
 , Gengqiu Liu 1,†, Zhu Liang 2,*
, Gengqiu Liu 1,†, Zhu Liang 2,* , Junhang Zhang 1,*,§
, Junhang Zhang 1,*,§1 Department of Thoracic Surgery, The Seventh Affiliated Hospital, Sun Yat-Sen University, 518107 Shenzhen, Guangdong, China
2 Department of Cardiothoracic Surgery, Affiliated Hospital of Guangdong Medical University, 524000 Zhanjiang, Guangdong, China
†These authors contributed equally.
§Lead contact.
Abstract
Adenocarcinoma of Lung (LUAD) remains a leading cause of cancer-related deaths across the globe, and patients harboring epidermal growth factor receptor (EGFR) mutations frequently develop resistance to targeted therapies. While aurora kinase A (AURKA) has been implicated in tumorigenesis, its involvement in regulating ferroptosis via the kelch-like ECH-associated protein 1 (KEAP1)/NF-E2-related factor 2 (NRF2)/heme oxygenase 1 (HO‑1) signaling axis in EGFR-mutant LUAD remains poorly understood.
We analyzed RNA-seq and clinical data from 594 LUAD samples from The Cancer Genome Atlas (TCGA) to explore associations between AURKA expression, EGFR mutation status, and immune cell infiltration. A ferroptosis-focused random forest algorithm was constructed to predict EGFR-mutant cases. In vitro, AURKA was silenced by siRNA in EGFR-mutant NCI-H1975 cells; subsequent assays included transcriptome profiling, measurements of intracellular Fe2⁺, malondialdehyde (MDA), glutathione (GSH), mitochondrial reactive oxygen species (ROS) levels, and ultrastructural examination by electron microscopy. Protein levels of NRF2, HO‑1, solute carrier family 7 member 11 (SLC7A11), glutathione peroxidase 4 (GPX4), and KEAP1 were assessed via western blot.
The ferroptosis gene–based random forest model distinguished EGFR-mutant LUAD with an area under the curve (AUC) of 0.84. Clinically, high AURKA expression was significantly associated with EGFR wild-type status (p = 0.035), reduced overall survival (p = 0.011), increased M1 macrophage infiltration, and decreased CD4⁺ T-cell infiltration. AURKA knockdown triggered hallmark features of ferroptosis—iron overload (p < 0.001), elevated MDA levels (p < 0.01), increased lipid peroxidation (p < 0.05), heightened mitochondrial ROS (p < 0.05), reduced mitochondrial membrane potential, GSH depletion (p < 0.05), and disruption of mitochondrial cristae. Mechanistically, loss of AURKA decreased KEAP1 (p < 0.01) and enhanced NRF2 (p < 0.001) and HO-1 (p < 0.01) and NRF2 nuclear translocation, while downregulating SLC7A11 (p < 0.05) and GPX4 (p < 0.01). Cell cycle analysis revealed G1-phase arrest (p < 0.001).
Our findings demonstrate that AURKA promotes ferroptosis resistance in EGFR-mutant LUAD by modulating the KEAP1/NRF2/HO-1 axis. Notably, this effect was validated in the gefitinib-resistant EGFR T790M-mutant NCI-H1975 cell model. Our results highlight AURKA as a potential therapeutic target for overcoming epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) resistance and suggest that disrupting the AURKA/KEAP1/NRF2/HO‑1 pathway may offer a novel strategy for treating EGFR-mutant LUAD.
Keywords
- adenocarcinoma of lung
- Aurora kinase A
- ErbB receptors
- ferroptosis
- NF-E2-related factor 2
Adenocarcinoma of Lung (LUAD), the most common form of non–small cell lung cancer (NSCLC), is the primary contributor to global cancer mortality, accounting for approximately 2.2 million new diagnoses and 1.79 million deaths annually, with a 5-year survival rate under 20% [1, 2]. In Asian populations, over 50% of NSCLC tumors harbor activating mutations in the ErbB receptors (EGFR) —most commonly exon 19 deletions or the L858R substitution—which drive aberrant proliferation and metastasis [3, 4, 5]. Although first-line EGFR inhibitors can delay disease progression by roughly 9–15 months on average [6, 7], resistance inevitably arises, often alongside upregulation of Aurora kinase A (AURKA) activity [8, 9].
Ferroptosis is a distinct, iron-dependent form of regulated cell death characterized by lethal lipid peroxides [10, 11], mitochondrial shrinkage, and loss of cristae architecture [12]. Tumor cells, with elevated reactive oxygen species (ROS) and dysregulated iron metabolism, are particularly prone to ferroptosis [13, 14, 15, 16], yet often evade it through the glutathione peroxidase 4 (GPX4)–mediated detoxification system and the kelch-like ECH-associated protein 1 (KEAP1)/NF-E2-related factor 2 (NRF2)/heme oxygenase 1 (HO‑1) antioxidant axis [17, 18]. In the cytoplasm, KEAP1 sequesters NRF2, preventing its nuclear translocation; upon release, NRF2 upregulates HO‑1 to counteract oxidative stress and inhibit ferroptosis [19, 20].
AURKA, a serine/threonine kinase critical for mitotic entry and spindle assembly [21], is overexpressed in multiple cancers, including LUAD, where it promotes proliferation, metastasis, epithelial–mesenchymal transition, and cancer stemness [22, 23]. Recent Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) screens and pharmacologic studies reveal that AURKA inhibition downregulates KEAP1 and activates NRF2, thereby tipping the balance toward ferroptosis in tumor cells [24]. Yet, its precise role in EGFR-mutant LUAD has not been fully defined.
Here, we investigate The Cancer Genome Atlas (TCGA)-based bioinformatics with in vitro functional assays to investigate how AURKA modulates the KEAP1/NRF2/HO‑1 pathway and ferroptosis in EGFR-mutant LUAD, aiming to uncover novel approaches to overcome epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) resistance.
Transcriptomic count data and matched clinical information from 594 LUAD patients were obtained from TCGA (https://portal.gdc.cancer.gov/). LUAD-specific gene mutation profiles were also obtained from the same source. Raw RNA-seq count were normalized into Reads Per Kilobase Per Million (RPKM) and Transcripts Per Million (TPM) using R software (version 4.1.1; https://cloud.r-project.org/) for downstream analysis. A curated list of ferroptosis-related genes was extracted from the FerrDb database (http://www.zhounan.org/ferrdb/).
For clinical validation, six LUAD tumor samples harboring confirmed EGFR mutations and matched adjacent normal tissues were collected from treatment-naïve patients undergoing surgical resection at our institution. All samples were flash-frozen on collection and kept at –80 °C in pre-cooled containers to preserve molecular integrity. EGFR mutation status was verified postoperatively by institutional pathologists. All patients were treatment-naive, with no history of radiotherapy, chemotherapy, targeted agents, or immunotherapy.
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee at Sun Yat-sen University’s Seventh Affiliated Hospital (Authorization Number: 2024-152). Informed consent was secured from each participant before collecting samples.
A random forest model was developed using the “Boruta” (https://cloud.r-project.org/web/packages/Boruta/index.html) R package, focusing on ferroptosis-related genes signatures. This ensemble learning method integrates multiple decision trees built on randomly selected data subsets to improve prediction robustness and reduce overfitting. The cohort was randomly split into training and validation sets at a 3:2 ratio. Feature importance was assessed and visualized using the “ggrepel” (https://cloud.r-project.org/web/packages/ggrepel/index.html) package . Model performance was evaluated by plotting the receiver operating characteristic (ROC) curve and calculating the area under the curve (AUC) using the “ggplot2” (https://cloud.r-project.org/web/packages/ggplot2/index.html) package. The ROC curve illustrated the sensitivity–specificity trade-off, with a higher AUC reflecting stronger discrimination capacity.
Differential expression between LUAD tumor and adjacent normal tissues was assessed using the “edgeR” (https://www.bioconductor.org/packages/release/bioc/html/edgeR.html) R package to identify significantly altered genes. A heatmap was generated using the “pheatmap” (https://cloud.r-project.org/web/packages/pheatmap/index.html) package to visualize expression patterns of differentially expressed genes (DEGs).
To evaluate prognostic value, patients were dichotomized into high- and low-expression groups based on median gene expression. Kaplan–Meier survival curves were generated using the “survival” (https://cloud.r-project.org/web/packages/survival/index.html) and “survminer” (https://cloud.r-project.org/web/packages/survminer/index.html) packages, and statistical differences were assessed to identify potential prognostic biomarkers.
Patients were stratified by EGFR mutation status (mutant vs. wild-type).
Expression differences of model genes were compared using the Wilcoxon rank-sum
test. Genes with p-values
Immune cell composition in tumor and normal tissues was estimated using the “CIBERSORT” algorithm. Patients were further grouped by median expression of selected genes to evaluate immune cell infiltration differences across subgroups.
To explore pathways associated with AURKA expression, GSEA was performed on
expression data from 594 LUAD cases. Patients were divided into high- and
low-expression groups based on the median AURKA level. A total of 1000
permutations were conducted to compute p-values. Significant pathways
were defined by p
A high-confidence protein–protein interaction (PPI) network of differentially expressed ferroptosis-related genes was constructed using the STRING database (https://string-db.org), applying a minimum interaction score threshold at 0.70 to ensure high-confidence associations. Network visualization and topological analysis were conducted in Cytoscape (version 3.10.2; https://cytoscape.org/) [25]. Functional enrichment analysis was performed through STRING using multiple ontologies and pathway databases, including Gene Ontology (GO: biological process [BP], cellular component [CC], molecular function [MF]), Reactome, Kyoto Encyclopedia of Genes and Genomes (KEGG), and WikiPathways. The enrichment analysis results were graphically presented using the “ggplot2” (https://cloud.r-project.org/web/packages/ggplot2/index.html) package in R. KEGG mapping identified significantly enriched signaling pathways [26], while GO terms highlighted key biological functions [27]. Reactome pathways provided expert-curated mechanistic insights [28], and WikiPathways served as a collaborative source for community-curated networks [29].
To detect densely connected regions within the network, the molecular complex detection (MCODE) plugin in Cytoscape (version 3.10.2; https://cytoscape.org/) was used [30]. MCODE identifies molecular complexes by assigning weights to nodes based on local connectivity and expanding from high-density seed proteins. Clustering was performed in directed mode to improve specificity. Modules and hub genes were identified using the following parameters: degree cutoff of 2, node score threshold of 0.2, k-core value of 2, and a maximum depth setting of 100.
