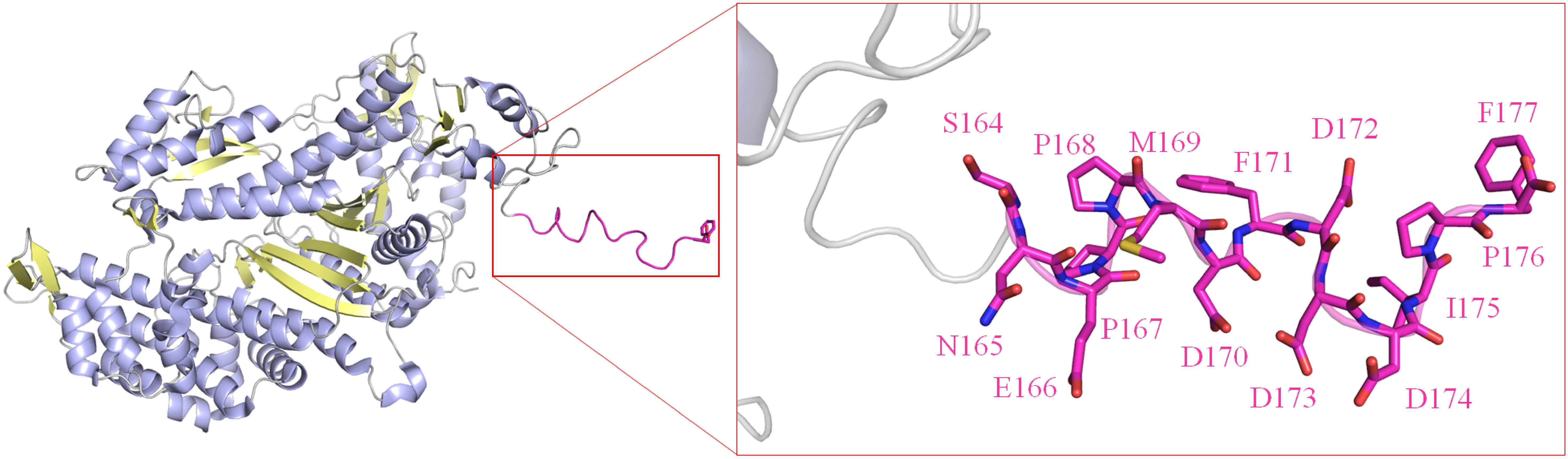 Fig. 5.
Fig. 5.
The structure of a myosin II-acidic tip fusion reveals the
structure of the C-peptide. In this structure (PDB ID 2XO8), the
Intrinsic to the tip model is the positioning of the acidic tip at the C-terminus of the protein. This enables the terminal phenylalanine to bind in the base of the pocket in the partner (Fig. 2D). While the positioning of this acidic sequence at the C-terminus occurs in a significant number of SSB sequences, it is not always the case including in the sequences of multiple E.coli SSB proteins (Fig. 6A, Ref. [69]). Analysis of 456 EcSSB sequences reveals that 161 proteins contain the acidic tip sequence N-DDDIPF-C. Of these, 23% contain additional sequence downstream of the acidic tip. Further, alignments show that the presence of additional residues downstream of the “acidic tip” sequence occurs in the SSB proteins from multiple bacterial species (Fig. 6B). These analyses indicate that it is the presence of the sequence that is critical for function, not its positioning at the extreme C-terminus of the protein. It is not surprising that at neutral pH the binding of the C-peptide to the EcSSB OB-fold is very weak with a Kd = 18.8 mM [81, 125]. This led to a proposal of a model by the Dixon group that instead of being the primary partner-binding site, this acidic sequence utilizes long-range electrostatic effects to regulate EcSSB functions [125]. A similar regulatory role for the acidic tip was proposed for Streptomyces coelicolor SsbA [126].
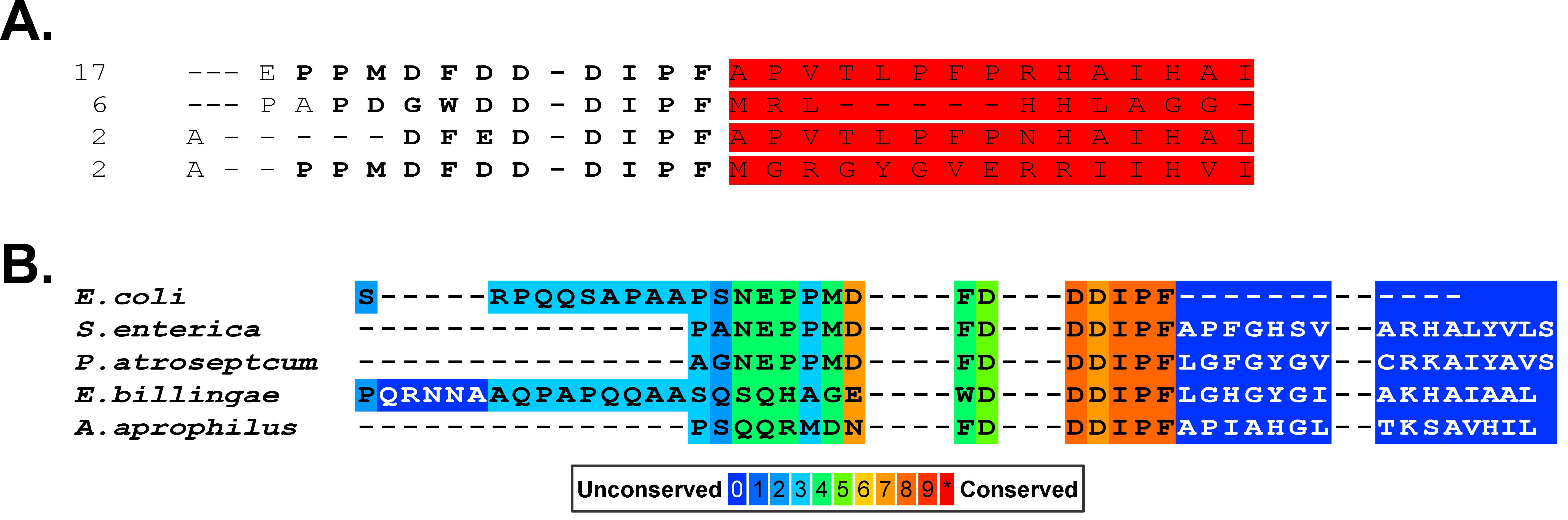 Fig. 6.
