




 , Kun Wang 3, Yili Hu 2, Yefu Xu 2, Jiangkai Yu 2, Chengming Li 1,*
, Kun Wang 3, Yili Hu 2, Yefu Xu 2, Jiangkai Yu 2, Chengming Li 1,*
1 Department of Spine Surgery, Zhongda Hospital Southeast University, 210009 Nanjing, Jiangsu, China
2 Medicine School, Southeast University, 210009 Nanjing, Jiangsu, China
3 Department of Spine Surgery, Shanghai Sixth People’s Hospital, 201306 Shanghai, China
Abstract
After spinal cord injury (SCI), pro-inflammatory microglia accumulate and impede axonal regeneration. We explored whether secreted protein acidic and rich in cysteine (Sparc) restrains microglial inflammation and fosters neurite outgrowth.
Mouse microglial BV2 cells were polarized to a pro-inflammatory phenotype with lipopolysaccharides (LPSs). Sparc mRNA and protein were quantified by reverse transcription quantitative PCR (RT-qPCR). Sparc was overexpressed via plasmid transfection, then inflammatory cytokines, mitochondrial membrane potential (Δψm), reactive oxygen species (ROS), and oxidative-phosphorylation proteins, including voltage-dependent anion channel 1 (VDAC1), cytochrome c oxidase subunit 1 (COX1), and ATP synthase α subunit (ATP5A), were assayed by Western blot, enzyme-linked immunosorbent assay (ELISA), and flow cytometry. Immunoprecipitation plus mass spectrometry, co-immunoprecipitation, and immunofluorescence confirmed the interaction between Sparc and ubiquitin A-52 residue ribosomal protein fusion product 1 (Uba52). Effects of Sparc overexpression alone or combined with Uba52 small interfering RNA (si-Uba52) were compared in LPS-induced BV2 cells. Finally, BV2 cells and a mouse hippocampal neuron (HT-22) were co-cultured in the Transwell chamber, and the changes in proliferation, apoptosis, and III-tubulin content of the latter were detected.
In LPS-induced BV2 cells, the tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and ROS levels were elevated, while the IL-10 and transforming growth factor-β (TGF-β) levels, Δψm, and the proteins levels of the VDAC1, COX1, ATP5A, and Sparc decreased. Sparc overexpression reversed these changes. Mechanistically, Sparc bound Uba52 and upregulated its expression; Uba52 knockdown abolished the anti-inflammatory and mitochondrial-protective effects of Sparc. In co-culture, Sparc overexpression rescued HT-22 neurons apoptosis and enhanced axonal growth, but the effects were also reversed by Uba52 knockdown.
Sparc may maintain mitochondrial homeostasis by interacting with Uba52 to inhibit LPS-induced BV2 inflammatory response, thereby promoting neuronal axonal regeneration. This suggests that Sparc may play a potential role in SCI repair.
Keywords
- spinal cord injury
- microglial
- inflammation
- axonal regeneration
- Sparc
- Uba52
Spinal cord injury (SCI) involves complete or partial disruption of spinal cord structural integrity and neurological functions resulting from traumatic or non-traumatic pathogenic factors, precipitating pathological cascades characterized by neuronal loss and axonal rupture [1, 2, 3]. In the acute phase following SCI, resident microglia are rapidly recruited and activated at the lesion site. Functionally distinct microglial subtypes emerge: pro-inflammatory microglia drive neurotoxic effects including amplified neuroinflammation, heightened neuronal death, and aggravated tissue damage, whereas anti-inflammatory microglia enhance neural tissue tolerance, mediate blood–spinal cord barrier repair, support neurovascular regeneration, and restore microenvironmental homeostasis [4, 5]. Within hours post-injury, pathological stimuli trigger microglial polarization. Persistent exposure to the dysregulated microenvironment subsequently promotes progressive dominance of pro-inflammatory microglial populations. While suppression of pro-inflammatory microglial expansion has been shown to improve neurological recovery post-SCI, the precise regulatory mechanisms remain poorly defined [6, 7].
Emerging evidence implicates mitochondria as central signaling platforms that govern immune cell phenotypic specification and modulate microglial inflammatory responses [8]. Mitochondrial dysfunction during neuroinflammation critically alters microglial metabolic states, thereby influencing inflammatory progression. Our prior work revealed concurrent activation of mitophagy during microglial polarization, with experimental manipulation of mitophagic flux directly modulating phenotypic switching [9].