LUAD cell lines NCI-H1975 (harboring the EGFR L858R/T790M mutation conferring intrinsic gefitinib resistance), A549, NCI-H1650, and PC‑9 were obtained from the American Type Culture Collection (ATCC) (https://www.atcc.org/). Cell authentication was verified via short tandem repeat (STR) profiling, and all lines were confirmed to be mycoplasma-free. Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and maintained at 37 °C in a 5% CO2 humidified incubator (Thermo Scientific, MA, USA, cat#51033582).
Before seeding, cells were confirmed to be 80–90% confluent by microscopy
(Olympus,Tokyo, Japan, cat#CX23-1RH2) . Cells were washed with PBS, digested
with trypsin, and resuspended in complete medium. A 10 µL aliquot of the
suspension was used for cell counting with a hemocytometry (Hausser Scientific,
Horsham, PA, USA, cat#15170-172). Cells (8
Gene knockdown was achieved using small interfering RNA (siRNA) transfection. Specific siRNAs targeting AURKA mRNA were synthesized (GEMA Genetics, Shanghai, China). A negative control (NC) siRNA was included to control for off-target effects.
Cells were plated in 12-well plates and transfected after a 24-hour incubation. A mixture of siRNA, transfection reagent, and PolyJet (as per manufacturer’s protocol) was prepared and incubated for 15 minutes. Culture medium was replaced with 1 mL of antibiotic-free 1640 medium with 10%. Subsequently, 100 µL of the transfection mixture was added to each well, yielding a final siRNA concentration of 20 nM. Cells were incubated for 72 hours before harvesting protein analysis.
siRNA sequences:
si-NC: 5′-UUCUCCGAACGUGUCACGUTT-3′; 5′-ACGUGACACGUUCGGAGAATT-3′.
si-AURKA: 5′-GCAAUUUCCUUGUCAGAAUTT-3′; 5′-AUUCUGACAAGGAAAUUGCTT-3′.
Forty-eight hours after siRNA-induced AURKA silencing, total RNA was isolated
from NCI-H1975 cells using TRIzol reagent (Sangon Biotech, Shanghai, China,
cat#B511311). RNA sequencing and primary data processing was carried out by
Sangon Biotech (Shanghai, China, https://www.sangon.com/). Raw read counts were
converted to TPM for downstream analysis using R software (v4.1.1; https://cloud.r-project.org/).
Differential expression analysis was performed using the “DESeq2” (https://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) package in R.
The Benjamini-Hochberg method was applied to adjust p-values for multiple comparisons,
and genes with an adjusted p-value (q-value)
To assess the long-term proliferative capacity, colony formation assays were performed. NCI-H1975 cells were seeded into 12-well plates at 2000 cells per well and cultured at 37 °C with 5% CO2 for 2 weeks. Colonies were then washed with PBS, fixed with 4% paraformaldehyde (MACKLIN, Shanghai, China, cat#P804536) for 20 minutes, and stained with 1% crystal violet (Solarbio, Beijing, China, cat#C8470) at room temperature for 10 minutes. After washed off excess dye, the plates were air-dried and imaged.
Cell migration following AURKA silencing was assessed using the Transwell assay. After 48 hours of siRNA transfection, NCI-H1975 cells were washed, digested with trypsin, and resuspended in serum-free 1640 medium. Approximately 30,000 cells were added to the upper compartment of Transwell inserts, which were positioned in 24-well plates filled with 700 µL RPMI-1640 medium containing 15% FBS. After 48 hours of incubation at 37 °C with 5% CO2, cells on the upper surface of the membrane were removed, and migrated cells on the underside were fixed in formaldehyde, stained with 1% crystal violet, and imaged under a stereomicroscope (Olympus,Tokyo, Japan, cat#SZX16).
Intracellular Fe2+ levels were assessed by treating siRNA-transfected NCI-H1975 cells with 1 µM FerroOrange (Dojindo, Kumamoto, Japan, cat#F374) at 37 °C for 30 minutes. Fluorescence signals indicating ferrous iron accumulation were visualized using a fluorescence microscope (BioTek Cytation 5, BioTek, Winooski, VT, USA).
Lipid peroxidation, a key marker of ferroptosis, was assessed using the fluorescent probe C11-BODIPY 581/591 (10 µM; ABclonal, Wuhan, China, cat#RM02821). NCI-H1975 cells were treated with siRNA for 72 hours and incubated with the probe for 1 hour at 37 °C in 5% CO2. After washing with PBS, cells were harvested and resuspended for flow cytometry analysis. To investigate ferroptosis-specific responses, cells were pretreated 24 hours in advance with ferroptosis inhibitors or inducers, including Ferrostatin-1 (Fer-1, 10 µM, cat#S7243), Liproxstatin-1 (LIP-1, 10 µM, cat#S7699), Deferoxamine (DFO, 10 µM, cat#S5742), Necrostatin-1 (10 µM, cat#S8037), and Sorafenib (20 µM, cat#S7397) —all from Selleck (TX, USA).
Following 72 hours of siRNA treatment in NCI-H1975 cells, Fer-1 (10 µM, Selleck, TX, USA, cat#S7243) was added 24 hours prior to the assay. Mitochondrial membrane potential was assessed using Mito-Tracker Red FM (Beyotime Biotechnology, Shanghai, China, cat#C1032) following the supplier’s instructions. Fluorescence intensity was recorded using the Cytation5 cell imaging multi-mode reader (BioTek Cytation 5, BioTek). A reduction in mitochondrial membrane potential was interpreted as a hallmark of ferroptosis.
After 72 hours of AURKA knockdown and 24-hour Fer‑1 pre-treatment (10 µM), NCI‑H1975 cells were fixed in 2.5% glutaraldehyde in 0.1 M Sorenson buffer (pH 7.2) for 1 hour, followed by postfixation in 1% osmium tetroxide for another hour. Samples were dehydrated, embedded, and sections into 60 nm ultrathin slices using an MT-7000 ultramicrotome (RMC, Tucson, AZ, USA, cat#MT-7000). Sections were stained with uranyl acetate and lead citrate, and mitochondrial ultrastructure was observed using a JEOL JEM-1200 EXII transmission electron microscope (JEOL, Tokyo, Japan, cat#JEM-1200 EXII).
To assess lipid peroxidation and antioxidant status, malondialdehyde (MDA) and glutathione (GSH) levels were measured in NCI-H1975 cells after 72 hours of siRNA transfection, with Fer-1 (10 µM) pre-treatment for 24 hours. Commercial kits (Beyotime Biotechnology, cat#S0131M for MDA and cat#S0053 for GSH) were used following the supplier’s instructions. Elevated MDA levels were indicative of increased lipid peroxidation, while decrease GSH levels reflected consumption during oxidative stress.
ROS were quantified using the MitoSOX™ Red mitochondrial superoxide indicator (Beyotime Biotechnology, cat#S0061S). After 72 hours of AURKA knockdown and 24-hour Fer‑1 treatment, cells were incubated with the probe following the manufacturer’s protocol. ROS levels were analyzed via flow cytometry. A marked increase in mitochondrial ROS was considered evidence of ferroptotic oxidative injury.
NCI-H1975 cells transfected with AURKA-targeting siRNA were collected, washed with PBS and stained with Annexin V–FITC and propidium iodide (PI) using a commercial apoptosis detection kit (Beyotime Biotechnology, cat#C1052). After a 15-minute incubation in the dark at room temperature, followed by additional buffer wash and repeat staining, samples were analyzed by flow cytometry to determine apoptotic and necrotic cell populations.
For cell cycle distribution, siRNA-treated cells were fixed in pre-cooled 70% ethanol at 4 °C for at least 30 minutes. After fixation, cells were washed in PBS and resuspended in 0.5 mL of PI/RNase staining buffer (50 µg/mL PI, 200 µg/mL RNase). Samples were incubated at 37 °C for 30 minutes and analyzed within 1 hour using flow cytometry to determine the percentage of cells in G0/G1, S, and G2/M phases.
Western blotting was conducted to assess protein expression following pharmacologic and genetic interventions. NCI-H1975 cells were pretreated with the AURKA inhibitor LY3295668 (500 nM, Selleck, TX, USA, cat#S8782) for 72 hours and the NRF2 inhibitor ML385 (10 µM, Selleck, cat#S8790) for 24 hours before lysis. Cell lysates were prepared using PMSF-supplemented RadioImmunoPrecipitation Assay (RIPA) buffer (Solarbio, China, cat#R0010), and protein concentrations were determined using a bicinchoninic acid (BCA) assay kit (Sangon Biotech, China, cat#C503021). Subsequently, 10 µg of total protein per sample was separated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked in 5% BSA for 1 hour, incubated overnight at 4 °C with primary antibodies, washed with Tris-buffered saline with Tween®20 (TBST), and probed with horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA, USA, cat#7074P2/cat#7076P2, 1:4000) for 1 hour at room temperature. Chemiluminescent detection was performed using BeyoECL Moon (Beyotime Biotechnology, Shanghai, China, cat#P0018FS).
The following primary antibodies were used: 4-HNE (Biosynthesis Biotechnology, Beijing, China, cat#bs6313R, 1:1000), GPX4 (Cell Signaling Technology, cat#52455, 1:1000), solute carrier family 7 member 11 (SLC7A11; ABclonal, China, cat#A13685, 1:1000), NRF2 (Cell Signaling Technology, cat#12721, 1:1000), KEAP1 (Cell Signaling Technology, cat#8047, 1:1000), HO-1 (Cell Signaling Technology, cat#86806, 1:1000), AURKA (Cell Signaling Technology, cat#91590, 1:1000), Cyclin D1 (Cell Signaling Technology, cat#55506, 1:1000), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Sangon Biotech, Shanghai, China, cat#D110016, 1:1000). Secondary antibodies included anti-rabbit IgG (Cell Signaling Technology, cat#7074, 1:3000) and anti-mouse IgG (Cell Signaling Technology, cat#5127, 1:3000).