Fig. 6.
The presence of the acidic residue patch is not always at the extreme C-terminus of prokaryotic SSB proteins. (A) An alignment of 467 EcSSB sequences was done and those with tails longer than the canonical sequence are shown. The regions in red are the additional sequence downstream. The numbers to the right of each sequence indicate the number of times this extension appeared in the alignment. (B) Representative SSB tail sequences taken from an alignment to show the variation in sequences positioned after the acidic stretch. The sequence alignment was done with PRALINE [69]. The figure was made using Corel Designer v2018.
Instead of the acidic tip or C-peptide functioning as the primary partner binding domain, the linker/OB-fold model provides a convincing rationale for the mechanism of action of SSB and its cross-talk with the interactome members containing OB-folds (Fig. 1 and Fig. 2C) [3, 84]. Here the interface between SSB and a partner is comprised of the PXXP motifs of the linker on one side and a structurally conserved OB-fold on the other. It is well known that the primary amino acid sequence of OB-folds is not conserved [127]. However, the structure is conserved and more than 80% of interactome partners contain OB-folds [3, 127, 128, 129, 130].
This mechanism of linker/OB-fold binding is comparable to the binding of
PXXP-ligands by SH3 domains in eukaryotic systems [131, 132, 133]. The importance of
this result for interactome function became clear when it was realized that SH3
domains are structurally almost identical to OB-folds [134]. When these domains
are superimposed, they differ by less than 2Å for the
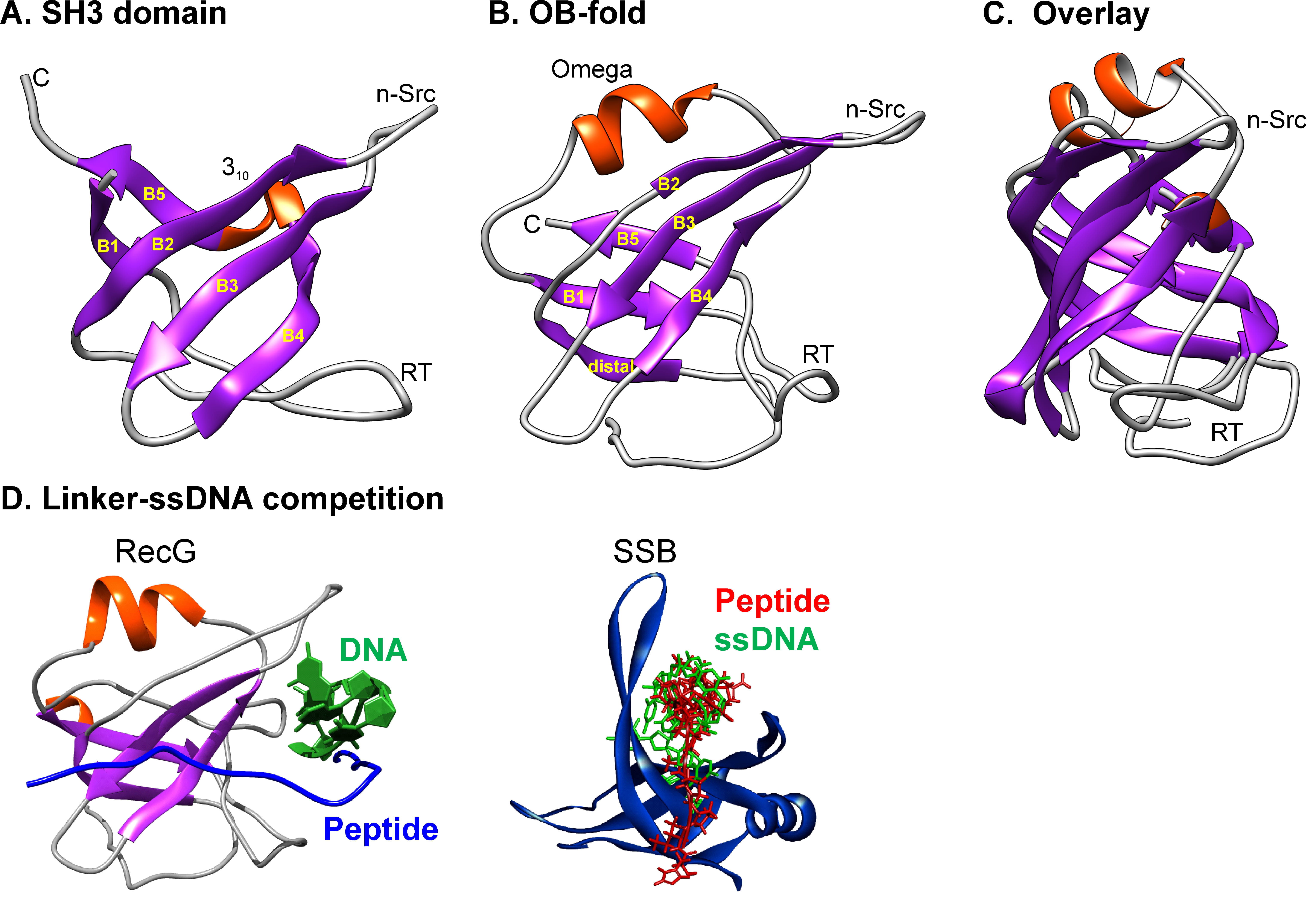 Fig. 7.