Secreted protein acidic and rich in cysteine (Sparc), a matricellular protein, regulates cytokine activity, extracellular matrix dynamics, and tissue repair processes. During central nervous system (CNS) development, Sparc is abundantly expressed in microglia, yet its expression diminishes significantly in mature CNS microglia [10]. Sparc deficiency has been demonstrated to exacerbate microglial macrophage activation [11, 12], suggesting its potential role in constraining pro-inflammatory phenotypic conversion. To address current knowledge gaps, this study investigates Sparc-mediated regulation of pro-inflammatory microglial activation, delineates underlying molecular mechanisms, and evaluates its neuroprotective effects through suppression of microglial-driven neuroinflammation. These findings may advance therapeutic strategies for axonal regeneration and functional recovery following SCI.
BV2 microglial cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. HT-22 neuronal cells were maintained in high-glucose dulbecco’s modified eagle medium (DMEM) containing 10% FBS. Subculture protocol: After aspirating the spent medium, cells were gently washed twice with 37 °C pre-warmed PBS, followed by digestion with 0.25% Trypsin-EDTA (45 seconds for BV2; 2.5 minutes for HT-22). Upon observation of intercellular gap widening under a microscope, serum-containing medium was added to neutralize trypsin. Cells were collected by centrifugation at 1000 rpm for 5 minutes and subcultured at a 1:3 ratio. Complete medium replacement was performed every 2 days, with cell density maintained between 30% and 80%. BV2 and HT-22 cells were purchased from Jiangsu KeyGENE Technology Co., Ltd. (Nanjing, China). All cell lines were validated by short tandem repeat (STR) profiling and tested negative for mycoplasma.
The Sparc coding DNA sequence (CDS) region sequence was searched through the national center for biotechnology information (NCBI) online website (https://www.ncbi.nlm.nih.gov/), and the CDS region sequence was imported into pcDNA3.1 expression plasmid to construct the Sparc overexpression recombinant plasmid. Sparc overexpression plasmid and Uba52 small interfering RNA (si-Uba52) sequences were synthesized by KeyGEN BioTECH Co., Ltd. (KeyGEN Biotech, Nanjing, China). When the fusion degree of BV2 cells reached about 70%, the cells were divided into 3 groups: control, lipopolysaccharides (LPS), and LPS + Sparc. Cells in the LPS group were treated with 200 ng/mL LPS (Sigma, St. Louis, MO, USA) for 24 hours. LPS + Sparc cells were transfected with Sparc overexpression plasmid by Lipofectamine 3000 (Thermo Fisher, Waltham, MA, USA), cultured for 24 hours and then treated with 200 ng/mL LPS for 24 hours. Cells in the control group were cultured normally for 24 hours.
In addition, the cells were divided into three groups again: LPS, LPS + Sparc, and LPS + Sparc + si-Uba52. Cells in the LPS and LPS + Sparc groups were treated as above. Cells in the LPS + Sparc + si-Uba52 group were co-transfected with the Sparc overexpression plasmid and si-Uba52 by Lipofectamine 3000. After 24 hours of culture, cells were treated with 200 ng/mL LPS for 24 hours.
Lysis conditions: 1
Immunoprecipitation: 1 mg total protein was incubated with Sparc antibody overnight at 4 °C with rotation, followed by 4 hours of incubation with Protein G agarose beads. Beads were washed 5 times with lysis buffer, and bound proteins were eluted using low-pH elution buffer. Target bands excised from sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels were trypsin-digested at 37 °C for 16 hours for subsequent mass spectrometry analysis.
The transfected cells and untransfected cells were digested, counted, and
prepared into cell a suspension with a concentration of 1
The transfected cells and untransfected cells were digested, counted, and
prepared into a cell suspension with a concentration of 1
After centrifugation, the colorless supernatant water was drawn and transferred
to another centrifuge tube. After centrifugation, an equal volume of isopropanol
and 70% ethanol was added. After centrifugation, RNase-free water was added to
dissolve the RNA precipitation. After the concentration and purity of RNA were
determined, 0.2 mL polymerase chain reaction (PCR) tubes that had been sterilized
and had no nuclease were taken, and RNA (2 µg), 5
Cells were collected, lysates were added, protein samples were extracted, protein concentration was calculated according to the standard curve, 10% SDS-PAGE gel was prepared, electrophoresed, transferred to polyvinylidene fluoride (PVDF) membrane, blocked with 1% bovine serum albumin (BSA) for 1 hour, added primary antibody, incubated at 4 °C overnight, wash with PBS, add secondary antibody and incubate at room temperature for 2 hours. The obtained bands were imaged by ChemiDoc MP Imaging System 3.0.1 version (Bio-Rad, Hercules, CA, USA), and the gray level was analyzed by the Gel-Pro32 software 6.0 version (Media Cybernetics, Inc., Rockville, MD, USA).