Paraffin-embedded tissue sections were baked at 60 °C for 1, deparaffinized in xylene, and rehydrated through graded ethanol. Antigen retrieval was performed in citrate buffer. Endogenous peroxidase activity was quenched with 3% H2O2 for 30 minutes, followed by blocking with 5% goat serum for another 30 minutes. Sections were incubated overnight at 4 °C with anti-AURKA (Cell Signaling Technology, cat#91590, 1:300), followed by 30-minute incubation with HRP-conjugated secondary antibody at 37 °C. Immunoreactivity was visualized with the 3,3′-diaminobenzidine (DAB, Sangon Biotech, Shanghai, China, cat#A600140), and nuclei were counterstained with hematoxylin.
For gene expression analysis, NCI-H1975 cells were plated in 6-well plates and treated with ML385 (10 µM) for 24 hours and AURKA siRNA for 72 hours. Total RNA was extracted using TRIzol reagent (Solarbio, China, cat#10296010) and reverse transcribed with M-MLV Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA, cat# 28025013) from 1 µg of RNA in a 20 µL reaction. qPCR was performed with SYBR® Green (Promega, Madison, WI, USA, cat# A6001) on the ABI 7500 system. GAPDH was used as an internal control, and relative gene expression was calculated via the 2-ΔΔCt method [20]. All reactions were performed in triplicate. Primer sequences for GAPDH, AURKA, and NRF2, are listed in Table 1.
| Gene | Primer sequence 5′-3′ |
| NRF2 | F: CAAGTCCCAGTGTGGCATCA |
| R: CCCCTGAGATGGTGACAAGG | |
| GAPDH | F: ATGGGGAAGGTGAAGGTCG |
| R: GGTCATTGATGGCAACAATATC | |
| AURKA | F: AAGACTTGGGTCCTTGGGTC |
| R: CAAAGGAATGCGCTGGGAAG |
NRF2, NF-E2-Related Factor 2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; AURKA, Aurora kinase A.
Cells were seeded on coverslips in 6-well plates and treated with ML385 (10 µM) and siRNA as described. After fixation with 4% paraformaldehyde for 20 minutes, cells were permeabilized with 0.5% Triton X-100 buffer (Beyotime, Shanghai, China, cat#P0096) and blocked with 5% BSA for 30 minutes. Primary antibody against NRF2 was applied overnight at 4 °C. After PBS washes, fluorescent secondary antibody (CST, USA, cat#7074, 1:800) were added for 1 hour at room temperature. Nuclei were counterstained with DAPI (Cell Signaling Technology, Danvers, USA, cat#4083). Images were acquired using the Cytation 5 system and quantified using ImageJ.
All data are presented as mean
To explore genetic factors related to LUAD mutations, we analyzed the somatic mutation profiles of individual samples from the TCGA-LUAD cohort. As shown in Supplementary Fig. 1A, the top frequently mutated genes included titin (TTN), mucin 16 (MUC16), ryanodine receptor 2 (RYR2), CUB and Sushi multiple domains 3 (CSMD3), tumor protein p53 (TP53), low density lipoprotein receptor related protein 1B (LRP1B), usherin (USH2A), zinc finger homeobox 4 (ZFHX4), xin actin binding repeat containing 2 (XIRP2), and KRAS proto-oncogene (KRAS). Further assessment of commonly altered oncogenic drivers in lung cancer revealed high mutation frequencies in KRAS, EGFR, and B-Raf proto-oncogene (BRAF) (Supplementary Fig. 1B).
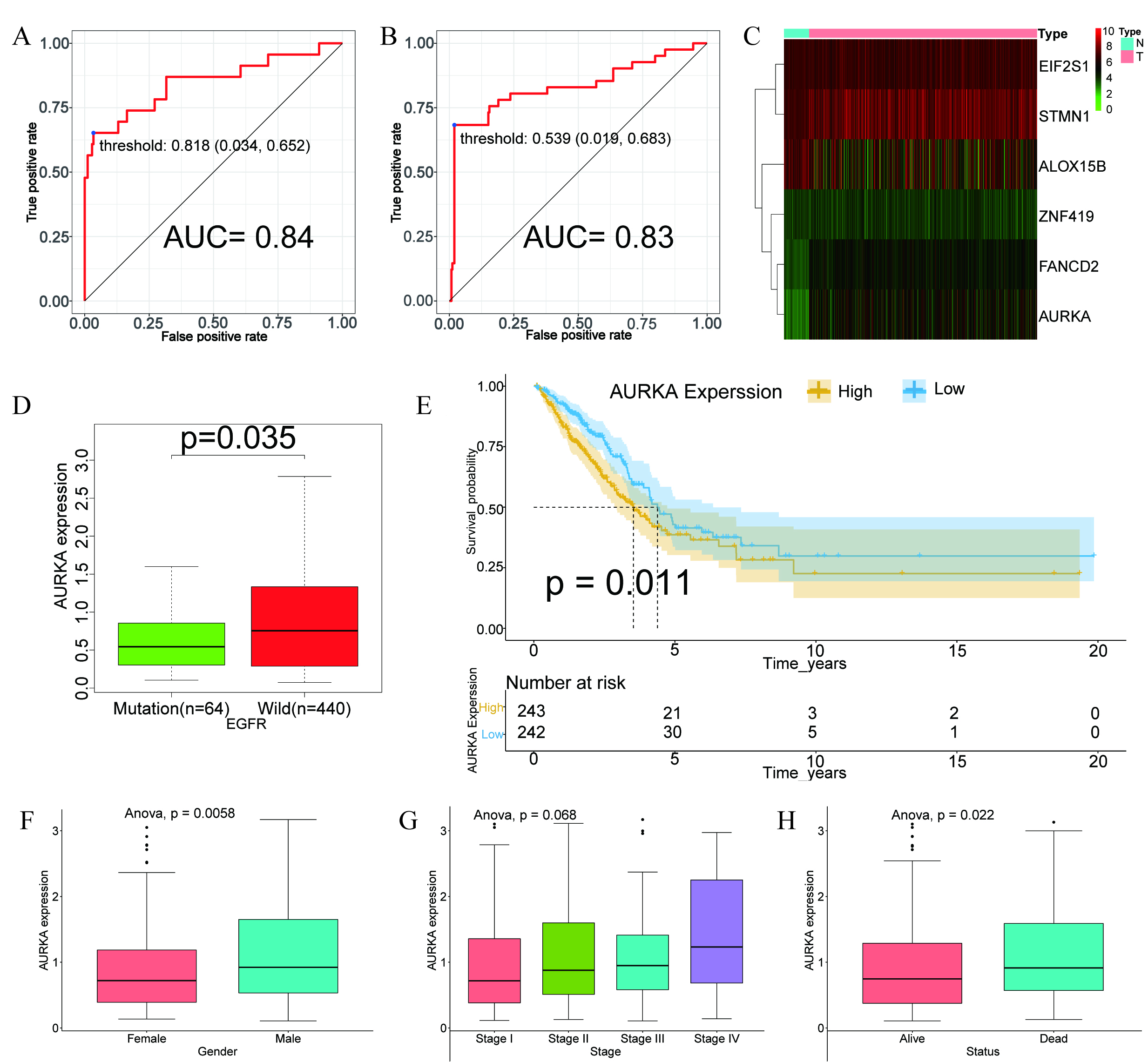
Considering the higher frequency of EGFR mutations in Asians populations [3, 4] and the widespread clinical use of EGFR‑targeted therapies—despite rising resistance rates [31, 32]—we developed a ferroptosis gene—based random forest model to predict EGFR mutation status. The model’s convergence is illustrated in Supplementary Fig. 2A, and the ranked variable importance is displayed in Supplementary Fig. 2B. The model demonstrated strong predictive power, with AUC values of 0.84 in the training set (Fig. 1A) and 0.83 in the validation set (Fig. 1B), indicating robust performance and generalizability.
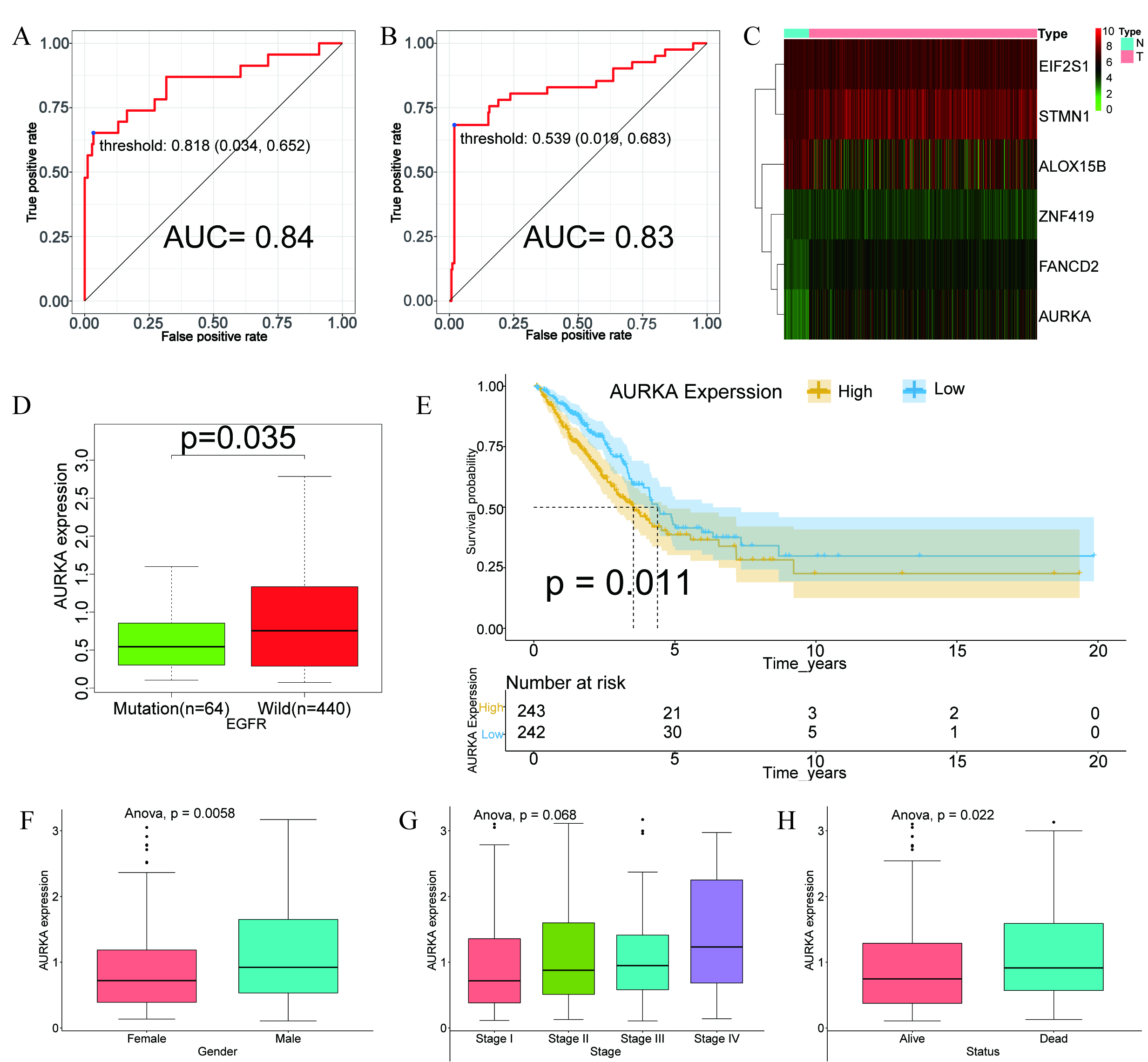 Fig. 1.