Fig. 7.
The similarity in structure of SH3 domains and OB-folds provides a mechanism for SSB interactome regulation. (A) The SH3 domain form PDB file 2KXC. (B) The OB-fold from 1GM5. (C) Structural alignment was done with TM-align. (D) The structures of the OB-folds of two interactome members are shown with the DNA and aligned PXXP ligand indicated. DNA and ligand (SSB linker) occupy the same or overlapping binding site suggesting competitive binding and a mechanism for interactome regulation. Adapted from [71, 128]. The figure was made using Corel Designer v2018.
Evidence for this model comes from both in vivo and in vitro studies, using full-length SSB proteins, not isolated peptides, which can behave differently than when attached to the rest of the protein. The first requirement for this model is that an OB-fold be present in the partner. To test this, the OB-folds in PriA, RecG, and RecO were deleted resulting in a loss of SSB binding in vivo [79]. Next, carefully selected residues in the RecG OB-fold were mutated, and SSB binding in vivo was assessed. The results showed that mutation of amino acids in the ssDNA binding site in the OB-fold of RecG eliminated SSB binding in vivo [6]. Notably, RecG F97 mutants which are defective in vivo are unable to bind either SSB or DNA in vitro, consistent with the model that these binding sites overlap and that DNA and SSB binding is competitive [143, 144]. Additional support for this competition comes from an atomic force microscopy study that showed that during loading of RecG by SSB, the RecG OB-fold which is essential for fork binding is occluded and the helicase domains of the enzyme are used to bind to parental duplex DNA [6, 139, 144]. The binding of the acidic tip to RecG at a site distant from the OB-fold cannot explain the loading and remodeling of the helicase mediated by SSB nor does the acidic tip occlude the OB-fold to ensure DNA binding is provided by the helicase domains [145].
The second requirement for this model is that the linker contains the necessary information for partner binding. To test this, deletions in the EcSSB linker were made and the binding to RecG and separately, RecO was assessed in vivo [84]. Although each linker deletion mutant retained the acidic tip, they were unable to bind to either partner. To further define the SSB side of the linker/OB-fold interface, single residues were mutated. This included the insertion of an asparagine into the center of each motif as this disrupts spacing between the prolines of the PXXP motif and separately, the mutation of the proline residues. Results show that the insertion of Asn into motifs I and III or mutation of the first motif proline results in loss of RecG binding in vivo [79]. This also produces a 3-fold increase in Kd in vitro (Bianco, unpublished). Asn insertion in motif I also eliminated cooperative ssDNA binding by SSB, indicating that the linker/OB-fold mechanism applies to the essential protein regulating interactome function as well [79]. The addition of a single residue in the linker resulting in loss of binding demonstrates that the PXXP motifs are critical for OB-fold binding, similar to what is observed for SH3 domains [146]. This result also shows that the intrinsically disordered linker is not an unimportant spacer sequence. Using a two-hybrid approach, E.coli Alkylation protein B was also shown to require the linker region to bind to SSB and not the acidic tip [21]. A similar conclusion was reached for EcPriA as explained in more detail below [88]. These data demonstrate the linker/OB-fold model applies to SSB partner binding and interactome regulation.
A recent in vivo study found that the SSB linker was not required for protein function only the acidic tip [145]. The implications of this finding are unclear for several reasons. First, SSB proteins containing deletions of the linker as short as 20 residues but with functional acidic tips, do not bind interactome partners in vivo [84]. Second, point mutations in the linker eliminate RecG binding in vivo [79]. Therefore, the linker is essential for partner binding, consistent with the linker/OB-fold model. Consequently, an alternative explanation must apply to explain the data of Bonde et al. [145]. In this study, cells were grown in Luria Broth, which contains 86 mM NaCl, and, the strains used were wild type for Rep, an accessory helicase at DNA replication forks [147, 148, 149]. The presence of NaCl in the media and rep+ have been shown to separately suppress ssb mutations and Rep binds to SSB in vivo [150, 151, 152]. Further, acidic tip peptides bound to beads were able to pull down Exo I and separately, RecO when added to cell lysates [153]. However, full-length SSB proteins do not bind partners when lysates are mixed [120]. Instead, linker/OB-fold binding of SSB to RecG, PriA, or both, occurs only in vivo and not when lysates containing each protein expressed at high levels are mixed [120]. Thus the linker deletion strains used by Bonde et al. [145] may be producing truncated SSB proteins that act as “in vivo beads” comparable to those used by Tököli et al. [153], presenting as many as 8000 unregulated acidic tips, capable of binding multiple proteins with the ssb phenotype suppressed by a combination of the media and Rep helicase [153]. Finally, mutations in the RecG OB-fold that eliminate SSB binding are also defective for DNA repair in vivo and this defect is independent of the growth medium and the presence of Rep [79, 144]. Thus binding of the SSB linker to partner OB-folds is intrinsic to interactome function.