The kits (Tumor necrosis factor-
Co-culture of BV2 cells and HT-22 cells was performed using Transwell chambers. BV2 cells were seeded in the upper chamber of Transwell, and HT-22 cells were seeded in 24-well plates. The co-culture cells were divided into 4 groups: Control/HT-22, LPS/HT-22, LPS + Sparc/HT-22, LPS + Sparc + si-Uba52/HT-22. BV2 cells in each group were treated as above and co-cultured with HT-22 cells for 24 hours.
24 hours after transfection, BV2 cells were digested, counted, and prepared into
a cell suspension with a concentration of 1
The co-cultured HT-22 cells were collected by 0.25% trypsin (without EDTA)
digestion. Cells were washed twice with PBS (centrifuged at 1000 rpm for 5
minutes) to collect 1
The lower layer of HT-22 cells was removed, the medium was discarded, and the
cell samples were allowed to dry naturally. The cells were immersed in 4%
paraformaldehyde fixator for 30 min or overnight to improve cell permeability,
and the cells were immersed in PBS for 3 minutes
For dual-colour immunofluorescence co-localisation of Uba52 and Sparc, BV2 cells seeded on glass coverslips were fixed with 4% paraformaldehyde for 30 minutes, permeabilised with 0.2% Triton X-100, and blocked with 3% BSA for 30 minutes. Sequential tyramide signal amplification (TSA) labelling was performed on the same slide. First, rabbit anti-Uba52 (Abclonal, Wuhan, China, A20876, 1:500) was applied overnight at 4 °C, followed by goat anti-rabbit IgG-HRP (KeyGEN Biotech, Nanjing, China, KGC6202, 1:5000) and Alexa Fluor 488-TSA. After stringent antibody elution (37 °C, 15–30 minutes in citrate-based buffer), the second cycle was executed with rabbit anti-Sparc (Proteintech, Wuhan, China, 15274-1-AP, 1:1000), identical HRP-secondary and Alexa Fluor 594-TSA. Nuclei were counterstained with 4′,6-Diamidino-2′-phenylindole (DAPI). Coverslips were mounted with anti-fade medium and imaged on a confocal laser-scanning microscope (Olympus, Tokyo, Japan).
All experimental data are presented as mean
Mitochondrial dysfunction during neuroinflammation exacerbates inflammatory
progression by reprogramming microglial metabolism. In LPS-induced BV2 cells.
Sparc overexpression significantly restored mitochondrial membrane
potential (Fig. 1A), attenuated ROS production (Fig. 1B), and upregulated
mitochondrial oxidative phosphorylation (mt-OXPHOS)-related proteins
voltage-dependent anion channel 1 (VDAC1), cytochrome c oxidase subunit 1 (COX1),
and ATP synthase
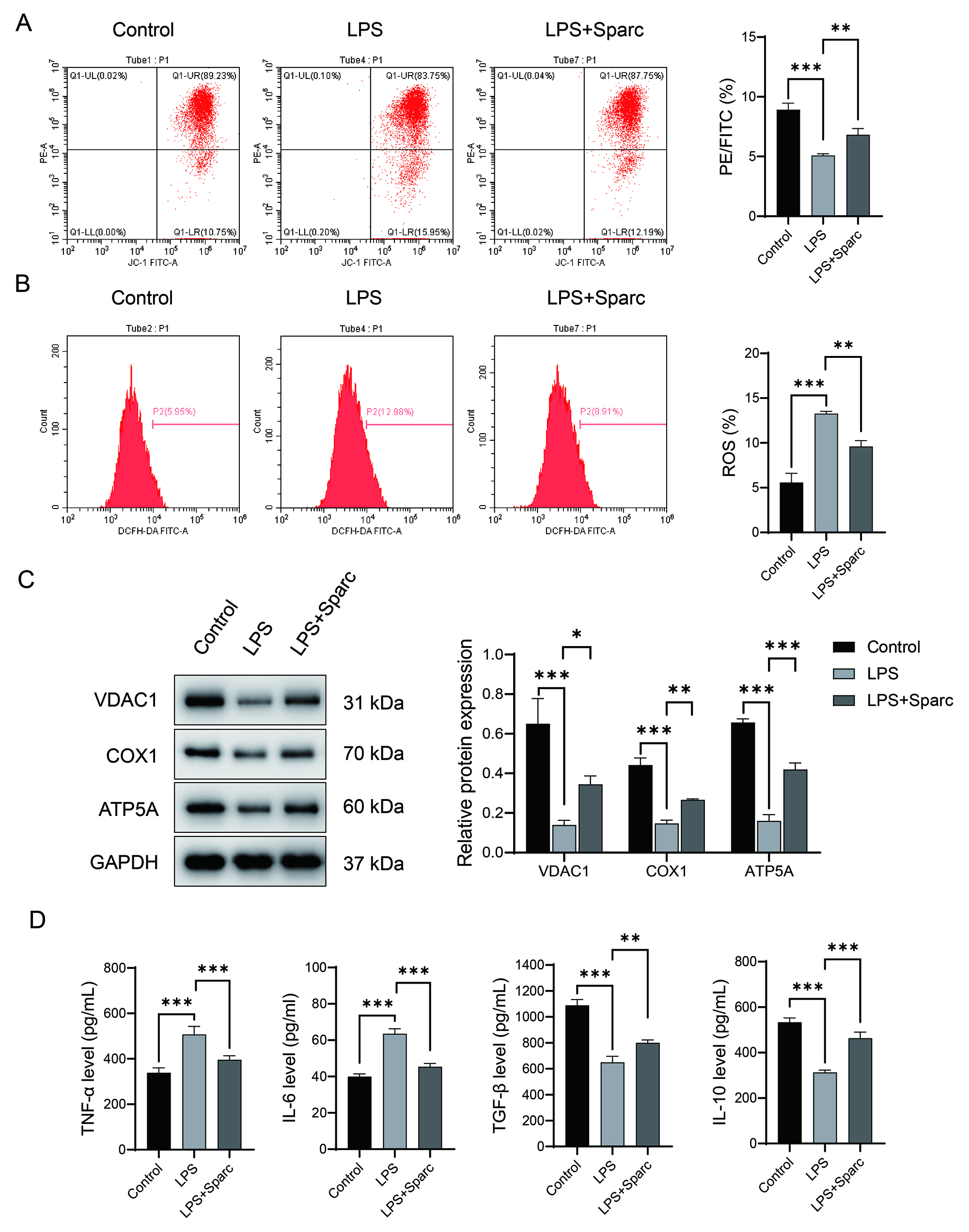 Fig. 1.
Fig. 1.
Sparc overexpression suppresses pro-inflammatory responses and
regulates mitochondrial homeostasis in lipopolysaccharides (LPS)-induced BV2
cells. (A) JC-1 method for detecting mitochondrial membrane potential. (B)
2′,7′-Dichlorofluorescin diacetate (DCFH-DA) probe for detecting reactive
oxygen species (ROS) levels. (C) Western blot detection of mitochondrial
functional proteins (voltage-dependent anion channel 1 (VDAC1), cytochrome c
oxidase subunit 1 (COX1), ATP synthase