Fig. 1.
Construction of the ferroptosis-based random forest model and prognostic analysis of AURKA. (A,B) Receiver operating characteristic (ROC) curves illustrating the performance of the random forest model in the training (A) and validation (B) datasets. (C) Heatmap showing differential expression of model-related genes in LUAD versus normal tissues. (D) Box plot illustrating the association between AURKA expression and EGFR mutation status. (E) Kaplan–Meier survival curve showing that high AURKA expression correlates with poor prognosis (p = 0.011). (F–H) Correlation of AURKA expression with gender (p = 0.0058), tumor stage (p = 0.068) and survival status (p = 0.022). AURKA, Aurora kinase A; LUAD, adenocarcinoma of Lung; EGFR, epidermal growth factor receptor; EIF2S1, eukaryotic translation initiation factor 2 subunit 1; STMN1, stathmin-1; ALOX15B, arachidonate 15-lipoxygenase type B; ZNF419, zinc finger protein 419; FANCD2, fanconi anemia group D2 protein; AUC, area under the curve.
Heatmap analysis comparing tumors with adjacent normal tissues showed downregulation of arachidonate 15-lipoxygenase type B (ALOX15B), alongside upregulation of fanconi anemia group D2 protein (FANCD2), eukaryotic translation initiation factor 2 subunit 1 (EIF2S1), stathmin-1 (STMN1), zinc finger protein 419 (ZNF419), and AURKA in tumor samples (Fig. 1C).
To identify genes linked to EGFR status, we examined their expression in EGFR-mutant versus wild-type subgroups. Notably, AURKA expression was significantly elevated in EGFR wild-type (p = 0.035; Fig. 1D), suggesting a potential inverse association between AURKA activity and EGFR mutations.
To evaluate AURKA’s prognostic relevance, LUAD patients were categorized into high- and low-expression cohorts using the median expression as a cutoff. Kaplan–Meier analysis revealed that elevated AURKA levels were significantly linked to reduced overall survival (p = 0.011; Fig. 1E). Additionally, high AURKA expression correlated with more advanced pathological stage, male sex, and increased mortality (Fig. 1F–H), supporting its potential role as a negative prognostic biomarker in LUAD.
To investigate how AURKA expression correlates with immune cell infiltration in
LUAD, we utilized the CIBERSORT algorithm to estimate the proportions of 22
distinct immune cell types. Comparative analysis between tumor and adjacent
normal tissues revealed that 17 immune cell types exhibited significant
differences. Specifically, M2 macrophages, M0 macrophages, resting CD4 memory T
cells, resting mast cells, activated dendritic cells, monocytes, neutrophils,
eosinophils, and resting NK cells showed decreased infiltration in tumor
(p
Patients were divided into high- and low-AURKA expression cohorts according to
the median transcript level. Among the 22 immune cell types, 14 showed
significant infiltration differences between the two groups. High AURKA
expression was associated with elevated infiltration of M0 and M1 macrophages,
follicular helper T cells, activated CD4 memory T cells, activated mast cells,
resting NK cells, and
Conversely, resting CD4 memory T cells, resting mast cells, resting dendritic
cells, Tregs, activated dendritic cells, monocytes, and memory B cells were more
abundant in the low AURKA expression group (p
Further analysis showed that AURKA expression was positively correlated with programmed Cell Death 1 (PDCD1) (p = 0.022), while no significant association was observed with CD274 (p = 0.066) or cytotoxic T-lymphocyte associated protein 4 (CTLA4, p = 0.22; Supplementary Fig. 3C–E). These results indicate that AURKA may play a pivotal role in shaping the immune microenvironment of LUAD, potentially affecting tumor progression through immune modulation. Collectively, the data support the classification of AURKA as a high-risk gene in LUAD, given its associations with EGFR mutation status, immune infiltration patterns, and unfavorable prognosis.
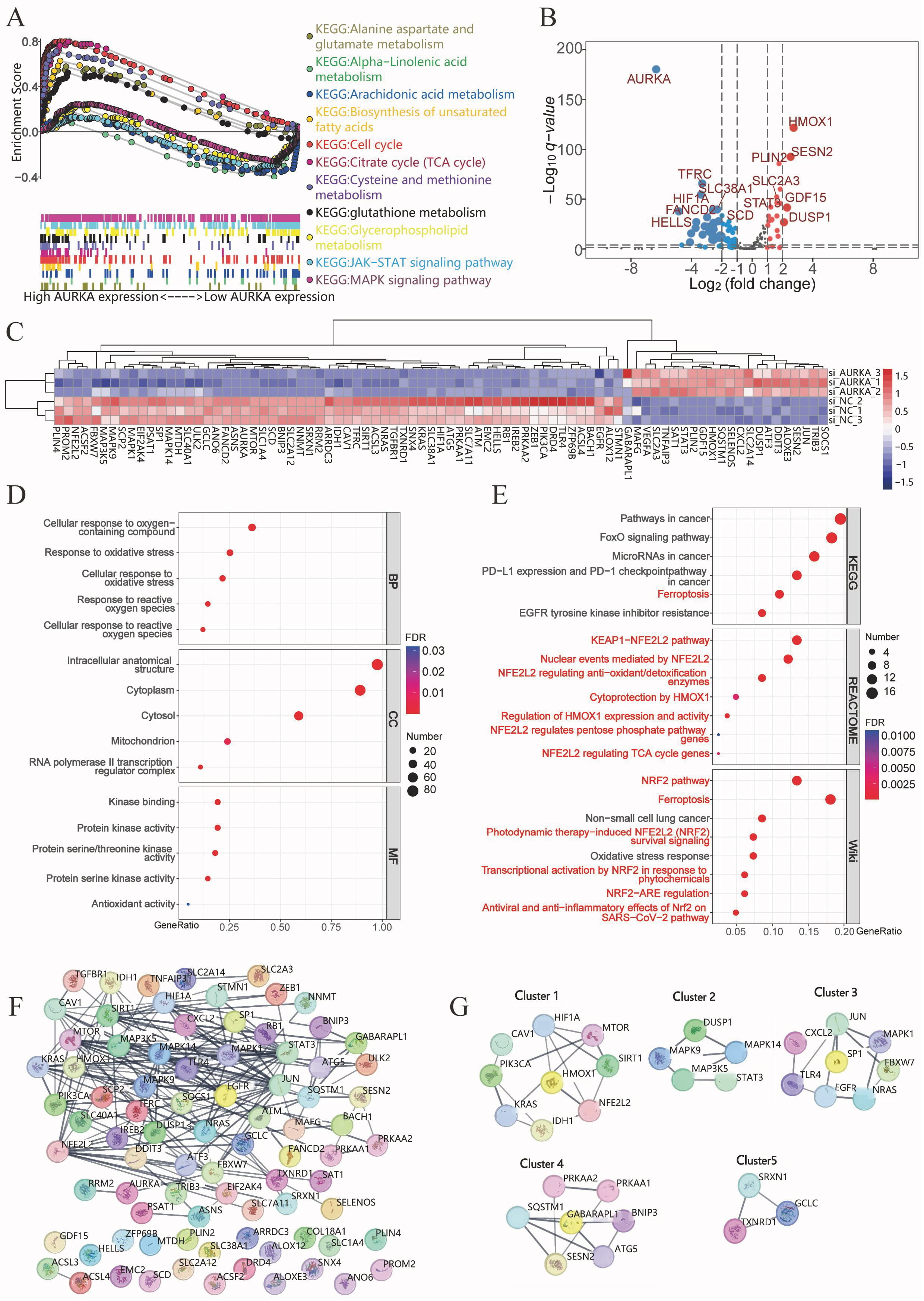
To uncover the biological pathways associated with AURKA, GSEA was performed
using TCGA-LUAD transcriptomic data. Among 177 pathways analyzed, 124 showed a
positive correlation with AURKA expression, with 42 significantly enriched
(p
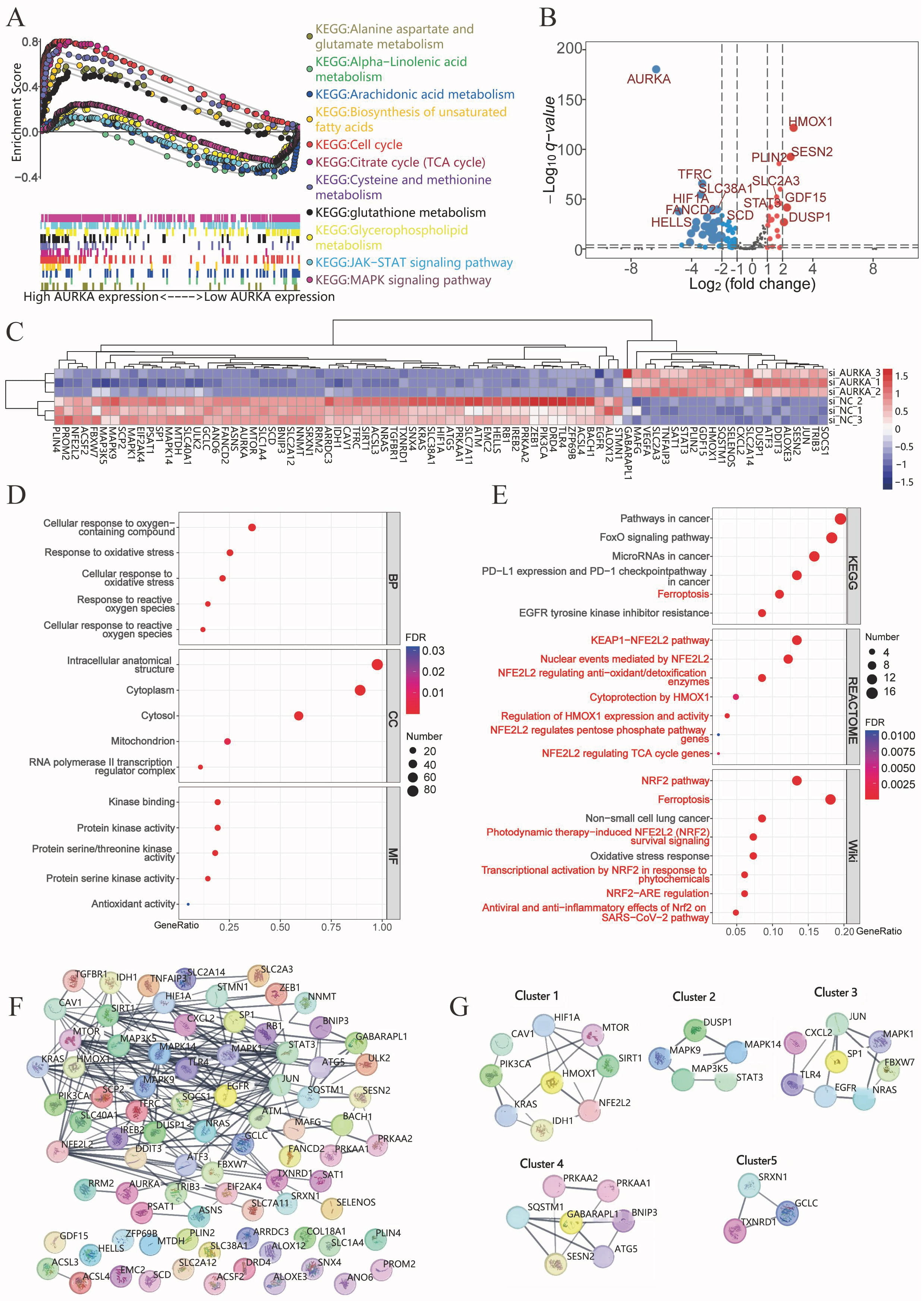 Fig. 2.