Alignments of bacterial SSB sequences show that significant homology is restricted to the N-terminal 1~120 residues and the C-terminal 8–10 amino acids [1, 54, 71, 73, 154, 155]. As there is minimal homology in the intrinsically disordered linkers in these alignments, only the C-termini of 251 Proteobacterial SSB sequences was done. A summary of the alignment beginning at residue 110 (E. coli numbering) is shown in Fig. 8A (Ref. [71, 156]).
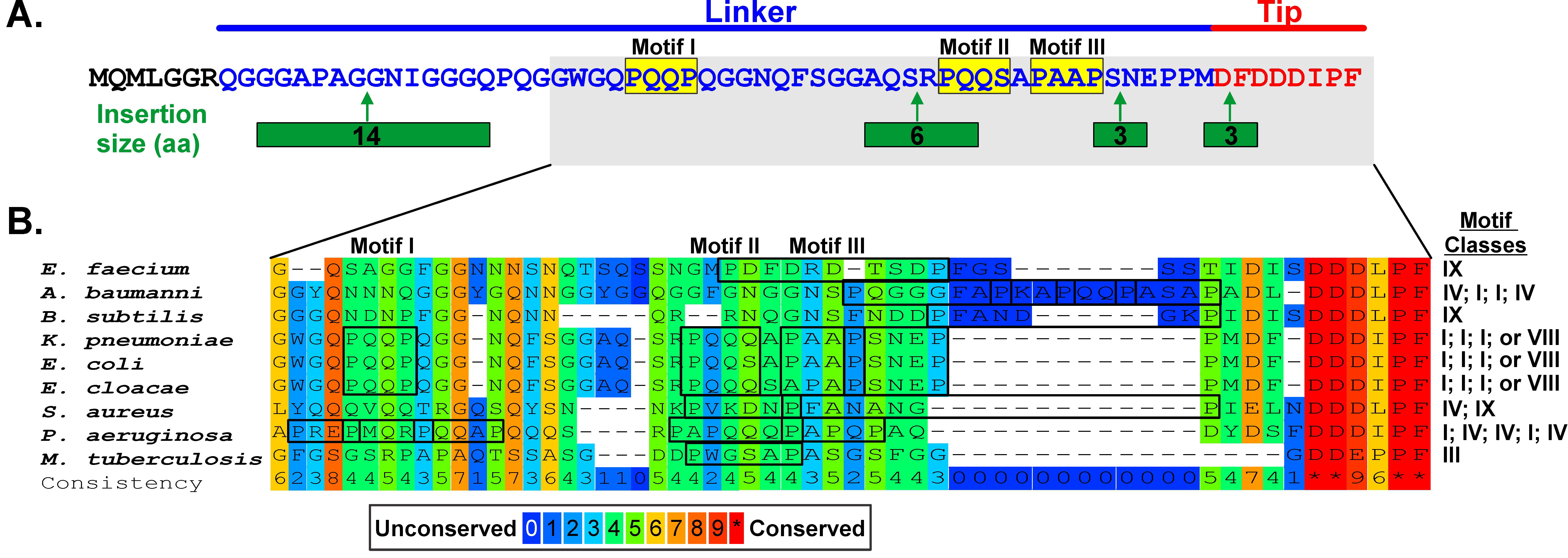 Fig. 8.
Fig. 8.
SSB linkers contain genus-specific PXXP motifs. (A) The result from an alignment of 250 Proteobacterial sequences is shown to indicate how insertion can produce poor alignments. The sequence of EcSSB is shown. This is adapted from [71]. (B) An alignment of the representative SSB proteins from clinically relevant and laboratory model organisms was done with PRALINE. Only the tails are shown with the positions of EcSSB motifs indicated as a reference. Potential PXXP motifs are highlighted in boxes and their classification according to Teyra indicated to the right [156]. The figure was made using Corel Designer v2018.
This analysis shows that for the entire C-terminal domain, sequences are 40% identical. As expected, the homology is highest within the first seven and last 5 residues and D171, corresponding to the C-terminal end of the core domain and acidic tip, respectively. Between these conserved ends, it initially appears that there is little homology. However, this is due to the presence of as many as four insertions. When the alignments are corrected for the insertions, the presence of canonical PXXP motifs is revealed [71]. When alignments are extended to include additional gram-negative and also gram-positive bacteria the presence of PXXP motifs becomes evident, although at first glance the canonical motif is present in only four of the 9 tails shown (Fig. 8B).
Using the classification of Teyra who identified 9 classes of “PXXP” motifs in eukaryotic SH3 domain ligands, different classes of motifs in ESKAPE-SSB linkers can also be identified, with the number and class of motifs ranging from one (E. faecium; class IX) to five for P. aeruginosa [156]. Collectively, this brief analysis suggests that the linker/OB-fold model may apply to multiple organisms including clinically relevant bacteria such as Mycobacterium and the ESKAPE pathogens, with a single SSB binding to multiple partners, each of which contains OB-folds like their E. coli homologs [1, 3]. Here, and as shown for EcSSB, different motifs within the linker enable binding to different partners. This makes sense as the structure of the OB-fold is conserved but the sequence is not [127, 128, 157]. More work is required to determine if this model applies to all prokaryotic SSB interactomes.