Immunoprecipitation combined with mass spectrometry confirmed potential binding between Sparc and Uba52 (Fig. 2A,B). Subsequently, the interaction of the Sparc-Uba52 was confirmed through co-localization of co-immunoprecipitation and immunofluorescence (Fig. 2C,D). LPS stimulation suppressed both Uba52 mRNA and protein levels, whereas Sparc overexpression rescued its expression (Fig. 2E,F).
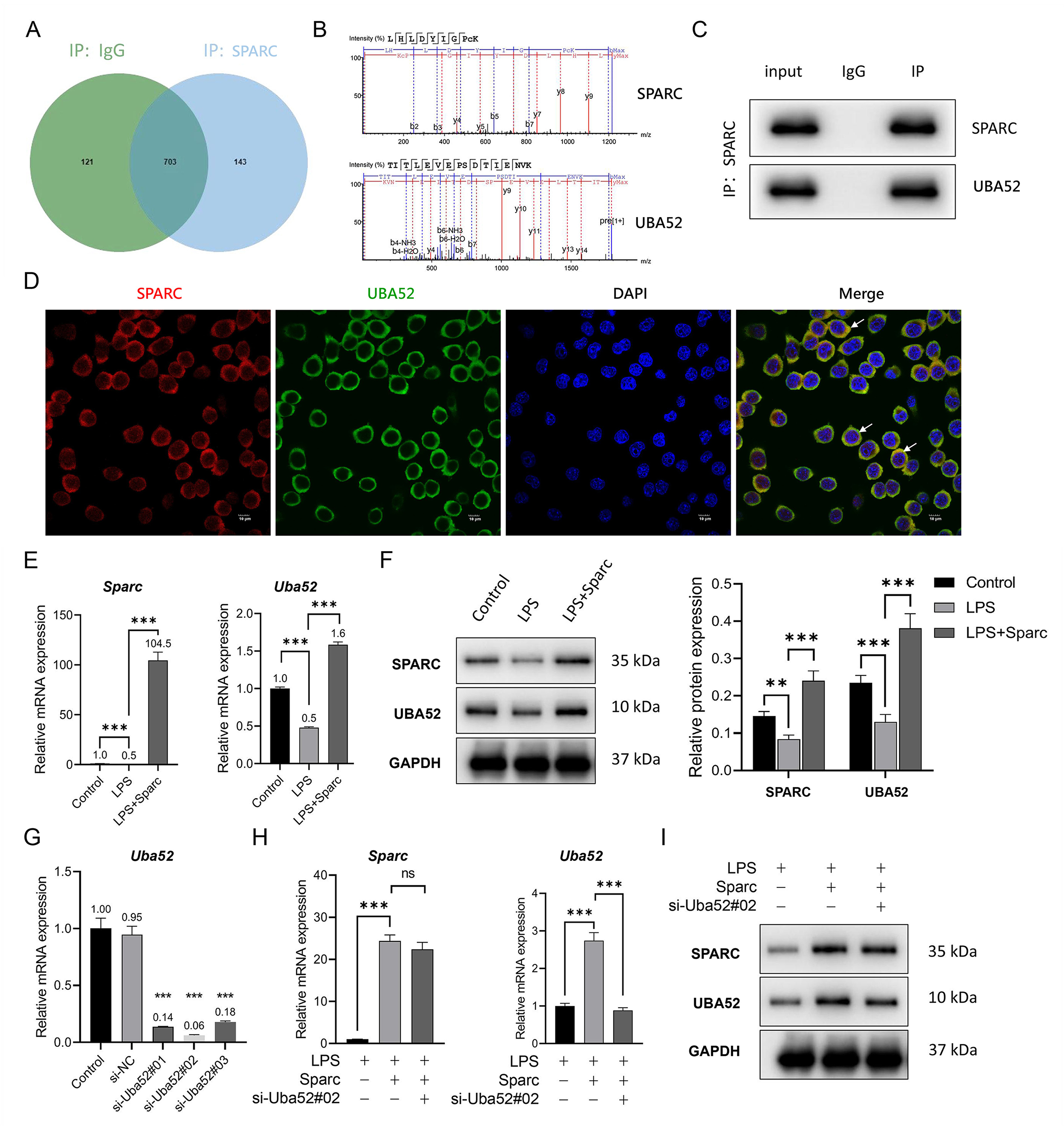 Fig. 2.
Fig. 2.
Sparc interacted with Uba52 and promoted Uba52
expression. (A–C) The interaction between Sparc and Uba52 protein was confirmed
through co-immunoprecipitation coupled mass spectrometry. Input: 10% total
lysate; Ig G: negative control; IP: anti-Sparc immunoprecipitated. (D)
Co-localization analysis of the Sparc and Uba52 was performed using
immunofluorescence. The white arrow represents the co localization area, where yellow
(red and green overlapping) indicates spatial overlap between Sparc and Uba52.
Scale bar: 10 µm. (E,F) Changes in expression levels of the Sparc
and Uba52 genes and proteins in BV2 cells under the intervention of Sparc
overexpression. (G) Uba52 knockdown validation was conducted using RT-qPCR. (H,I) Expression changes of the
Sparc and Uba52 genes and proteins after transfection of
si-Uba52 in BV2 cells. **p
To knockdown Uba52 in BV2 cells, three si-Uba52 were designed. The verification results of RT-qPCR showed that si-Uba52#02 had the best inhibition rate on the expression of the Uba52 gene (Fig. 2G). Therefore, si-Uba52#02 was selected for the Uba52 knockdown experiment. Uba52 knockdown reduced the Uba52 gene and protein without altering the Sparc gene and protein expressions (Fig. 2F,H,I), establishing Uba52 as a downstream effector of Sparc.