Fig. 2.
AURKA signaling pathway analysis in LUAD. (A) Gene set enrichment analysis (GSEA) showing AURKA-associated pathways in TCGA LUAD data. (B,C) Volcano plot (B) and heatmap (C) of differential expressed ferroptosis-related genes following AURKA knockdown in NCI-H1975 cells. (D) GO enrichment of differentially expressed genes. (E) KEGG, Reactome, and WikiPathways analyses indicating ferroptosis- and NRF2-related pathway enrichment. (F) PPI network constructed from 83 ferroptosis-related genes using STRING. (G) Identification of key gene modules via MCODE analysis in Cytoscape. TCGA, The Cancer Genome Atlas; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; NRF2, NF-E2-Related Factor 2; PPI, protein–protein interaction; MCODE, molecular complex detection; TCA, the tricarboxylic acid.
| KEGG pathway | ES | NES | NOM p-val | FDR q-val |
| KEGG_ALANINE_ASPARTATE_AND_GLUTAMATE_METABOLISM | 0.58 | 1.67 | 0.008 | 0.019 |
| KEGG_BIOSYNTHESIS_OF_UNSATURATED_FATTY_ACIDS | 0.63 | 1.67 | 0.019 | 0.019 |
| KEGG_CELL_CYCLE | 0.8 | 2.97 | 0 | 0 |
| KEGG_CITRATE_CYCLE_TCA_CYCLE | 0.8 | 2.33 | 0 | 0 |
| KEGG_CYSTEINE_AND_METHIONINE_METABOLISM | 0.71 | 2.16 | 0 | 0 |
| KEGG_GLUTATHIONE_METABOLISM | 0.52 | 1.73 | 0.001 | 0.011 |
| KEGG_MAPK_SIGNALING_PATHWAY | –0.27 | –1.43 | 0 | 0.078 |
| KEGG_JAK_STAT_SIGNALING_PATHWAY | –0.34 | –1.71 | 0 | 0.017 |
| KEGG_GLYCEROPHOSPHOLIPID_METABOLISM | –0.31 | –1.38 | 0.036 | 0.098 |
| KEGG_ALPHA_LINOLENIC_ACID_METABOLISM | –0.46 | –1.42 | 0.057 | 0.076 |
| KEGG_ARACHIDONIC_ACID_METABOLISM | –0.37 | –1.58 | 0.014 | 0.033 |
Abbreviation: FDR, false discovery rate; NOM p-val, nominal p value; GSEA, Gene Set Enrichment Analysis; TCGA, The Cancer Genome Atlas; LUAD, adenocarcinoma of Lung; KEGG, Kyoto Encyclopedia of Genes and Genomes; ES, enrichment score; NES, normalized enrichment score.
To validate these findings, RNA sequencing was performed on NCI-H1975 cells following AURKA knockdown via siRNA. Differential expression analysis revealed 61 downregulated and 22 upregulated ferroptosis-related genes in the knockdown group (Fig. 2B,C). Functional enrichment analyses were conducted using GO, Reactome, KEGG, and WikiPathways databases to investigate the biological relevance of these genes. GO analysis indicated significant enrichment in oxidative stress and ROS-related processes (Fig. 2D). KEGG pathways included ferroptosis and FoxO signaling pathways—both linked to ROS homeostasis (Fig. 2E). Reactome analysis highlighted the KEAP1/NRF2 axis, and regulation of HO-1 activity, while WikiPathways emphasized enrichment in the ferroptosis and NRF2 signaling pathways, reinforcing the connection between AURKA and ferroptosis through the NRF2 pathway (Fig. 2E).
A PPI network of 83 differentially expressed ferroptosis-related genes was generated using STRING. MCODE clustering analysis identified five functional modules with scores of 4, 3.5, 3.429, 3.333, and 3, respectively (Fig. 2F). Four key hub genes— HO-1 (cluster 1), SP1 (cluster 3), GABARAPL1 (cluster 4), and SRXN1 (cluster 5) —were identified as seed genes (Fig. 2G). These data strongly implicate AURKA in the regulation of ferroptosis, potentially via modulation of the KEAP1/NRF2/HO-1 signaling axis.
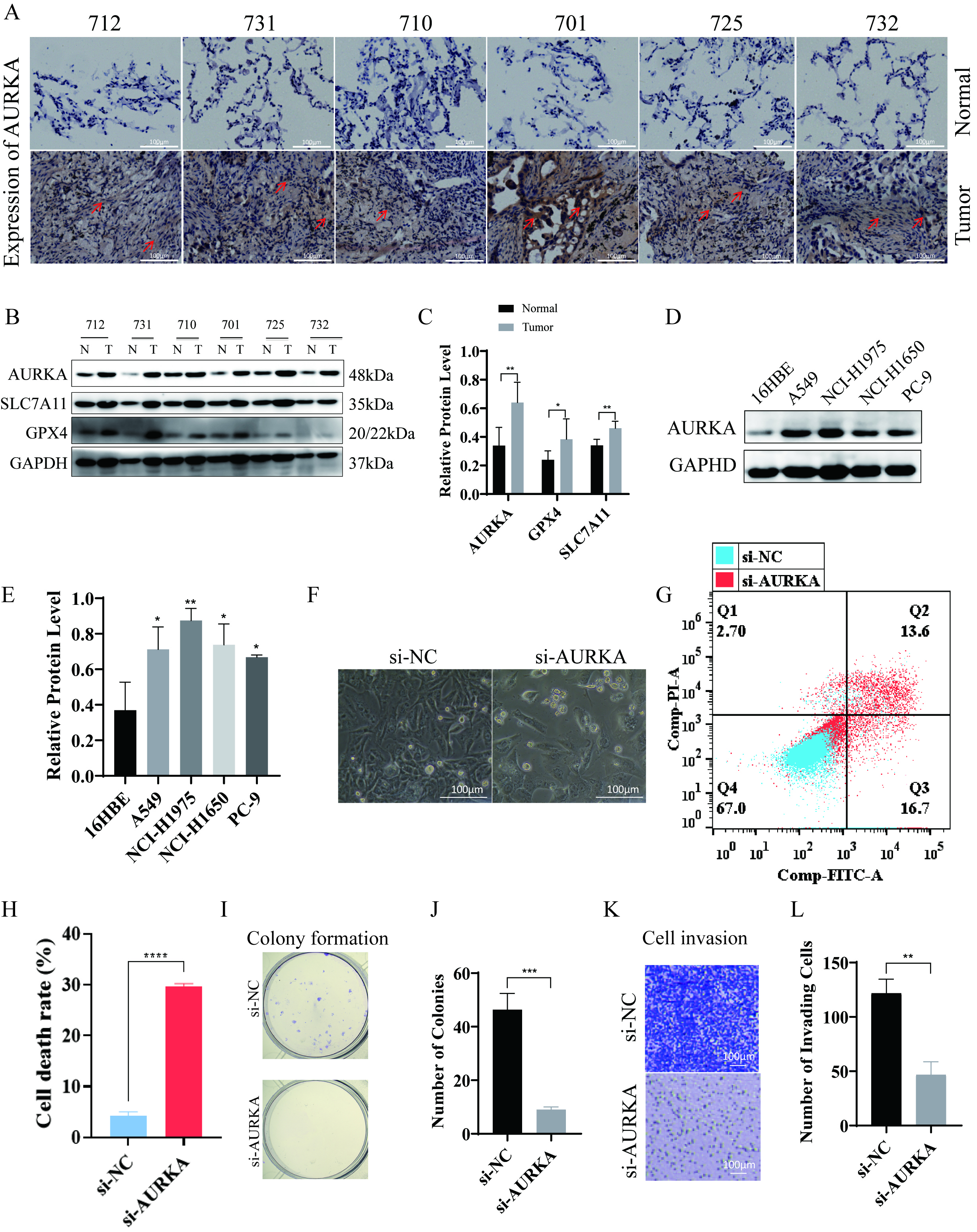
To evaluate AURKA expression in EGFR-mutant LUAD, Western blotting and immunohistochemical staining were performed on tumor specimens from six patients. Clinical and demographic characteristics—including sex, age, histological subtype, and TNM stage (classified according to the 9th edition of the AJCC Staging Manual) [33]—are detailed in Table 3. The results demonstrated strong expression of AURKA, along with ferroptosis-associated proteins GPX4 and SLC7A11, in tumor tissues (Fig. 3A–C), suggesting that LUAD with EGFR mutations may exhibit an upregulated ferroptosis-related protein profile, potentially indicating ferroptosis sensitivity.