For the linker/OB-fold model to apply, specificity must be afforded by sequence elements present in this region of SSB proteins. In E.coli, SSB interacts with PriA both physically and functionally to stimulate the ATPase and helicase activity of the enzyme [22, 120, 140, 158, 159]. This interaction involves linker/OB-fold binding and is genus-specific as shown for Klebsiella pneumoniae PriA [79, 88]. KpSSB and Salmonella typhi SSB stimulated KpPriA whereas Pseudomonas aeruginosa SSB did not (Fig. 9A, Ref. [88]). As the acidic tip sequences of these SSB proteins are almost identical, the ability of an SSB to function in this reaction is independent of the tip (Fig. 8 and Fig. 9B,C). Instead, the ability of an SSB to stimulate PriA is dependent on the intrinsically disordered linker. This follows because the PXXP motifs in Ec-, Kp-, and StSSB are identical, whereas PaSSB motifs are unique. Similarly, in a study of the ability of uracil DNA glycosylase (UDG) to remove uracil from a hairpin oligonucleotide containing dU at the second position in a tetraloop, only genus-specific SSBs stimulated their cognate UDG partners [160]. Even though Mycobacterium smegmatis SSB (MsmSSB) and MtuSSB are 86% identical, they do not enhance the activity of Mtu-, Msm- and EcUDG, respectively. As the acidic tips of Mtu- and MsmSSB are identical, the differences in their ability to stimulate cognate partners can be attributed to the variable sequence, linker regions in the SSB proteins (Fig. 9C). This results in a two-orders of magnitude lower affinity of MtuSSB for EcoUDG. Finally, in vivo, T. maritima RecG does not bind to EcSSB and, TmSSB does not bind to either EcRecG or EcRecO [84].
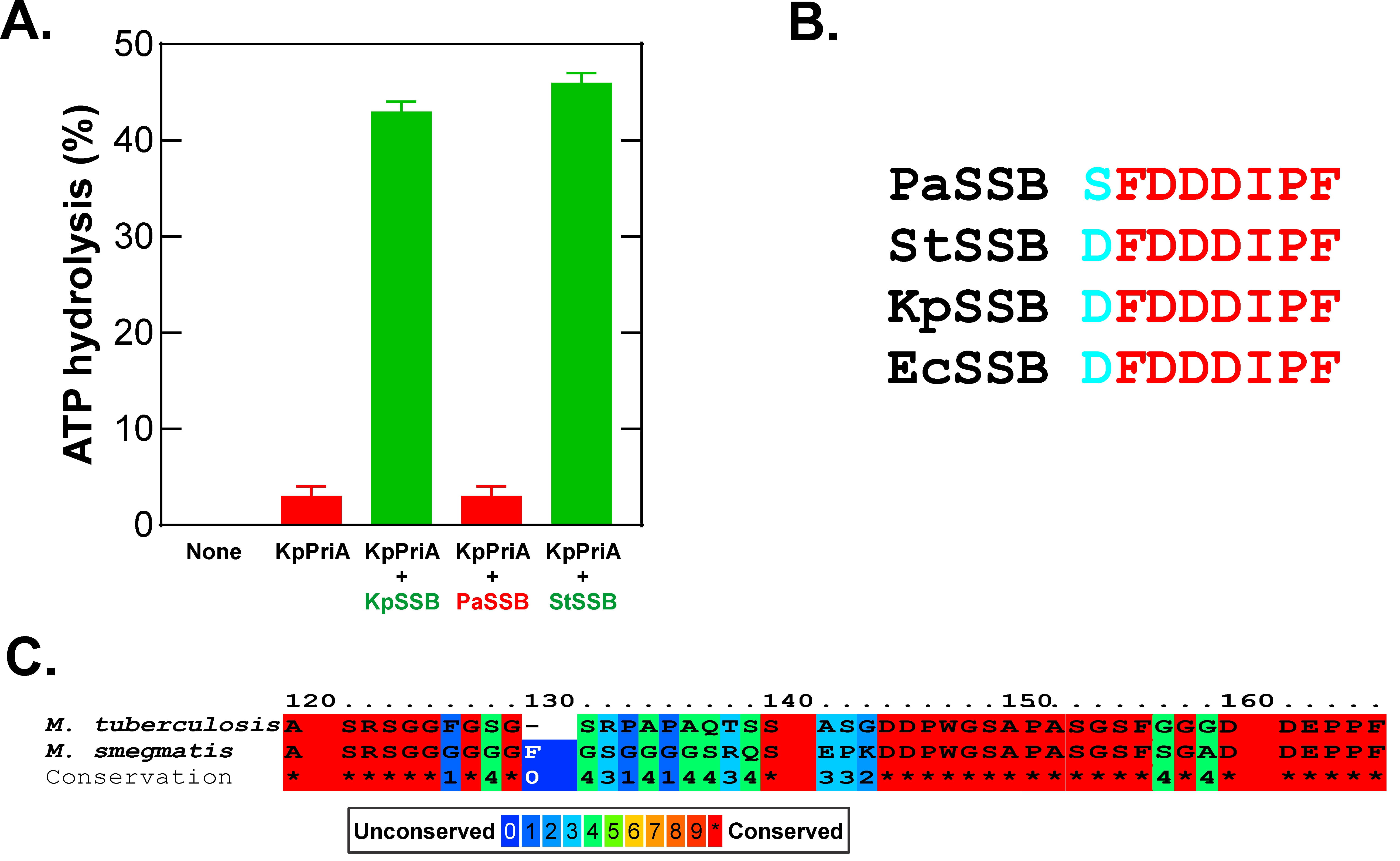 Fig. 9.
Fig. 9.
The linker/OB-fold mechanism is genus-specific. (A) Stimulation of KpPriA depends on the sequence of the linker of the SSB protein. Adapted from [88]. (B) The sequence of the acidic tips of the SSB proteins used in A. (C) An alignment of Mtu- and MsmSSB proteins was done and the C-termini are presented to highlight the differences in the linker regions while the acidic tips are virtually identical. The figure was made using Corel Designer v2018.