Sparc overexpression reversed LPS-induced mitochondrial depolarization
(Fig. 3A) and restored VDAC1, COX1, and ATP5A expressions (Fig. 3C), effects
abolished by Uba52 knockdown. Sparc suppressed ROS accumulation
(Fig. 3B), whereas Uba52 knockdown exacerbated it. Sparc
overexpression inhibited TNF-
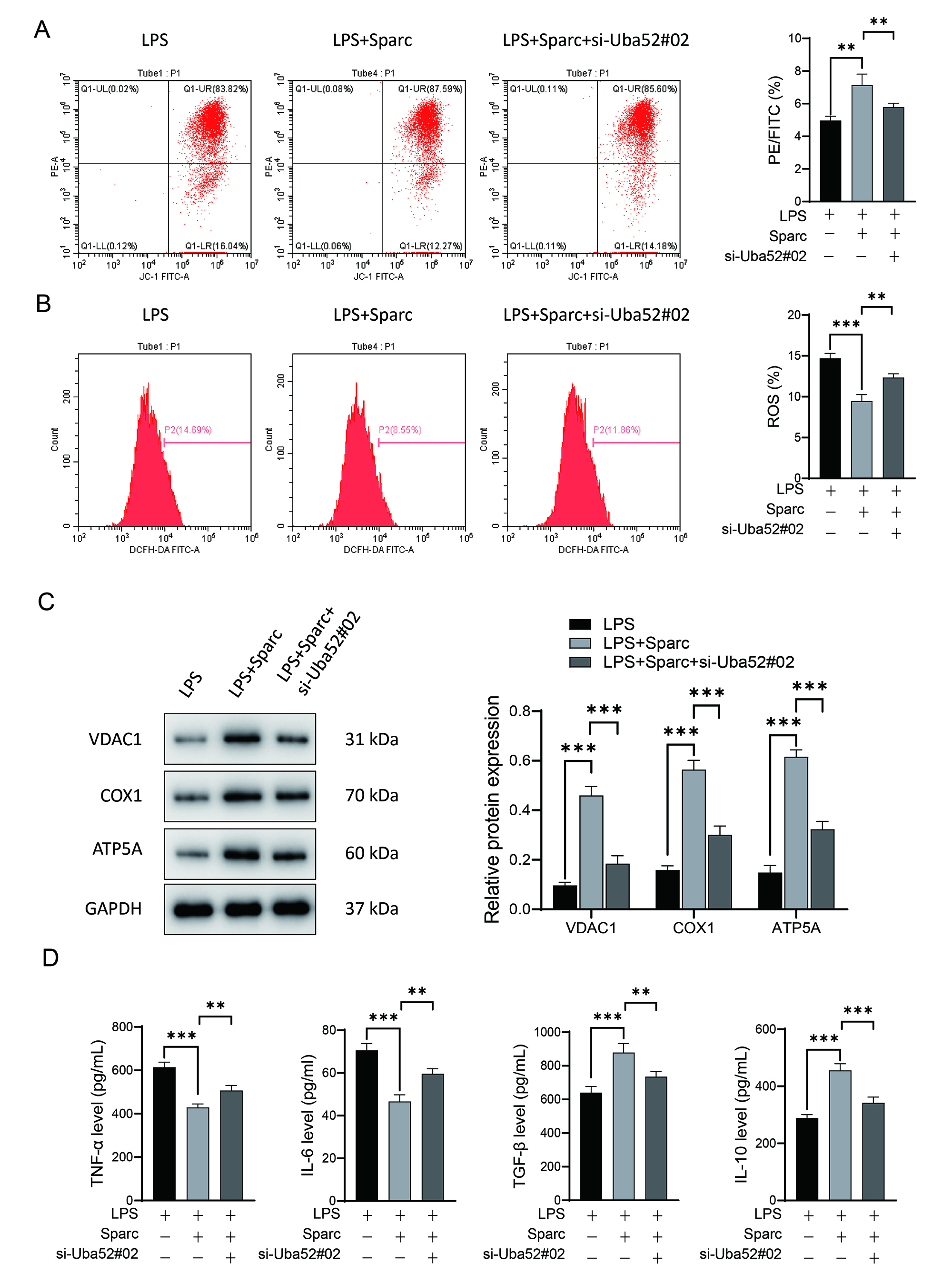 Fig. 3.
Fig. 3.
Sparc interacts with Uba52 to regulate mitochondrial respiration
and inhibit LPS-induced BV2 cell inflammatory responses. (A) Mitochondrial
membrane potential measured by JC-1 assay. (B) Intracellular ROS levels detected
by DCFH-DA probe. (C) Western blot analysis of mitochondrial respiratory chain
proteins (VDAC1, COX1, ATP5A). (D) mRNA levels of inflammatory factors by qPCR.
**p
Transwell co-culture assays demonstrated that pro-inflammatory BV2 supernatants strongly inhibited HT-22 neuronal proliferation (Fig. 4A,B). Sparc overexpression rescued neuronal proliferation (Fig. 4B), whereas si-Uba52 intensified suppression. Apoptosis analysis revealed that Sparc attenuated LPS-induced neuronal apoptosis (Fig. 4C), while Uba52 silencing amplified cell death (Fig. 4C). III-tubulin expression patterns corroborated these findings: Sparc restored neurite outgrowth capacity, whereas si-Uba52 diminished it (Fig. 4D).