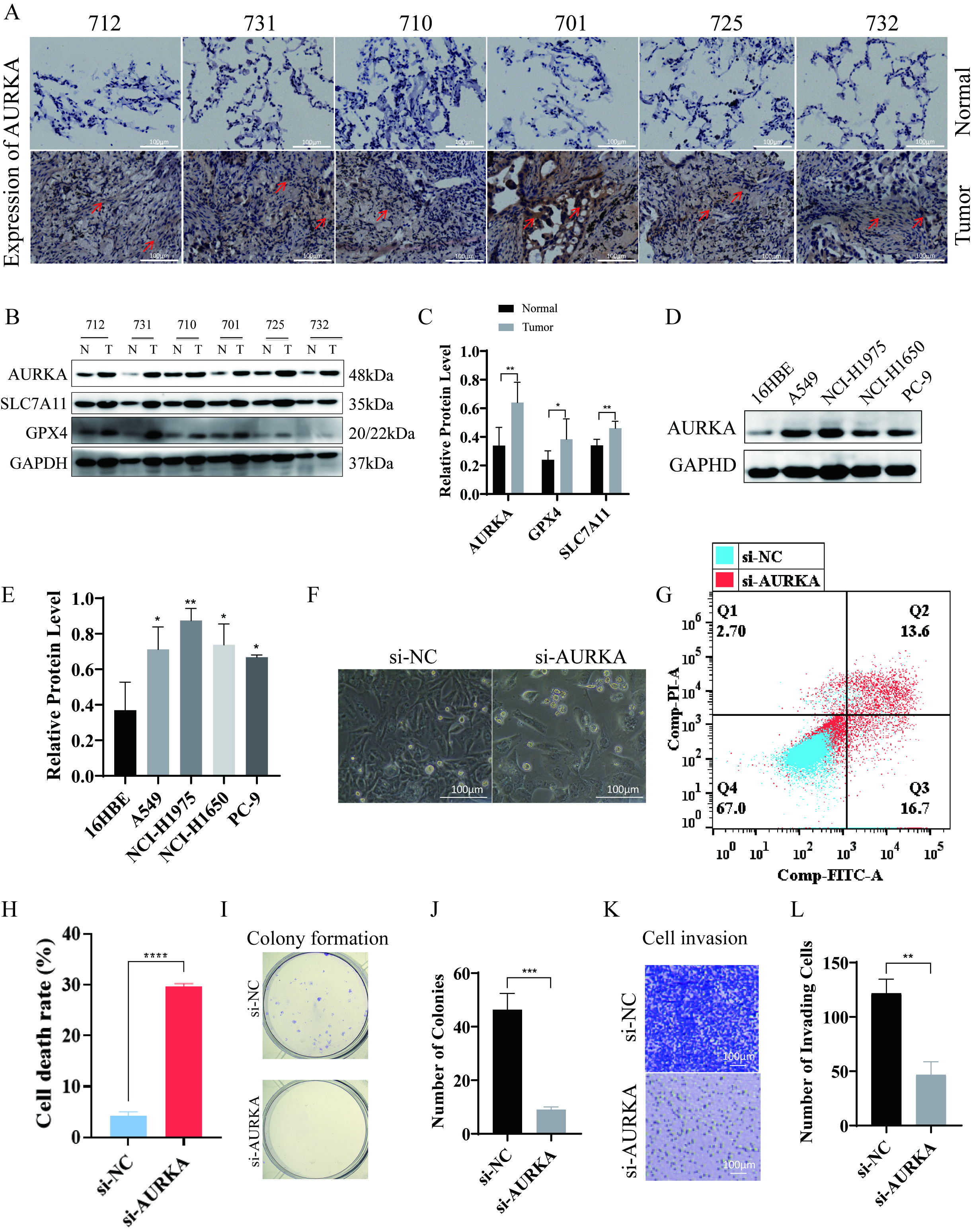 Fig. 3.
Fig. 3.
AURKA expression and its functional impact on ferroptosis and
proliferation in NCI-H1975 cells. (A) Immunohistochemical showing AURKA
expression in six LUAD tumor–normal tissue pairs (red arrows indicate elevated
staining), scale bar = 100 µm. (B,C) Western blot analysis (B) and quantification (C) of AURKA, GPX4,
and SLC7A11 in LUAD tumor vs. adjacent tissues. (D,E) AURKA expression across
LUAD cells lines, with quantification (E). (F) Morphological changes in NCI-H1975
cells after AURKA knockdown (20
| Patient ID | Sex | Age (years) | Histology type | TNM stage |
| 712 | Male | 61 | Adenocarcinoma | pT2a N1M0R0 (IIb) |
| 731 | Male | 60 | Adenocarcinoma | pT1c N0M0R0 (Ia3) |
| 710 | Male | 74 | Adenocarcinoma | pT2a N1M0R0 (IIb) |
| 701 | Female | 58 | Adenocarcinoma | pT2a N0M0R0 (Ib) |
| 725 | Female | 75 | Adenocarcinoma | pT2a N0M0R0 (Ib) |
| 731 | Female | 60 | Adenocarcinoma | pT1c N0M0R0 (Ia3) |
TNM staging follows the 9th edition of the American Joint Committee on Cancer (AJCC) Cancer Staging Manual.
To further investigate the functional relevance of AURKA in LUAD, we examined its expression in the human bronchial epithelial-like cells 16HBE and four LUAD cell lines: A549, NCI-H1975, NCI-H1650, and PC-9. Notably, NCI-H1975 cells harbor the EGFR T790M mutation, which confers primary resistance to first-generation EGFR-TKIs such as gefitinib. This makes NCI-H1975 an ideal model for investigating therapeutic strategies in TKI-resistant LUAD. The results revealed the highest AURKA expression in NCI-H1975 cells (Fig. 3D,E), leading us to select these cells for further experiments.
siRNA-mediated AURKA knockdown was then performed in NCI-H1975 cells.
Morphological assessment under a 20
Collectively, these results highlight the critical role of AURKA in promoting LUAD cell viability, proliferation, and metastatic behavior, underscoring its function as a key driver of malignancy in EGFR-mutant LUAD.
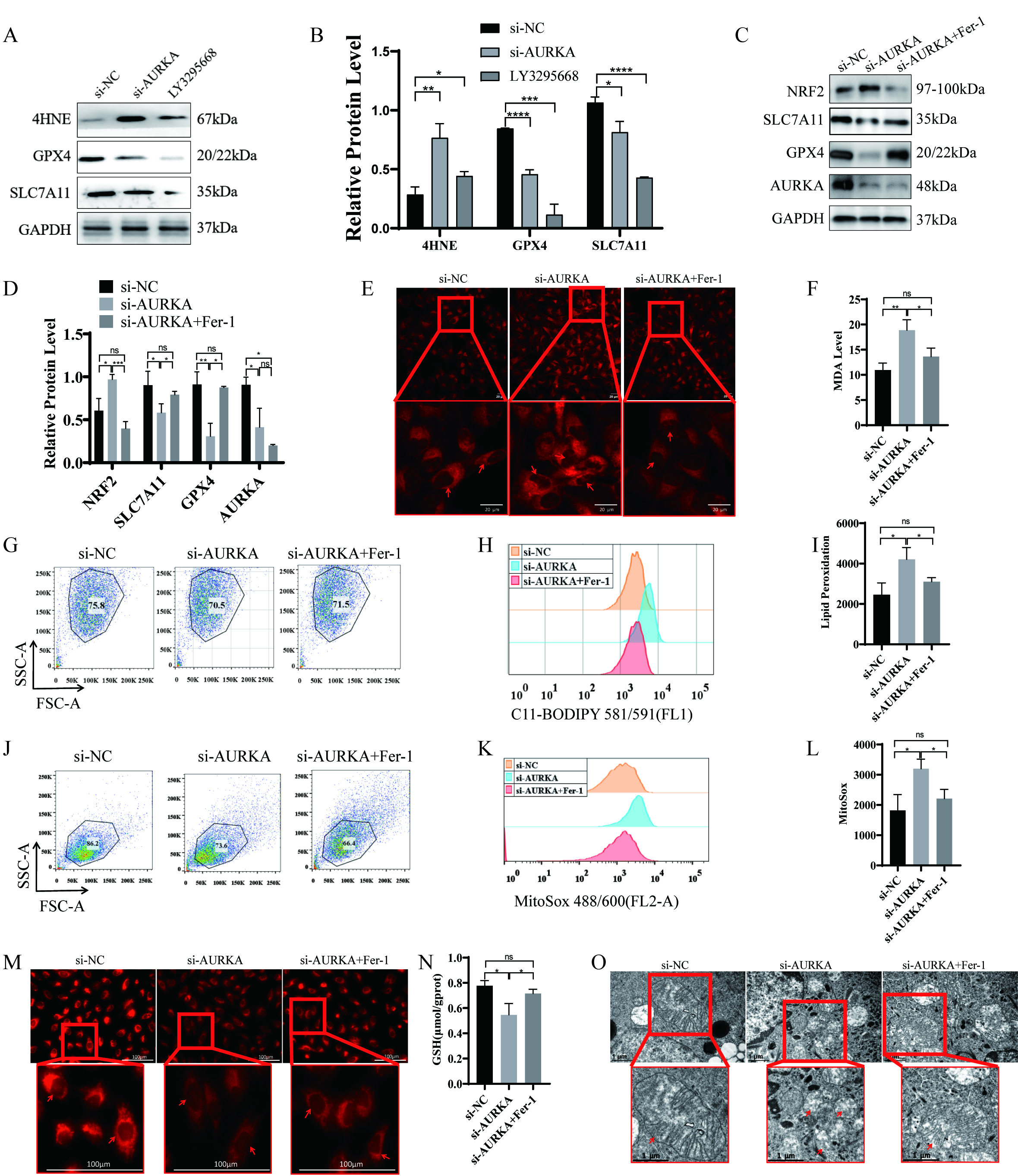
To determine whether AURKA regulates ferroptosis in lung cancer cells, we performed siRNA-mediated gene silencing and pharmacologic inhibition using the AURKA-specific inhibitor LY3295668. Both approaches resulted in decreased expression of the ferroptosis suppressors SLC7A11 and GPX4, along with elevated level of 4-Hydroxynonenal (4HNE) increased a marker of lipid peroxidation (Fig. 4A,B). These findings suggest that AURKA activity may negatively regulate ferroptosis in LUAD cells.