Further insight into the role of linker/OB-fold interactions in SSB interactome
function comes from studies using chimeric SSB proteins, consisting of the
OB-fold domain of one species and the tail of another. Kp/St-SSB stimulated PriA
whereas Kp/Pa-SSB did not [88]. Separately, chimeric SSB proteins were
constructed by exchanging parts of the MtuSSB linker with that of E.coli [23, 84, 161, 162]. As the acidic tips are almost invariant, dramatic changes in the
linker sequence result in an inability to complement
As the C-peptide has a critical role in regulating SSB tail function using
long-range electrostatic effects, it is not surprising that removal or mutation
of the acidic tip has profound effects on SSB. First, removal virtually
eliminates binding to target proteins [84, 120]. This has misled the field as
mutation of the tip inactivates SSB as the loss of electrostatic control enables
linkers to bind SSB OB-folds instead (see below). Second, it affects cooperative
binding to ssDNA [73]. Third, it affects the stability of the SSB tetramer.
In vitro in 10 mM ammonium acetate, SSB
As alluded to above, mutation or deletion of all four aspartates has dramatic
effects on the protein. The pI of SSB increases from 5.4 to 9.1, while that of
the C-terminal domain increases from 3.7 to 6.0. The loss of 80% of the charged
residues in the C-terminal tail allows the linkers to bind SSB OB-folds where
they are sequestered, cannot bind to partner proteins, and, are now more
efficient competitors for ssDNA binding. This is most clearly observed for the
SSB D4A4 protein which requires a 4-fold higher amount of ssDNA is
required to fully occlude the OB-folds, resulting in an increase in site size
from 10 nt/monomer for wild type to 35 [79]. This mutant, like SSB
This behavior of acid tip mutants is consistent with a model whereby the binding
of the intrinsically disordered linker to SSB OB-folds is in equilibrium with the
unbound state, with control afforded by the acidic tip. For WT at pH 7, the
equilibrium is in the direction of the unbound state [125]. For the mutants, the
bound state is favored [79]. When ssDNA binds, it outcompetes linkers for OB-fold
binding, exposing C-termini so that binding to partners can take place [74, 165].
Evidence for the conformational change is supported by data showing that the
binding of poly d(T) to SSB increases the sensitivity of the tail to proteolysis
[74, 85]. Furthermore, ssDNA binding to SSB enhances the affinity of the protein
for PriA and the
Finally, the addition of ssDNA to several SSB tip mutants restores RecG binding to wild-type levels by outcompeting linkers for binding to SSB OB-folds [79]. Therefore, while the acidic tip in E.coli SSB is not essential for partner binding, it has a critical role in C-terminal tail regulation and it functions as a secondary binding site. In the latter role, it may stabilize the interaction between SSB and partners other than itself. Consistent, the stoichiometry of binding of Exo IX and separately, RecG to SSB, is 2 per tetramer [19, 168]. For RecG this involves concurrent binding of the linker of one SSB subunit to the OB-fold of RecG and the acidic tip of SSB to a binding site located in the helicase domains of RecG (Bianco and Zeczycki, unpublished).
Unlike E. coli, which possesses a single type of SSB [54], many Gram-positive bacteria harbor two paralogous SSBs: SsbA and SsbB [169]. SsbA exhibits strong sequence similarity to E. coli SSB, particularly in the N-terminal DNA-binding domain and the C-terminal acidic tail, positioning it as a functional counterpart. While crystal structures suggest that both SsbA and SsbB bind ssDNA in a similar manner, their DNA-binding properties are distinct [15, 170, 171, 172]. Notably, several SsbBs lacks the linker and C-peptide that are typically involved in protein-protein interactions. For instance, Bacillus subtilis SsbB is missing the entire C-terminal domain (66 amino acid residues) present in SsbA (Fig. 10). In addition to B. subtilis, SsbB from Streptomyces coelicolor (150-PDPVPVG-156-C terminus) and Staphylococcus aureus (135-DFSDLPF-141-C terminus), but not Streptococcus pneumonia (125-EEEELSF-131-C terminus), also lack the recognizable C-terminal acidic tail of SSBs. Given that SsbB is known to interact with several partner proteins involved in chromosome segregation and transformation, the absence of this tail indicates that SsbB may interact with partner proteins in a unique way [170, 173]. This raises the possibility that its OB-fold in the N-terminal DNA-binding domain could play a role in these protein interactions. As little is known about the SsbB-partner binding, we instead focus below on the primary SSB from Bacillus subtilis, Deinococcus radiodurans, Staphylococcus aureus, and Mycobacterium tuberculosis, and how they interact with partner proteins.
 Fig. 10.
Fig. 10.
Alignment of SSBs from Gram-positive bacteria reveals unique C-terminal tails. The sequences of SaSsbA, SaSsbB, BsSsbA, BsSsbB, MtSSBa, and DrSSB were compared using CLUSTAL O. Despite being an SSB, BsSsbB lacks the typical C-terminal domain, including the linker and recognizable C-terminal acidic tail found in other SSBs. The figure was made using Corel Designer v2018.
B. subtilis is considered the best-studied Gram-positive bacterium and a model organism for studying bacterial chromosome replication and cell differentiation [174]. The SsbA protein from B. subtilis consists of 172 amino acids (BsSsbA), with a linker and a C-terminal acidic tail (DDDLPF) similar to EcSSB. Also like E. coli, BsSsbA can enlarge open complexes through coordinated action with the replication initiation protein DnaA at oriC (origin of replication). However, despite the conservation of DnaA in both organisms [175], the B. subtilis DnaA protein is toxic to the E. coli DnaA protein, and these proteins cannot be swapped for replication, suggesting a highly species-specific process [176].