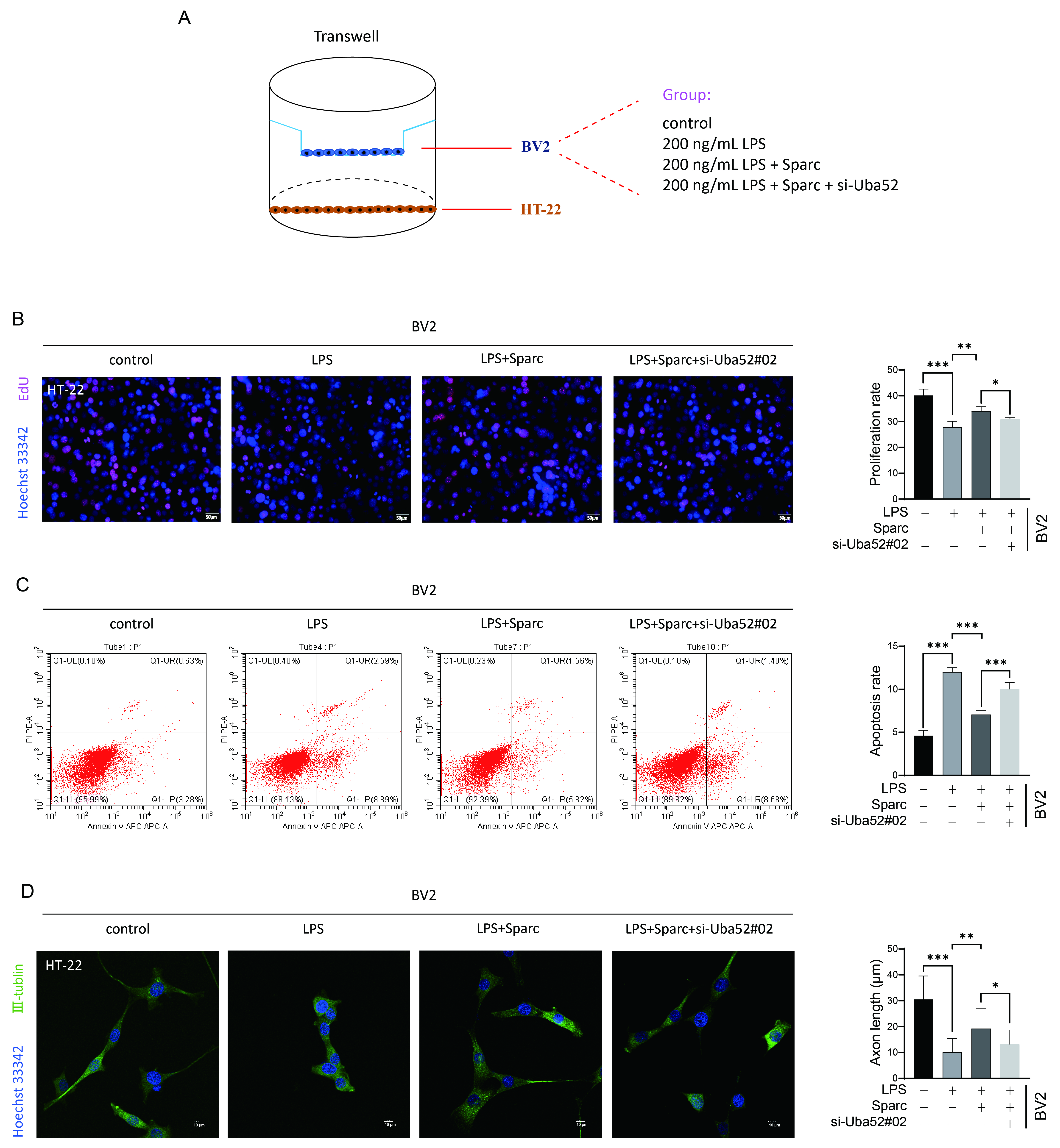 Fig. 4.
Fig. 4.