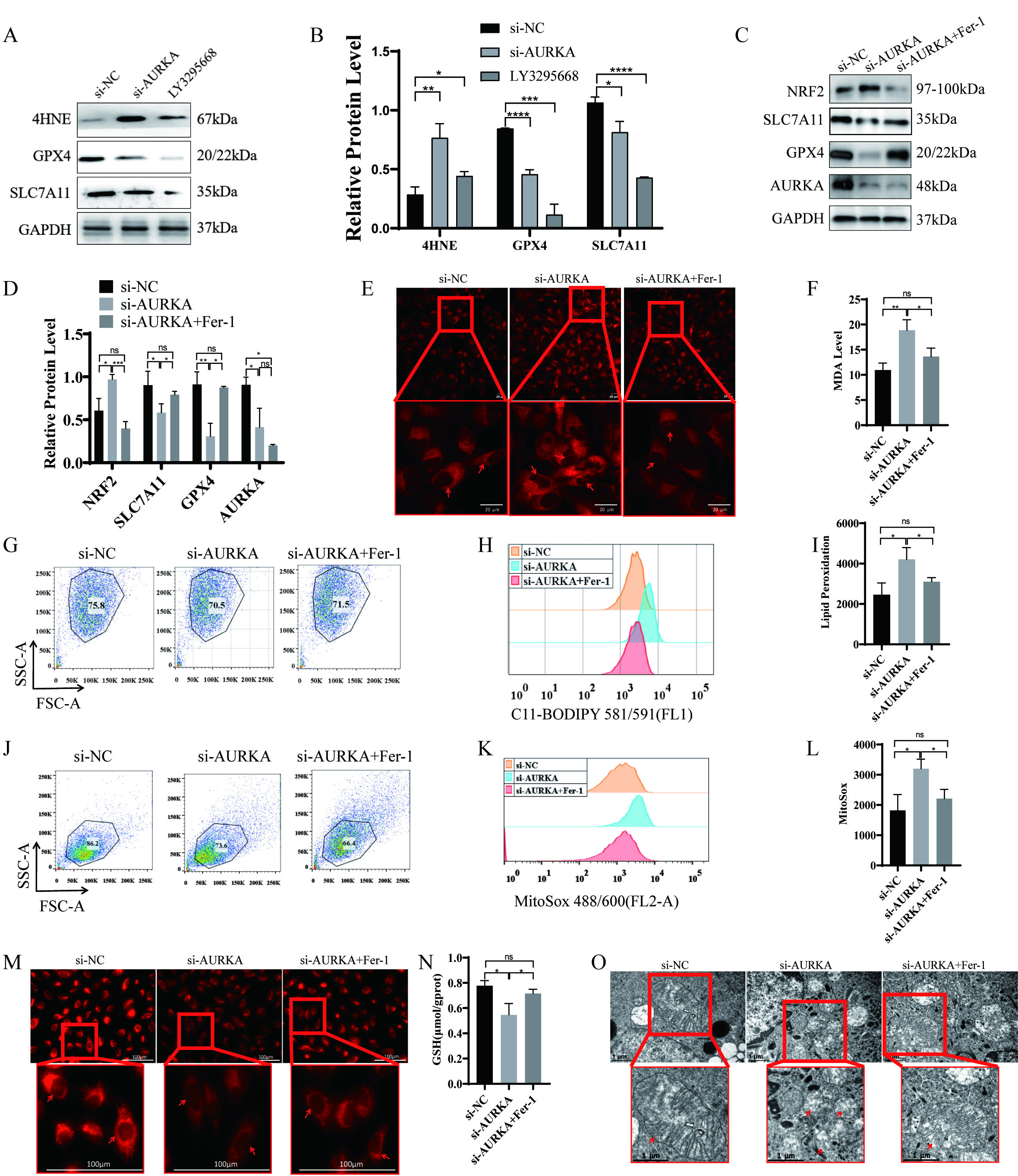 Fig. 4.
Fig. 4.
Ferroptosis induction following AURKA knockdown or inhibitor in
NCI-H1975 cells. (A,B) Western blot and quantification of ferroptosis-related
proteins (SLC7A11, GPX4, 4HNE) after AURKA knockdown or LY3295668 treatment.
(C,D) Western blot and quantification of ferroptosis markers after AURKA
knockdown with or without Fer-1 treatment. (E) Confocal imaging of intracellular
Fe2+ using FerroOrange (red intensity indicates iron levels, scale bar = 20
µm). (F) Quantification of intracellular MDA levels. (G–I) Lipid
peroxidation analysis using C11-BODIPY: dot plots (G), peak plots (H), and
statistical summary (I). (J–L) Mitochondrial ROS detection: dot plots (J), peak
plots (K), and quantification (L) following AURKA knockdown
To further validate this hypothesis, NCI-H1975 cells were treated with siAURKA in the presence or absence of ferroptosis inhibitor Fer-1. AURKA knockdown led to reduced protein levels of SLC7A11 and GPX4and upregulated NRF2 expression (Fig. 4C,D). Importantly, co-treatment with Fer-1reversed these changes, indicating that the observed molecular effects were ferroptosis-dependent.
Given that ferroptosis involves disrupted iron homeostasis, reduced GSH levels, and increased ROS, we subsequently assessed these key features. AURKA silencing led to increased intracellular ferrous iron (Fe2+; Fig. 4E), elevated MDA content (Fig. 4F), enhanced lipid peroxides as evidenced by C11-BODIPY staining (Fig. 4G–I), and increased mitochondrial ROS levels (Fig. 4J–L). Additionally, a significant decrease in mitochondrial membrane potential (Fig. 4M) and GSH levels (Fig. 4N) was observed. Transmission electron microscopy further confirmed classic ferroptotic morphological features, including mitochondrial shrinkage and cristae loss, in AURKA-silenced cells (Fig. 4O). All of these alterations were effectively reversed by Fer-1 co-treatment (Fig. 4E–O), confirming that AURKA knockdown induces ferroptosis in NCI-H1975 cells.
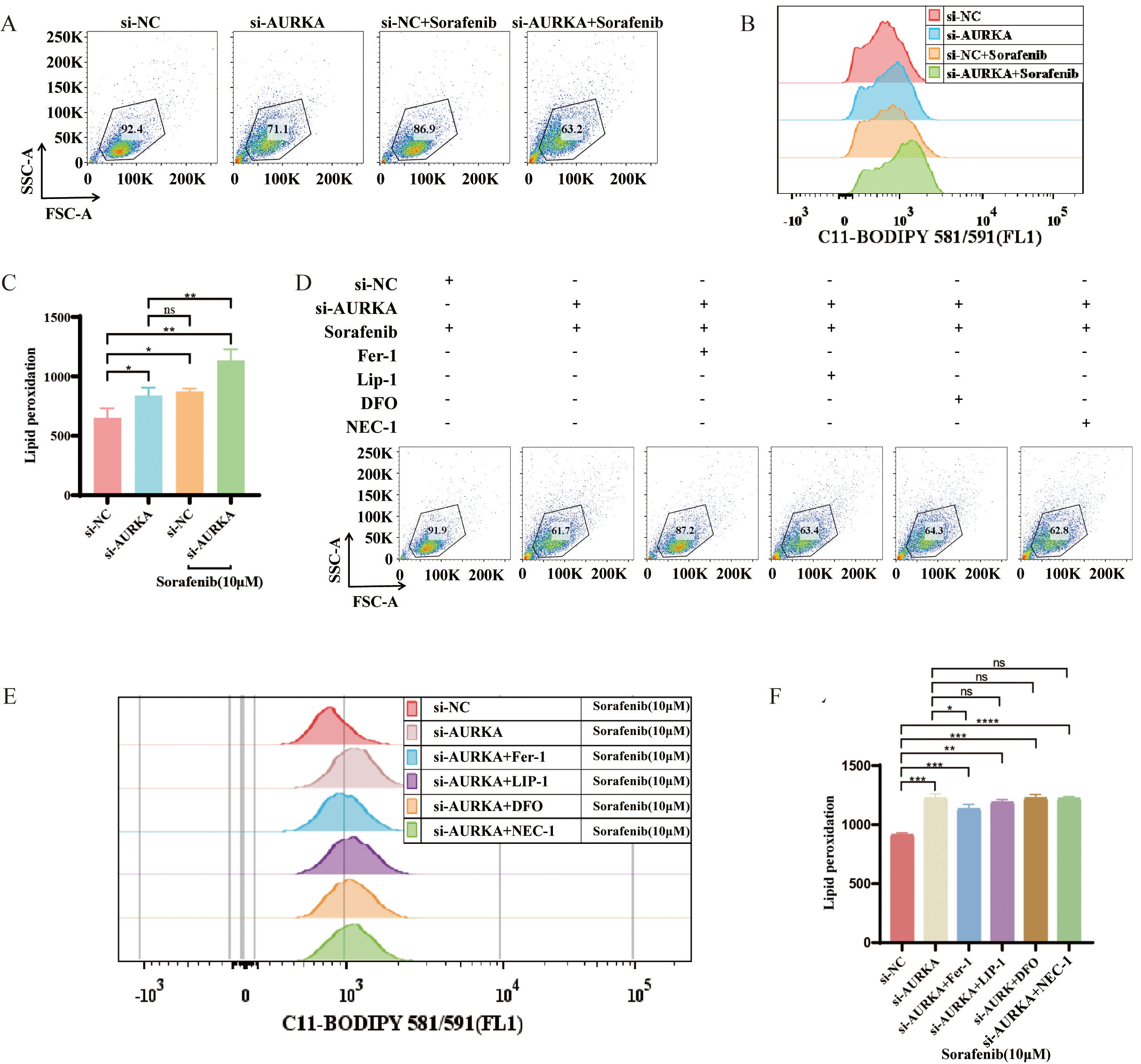
To further explore the interaction between AURKA and ferroptosis in the context
of pharmacologic induction, we assessed the effects of sorafenib, a
multi-targeted anti-cancer drug known to trigger ferroptosis [34, 35]. Compared
to sorafenib alone, AURKA knockdown significantly enhanced sorafenib-induced
lipid peroxidation (p
 Fig. 5.