For the replication restart initiation protein PriA, EcSSB physically interacts with this partner via the C-terminal acidic tail or the linker, enhancing PriA’s catalytic activity [22, 79, 114, 120]. In contrast, when BsSsbA is present, BsPriA shows a preference for binding to the ssiA sequence (the primosome assembly site) over other ssDNA sequences, but this SSB does not enhance PriA’s activity [177]. This indicates that BsSsbA-PriA binding may facilitate or enhance PriA specificity only. Many DNA binding properties of B. subtilis PriA, compared to E. coli PriA, such as displaying a higher affinity for ssDNA (with a 5-fold lower Kd value), differing ion effects on binding, and distinct binding behavior as judged by EMSA, suggest that the B. subtilis PriA-SsbA interaction is different from the E. coli system. This may partly explain why B. subtilis PriA does not complement a priA null mutant and cannot substitute for priA in vivo in E. coli [177]. Through green fluorescent protein (GFP)-facilitated fluorescence microscopy, the B. subtilis PriA-SSB interaction has been identified and shown to be mediated by the C-terminal region (35 amino acids) of BsSsbA, which is crucial for the localization and function of PriA at the replication fork [2]. If these 35 C-terminal amino acids of BsSsbA are deleted, B. subtilis PriA fails to interact and localize properly at the replication factory [2]. Thus, not only the C-terminal acidic peptide but also other regions in the C-terminal domain of BsSsbA, such as the flexible linker, may participate in interactions with B. subtilis PriA.
Not only PriA but also the replication fork reactivation SF2 (Superfamily 2) helicases, RecQ and RecG, interact with BsSsbA [2]. Co-purification experiments strongly support that the C-terminal domain of BsSsbA can directly interact with these two helicases. BsSsbA also directly interacts with B. subtilis RecD2 helicase [178]. The C-terminal domain of BsSsbA appears to serve as a recruitment platform for proteins, including DnaE, SbcC, RarA, RecJ, RecO, XseA, Ung, YpbB, and YrrC [53]. RecJ and RecO are part of the RecFOR pathway, a conserved DNA repair mechanism across bacteria that facilitates the loading of RecA onto ssDNA [179, 180]. RarA, SbcC, and XseA are also present in E. coli, suggesting that some interactions between BsSsbA and these proteins are conserved in E. coli. However, while the DNA damage-inducible DNA helicase DinG can form a stable complex with SSB in E. coli, PcrA, and DinG DNA helicases do not seem to belong to the SSB interactome in B. subtilis [24]. The essential DNA polymerase DnaE, as well as YrrC and the YpbB-RecS complex (RecS, a paralogue of RecQ not found in E. coli [181]), interact with BsSsbA and appear to be specific to B. subtilis [53, 182]. Additionally, given that the E. coli RNase HI-SSB interaction facilitates the hydrolysis of RNA in RNA/DNA hybrids to reduce replication fork stress, and considering that many Gram-positive bacteria, including B. subtilis, lack RNase HI, it suggests that B. subtilis has a somewhat different recruitment platform for proteins through BsSsbA compared to the SSB interactome in E. coli [25, 26, 183, 184, 185].
The C-terminal tail of BsSsbA interacts with RecO, and this interaction is crucial for RecO’s role in promoting the nucleation of RecA onto ssDNA, a necessary step in DNA recombination and repair [15, 186, 187, 188]. Notably, BsSsbB lacks the C-terminal tail and is unable to mediate the RecO recruitment process. However, despite the artificial insertion of the nine C-terminal residues of BsSsbA into BsSsbB, this modification is insufficient to promote RecO interactions in the same manner as SsbA, suggesting that SsbA interacts with RecO through more than just its C-terminal tail [15]. Given that DprA (DNA processing protein A) can interact with both SsbA and SsbB and recruits RecA onto ssDNA, it is likely that regions in the N-terminal OB-fold domain of these SSB proteins also interact with DprA, contributing to the mediation of nucleation activity [189, 190].
The bacterium Deinococcus radiodurans is the best-known extremophile
among the few organisms capable of surviving extremely high exposures to
desiccation and ionizing radiation, which shatter its genome into hundreds of
short DNA fragments [191, 192]. As a result, its DNA repair system may be
significantly different from that of other bacteria, enabling it to survive such
extreme conditions [193]. Indeed, unlike most bacterial SSBs, which contain a
single OB-fold domain, the protomer of D. radiodurans SSB (DrSSB) is
twice the size of other bacterial SSB proteins and contains two OB-fold domains
[194, 195]. DrSSB forms a dimer, so the overall quaternary structure is still
similar to most bacterial SSBs with four OB-folds [91]. Despite the similarity in
the DNA-binding domain and the sequence of the C-terminal acidic peptide, the
C-terminal acidic peptide in DrSSB, due to its dimeric structure, consists of
only two copies, unlike the four found in most bacterial SSBs. The flexible
linker in DrSSB is also significantly longer than in most bacterial SSBs (Fig. 10), although the reason for this is still unclear. Despite these many
differences between EcSSB and DrSSB, DrSSB can replace EcSSB in vivo and
can also interact with the
Similar to BsSsbA and EcSSB, DrSSB is known to interact with RecD2, RecA, RecJ, RecO, and uracil DNA glycosylase (PDB ID 3UFM) [115, 142, 196, 197, 198, 199]. Interestingly, D. radiodurans RecO does not bind the C-terminal domain of DrSSB, suggesting diverse mechanisms in the DNA repair pathways mediated by SSB-RecO in different organisms [8]. The differences in the C-terminal domain of DrSSB, particularly its lack of binding with RecO in contrast to EcSSB, highlight how SSB proteins can have species-specific roles in DNA repair [8]. This may be tailored to cope with extreme DNA damage, such as that caused by ionizing radiation, indicating that SSB proteins are highly adaptable and specialized for the unique needs of each organism.