Sparc interacts with Uba52 to regulate pro-inflammatory
microglia to promote neuronal cell axonal outgrowth. (A) Schematic diagram of
Transwell co-culture system. Created by Adobe Illustrator 2022. (B)
Quantification of HT-22 neuronal proliferation under different treatments. Left
panels: Representative images of EdU staining (red, proliferating cells) and
Hoechst 33342 (blue, nuclei). Scale bar: 50 µm. (C) Annexin
V-APC/Propidium Iodide (PI) dual staining was used to detect apoptosis. (D)
Quantification of axonal outgrowth in HT-22 neurons by III-tubulin
immunofluorescence. Left panels: Representative confocal images of III-tubulin
(green) and nuclei (blue, Hoechst). Scale bar: 10 µm. *p
This study elucidates a mechanism by which Sparc interacts with the ribosomal ubiquitination modifier Uba52 to maintain mitochondrial functional homeostasis and inhibit inflammation in LPS-induced BV2 cells, ultimately modulating neuronal function. Our findings reveal a previously unrecognized immunometabolic crosstalk network in neuroinflammatory regulation.
Our findings demonstrate that Sparc interacts with Uba52 to form a functional complex and upregulates Uba52 expression. Notably, while Uba52 is conventionally recognized as a ubiquitin precursor protein involved in ribosomal ubiquitination and protein quality control, this study unveils its potential role in mitochondrial function regulation. Specifically, Sparc can increase mitochondrial membrane potential and the expression of mt-OXPHOS-related proteins, and reduce the production of ROS and inflammation in LPS-induced BV2 cells. Critically, Uba52 knockdown partially abrogates Sparc-mediated protection of mitochondrial function and anti-inflammatory effect. It is reported that when macrophages transform into pro-inflammatory types, mitochondrial function changes, including OXPHOS and mitochondrial membrane potential decrease, while the production of ROS increases [13]. This metabolic-inflammatory coupling mirrors the “Warburg effect” observed in pro-inflammatory macrophages, where glycolytic flux predominates over oxidative phosphorylation for energy production [14, 15]. These studies suggest that Sparc may maintain mitochondrial functional homeostasis by interacting with Uba52 to inhibit the pro-inflammatory response of LPS-induced BV2 cells.
Although Sparc intervention significantly reduces total ROS levels, subcellular localization analysis is required to determine whether this reduction specifically targets mitochondrial ROS. Furthermore, the observed upregulation of ATP5A may enhance OXPHOS efficiency, potentially providing the energetic foundation necessary for anti-inflammatory polarization [16].
The study further reveals that pro-inflammatory BV2 cells suppress axonal growth
in HT-22 neurons, a phenomenon closely mirroring the “chronic
neuroinflammation-neuronal injury” vicious cycle characteristic of
neurodegenerative pathologies. We propose that Sparc promotes neuronal
repair via dual mechanisms: (1) Direct metabolic regulation: The Sparc-Uba52
complex sustains mitochondrial homeostasis and reduces pro-inflammatory cytokine
(TNF-
Moreover, conventional paradigms posit that microglial phenotypic switching is coordinately regulated by multiple signaling pathways [18]. Our study reveals that the Sparc–Uba52 axis independently governs metabolic reprogramming, suggesting the existence of parallel regulatory mechanisms. Extending the concept of mitochondria as immunometabolic regulators where mitochondrial-derived mitochondrial DNA (mtDNA) and ROS are known to activate the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome [19], this work identifies respiratory chain integrity itself as an inflammation-suppressive signal. While Uba52 typically participates in protein degradation, our findings demonstrate its protective role through mitochondrial protein stabilization, providing novel evidence supporting the emerging paradigm of bidirectional regulatory capacity in ubiquitination modifications.
Although this study revealed the potential mechanism by which the Sparc–Uba52
axis inhibits microglial inflammation by maintaining mitochondrial functional
homeostasis, the following key issues still need further validation: (1) Further
research is needed to determine whether the effect of Sparc–Uba52 axis on
mitochondrial protein expression depends on ROS clearance without the use of
antioxidants such as Mito-TEMPO; (2) In addition to inflammatory factors, the
toll-like receptor 4 (TLR4) mediated inflammatory pathway plays an important role
in the pro-inflammatory response and ROS of microglia. Subsequently, Western blot
will be used to detect the expression of key proteins on the toll-like receptor
4/myeloid differentiation factor 88/nuclear factor-
To sum up, this study demonstrates that Sparc modulates mitochondrial respiration and suppresses microglial pro-inflammatory responses through its interaction with Uba52, further promoting axonal regeneration. We initially elucidate the potential mechanisms by which the Sparc–Uba52 axis governs microglial inflammatory phenotype. These findings identify potential therapeutic targets for SCI.
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
HJ conducted experimental design, data analysis, and wrote this article. KW conducted experimental design. YH, YX and JY conducted experimental operations and data analysis. CL conducted experimental design, wrote this article and revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was funded by the Research Personnel Cultivation Programme of Zhongda Hospital Southeast University (No. CZXM-GSP-RC51).
The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.