Additionally, the dimeric structure of DrSSB allows for more complex interactions with DNA and other proteins, particularly due to its two OB-fold domains and significantly long flexible linker [200]. The two OB-fold domains in a single DrSSB monomer can evolve individually, unlike EcSSB, where the entire protein evolves together. This may offer advantages, with one OB-fold domain specifically binding ssDNA and the other serving a distinct function [200]. This structure may enable DrSSB to bind to more regions of DNA simultaneously, facilitating its participation in different DNA repair pathways. In contrast, EcSSB typically operates as a monomer with four identical OB-fold domains and may have a more streamlined mechanism of action in E. coli. The dimerization of DrSSB, with two different OB-fold domain sequences and a longer flexible linker within the C-terminal domain, could contribute to more complex interactions with other functional proteins, not seen in E. coli, enabling DNA repair after extreme stress such as radiation and desiccation.
Many interactions with SSB, not found in E. coli, have been identified in D. radiodurans with DrSSB, likely due to the research interest in understanding its high radiation resistance [201, 202, 203]. DdrB, an alternative SSB induced in D. radiodurans by ionizing radiation, can interact with DrSSB [201, 204, 205]. DR1088, highly conserved in the Deinococcus-Thermus phylum, exhibits some RecO-like biochemical properties, including single- and double-stranded DNA binding activity, and may also interact with DrSSB, showing ssDNA binding protein (SSB) replacement ability [206]. The interaction between DR0041 and the C-terminal acidic tail of DrSSB has also been assessed [207]. Due to the involvement of unique partners in DrSSB binding, as compared to EcSSB, the interactome of DrSSB should be studied more extensively.
S. aureus causes a wide variety of clinical diseases and is estimated to be responsible for approximately 19,000 deaths per year in the United States [208]. It has also been recognized as one of the ESKAPE pathogens by the Infectious Diseases Society of America, due to its ability to effectively ‘escape’ the effects of antibacterial drugs [209, 210]. Given the essential role of SSB in DNA metabolism and cellular growth, inhibitors targeting S. aureus SSB (SaSSB) are valuable for the development of new antibiotics [211].
Compared to EcSSB and BsSsbA, the interactome of SaSsbA has not been studied extensively; for example, only the interaction between S. aureus PriA and SsbA has been investigated. Although SaSsbA shares some structural similarities with BsSsbA and EcSSB, conflicting reports still exist regarding its binding to PriA. Unlike EcSSB, which can stimulate EcPriA through a physical interaction between EcPriA and the C-terminal tail of EcSSB, SaPriA can bind to SaSsbA but not to the C-terminal tail of SaSsbA [22, 87]. This difference may be explained by examining the C-peptide binding pocket (Trp82, Tyr86, Lys370, Arg697, and Gln701), which is not conserved in SaPriA [114]. Only Trp89 in SaPriA (equivalent to Trp82 in EcPriA) is present. Arg697 in PriA is known to play a critical role in altering the SSB35/SSB65 distribution, but this residue is replaced by Glu767 in SaPriA, which has an opposite charge to Arg. Due to differences in structure, protein length, and the SSB binding site of the partner protein (e.g., PriA), SaSsbA may have developed a unique interactome during evolution.
M. tuberculosis, the leading cause of death due to infection, should be addressed through drug target identification and drug development [212]. The main SSB in M. tuberculosis, commonly known as SSBa (MtSSBa), may serve as a molecular target for controlling the growth of the pathogen [213]. Similar to EcSSB, MtSSBa is also known to interact with RecA, DnaB, and uracil DNA glycosylases [23, 214, 215]. However, unlike EcSSB, which interacts with uracil DNA glycosylase only through the C-terminal tail, the C-terminal domain (34 amino acids) of MtSSBa is the predominant determinant for mediating this interaction. Although the crystal structure and ssDNA binding properties of MtSSBa are similar to those of EcSSB [105], MtSSBa cannot complement an EcSSB deletion strain of E. coli, suggesting species-dependent binding modes [160, 213]. Additionally, there are significant differences in RecA between M. tuberculosis and E. coli, indicating different SSB interaction modes and DNA repair mechanisms [216]. Given these differences compared to the EcSSB interactome, further investigation of the MtSSBa interactome is essential to better understand these species-dependent SSB interactomes.
The SSB interactomes of prokaryotes are essential to maintaining genome stability. The SSB proteins, not surprisingly, are essential to this process and interact with partners in unique ways. There are, however, common elements to the interaction and regulation of partners shared among these organisms. First, SSB proteins contain unique linkers that enable the protein to bind to different partners spatially and temporally. This requires a common binding domain that in this case, is the oligonucleotide-oligosaccharide binding fold. The structures of these domains are conserved but the sequences are not and this combined with the genus-specific linkers, assures specificity. To regulate binding and ensure that linkers do not bind to the SSB from which they emanate, C-peptide sequences are present. These are frequently, but not always, found at the extreme C-terminus of the protein and function to regulate C-terminal tails by long-range electrostatic effects and to serve as a secondary binding site locking the partner onto the SSB tetramer. In addition, the SSB proteins of multiple organisms also bind to partners through interactions mediated by residues within the OB-fold domains. Collectively, these interactions ensure specificity and maintain genome stability in ways unique to each organism.
PB and CH conceived and wrote the manuscript. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
Work in the Bianco laboratory is supported by NIH grant GM144414 to PRB.
The authors declare no conflict of interest. Piero R. Bianco is serving as one of the Guest editors of this journal. We declare that Piero R. Bianco had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Katrina F. Cooper.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.