Fig. 5.
AURKA regulates sorafenib-induced ferroptosis in NCI-H1975
cells. (A–C) Flow cytometry of lipid peroxidation in AURKA knockdown +
sorafenib vs. sorafenib alone. (D–F) Lipid peroxidation levels following
treatment with sorafenib alone or in combination with AURKA knockdown, Fer-1,
Lip-1, DFO, or necrostatin-1 (NEC-1); includes dot plots (D), peak plots (E), and
quantitative analysis (F). Fer-1, Ferrostatin-1; Lip-1, Liproxstatin-1; DFO,
Deferoxamine; Deferoxamine; *p
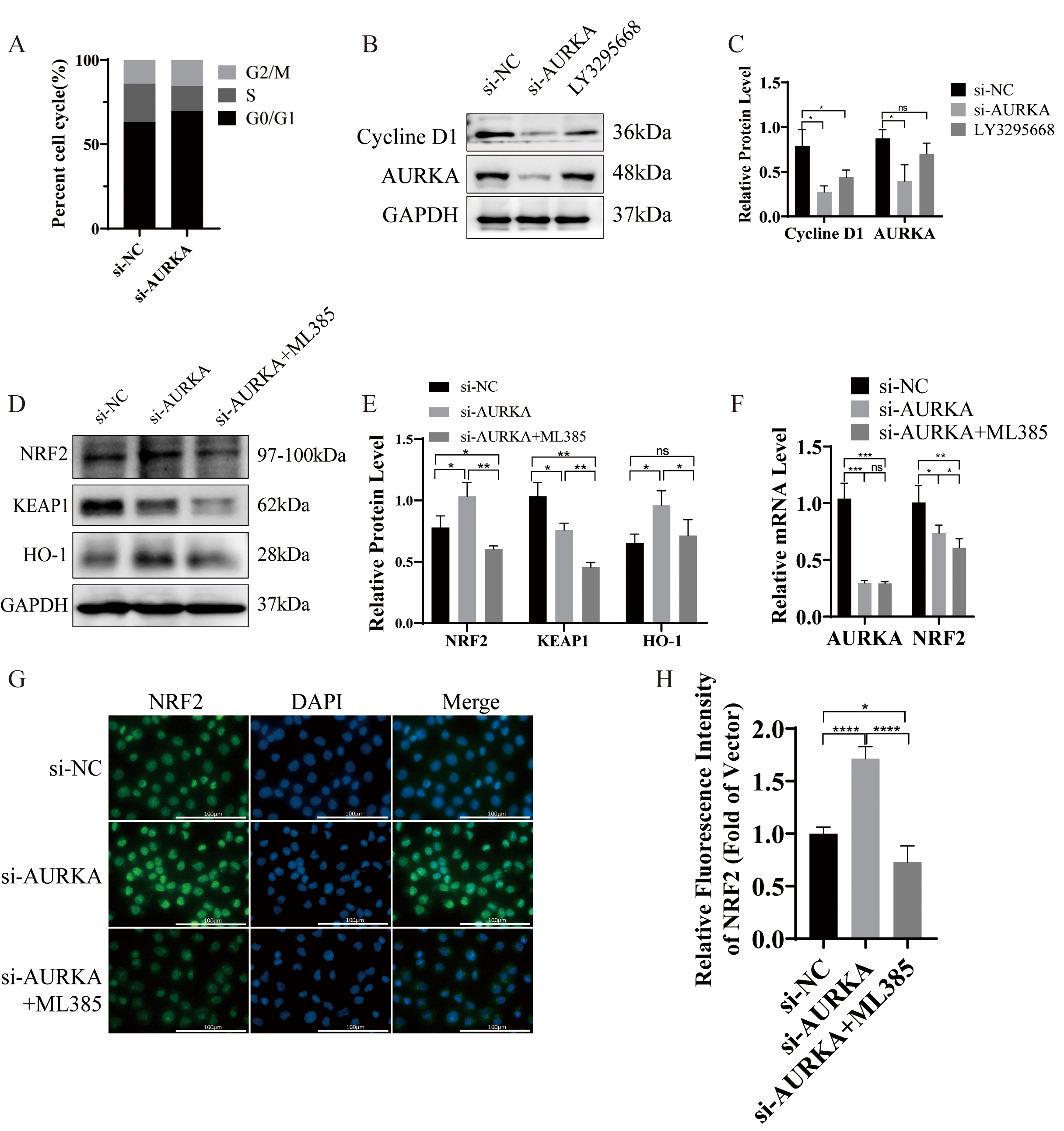
AURKA has been widely recognized for its essential function in driving mitosis and controlling the cell cycle [36, 37]. Consistent with this, GSEA analysis in our study identified a significant enrichment of the cell cycle pathway in association with AURKA expression (Fig. 2A; Table 2). To investigate whether AURKA knockdown disrupts cell cycle progression in LUAD cells, flow cytometry was employed to assess the cell cycle distribution, and Western blotting was used to evaluate cyclin expression.
Flow cytometric analysis showed that silencing AURKA resulted in significant G0/G1 phase enrichment, suggesting induction of cell cycle arrest (Fig. 6A). Additionally, Western blot analysis demonstrated reduced expression of cyclin D1 following either AURKA knockdown or treatment with the selective AURKA inhibitor LY3295668 (Fig. 6B,C). These findings collectively support that AURKA suppression induces G1 phase arrest in NCI-H1975 cells.
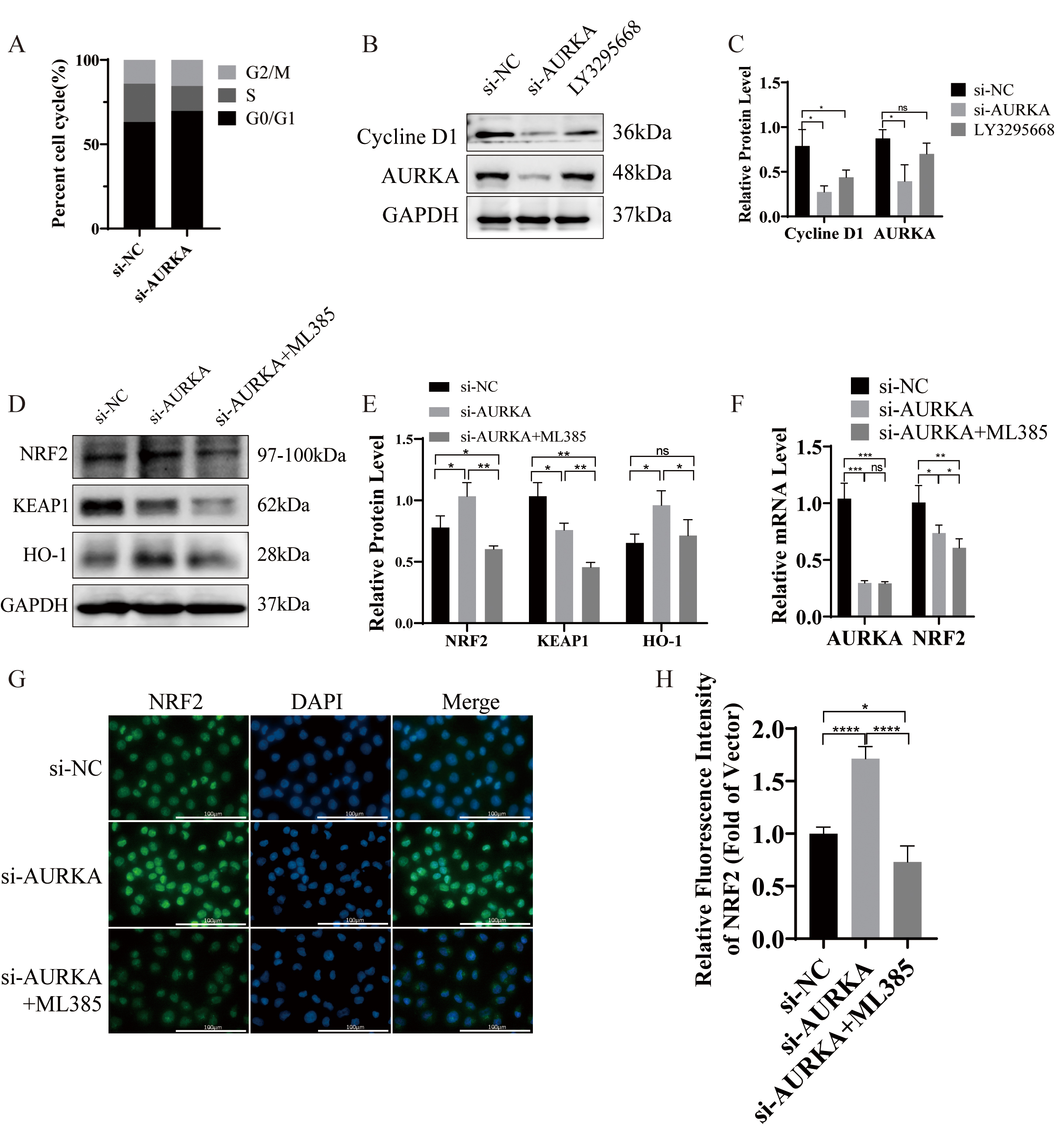 Fig. 6.
Fig. 6.
AURKA knockdown induces cell cycle arrest and activates
KEAP1/NRF2/H0-1 signaling in NCI-H1975. (A) Cell cycle analysis by flow
cytometry showing G0/G1 arrest after AURKA knockdown. (B,C) Western blot and
quantification of Cyclin D1 following AURKA knockdown or LY3295668 treatment.
(D,E) Western blot and quantification of KEAP1, NRF2, and HO‑1 expression after
AURKA knockdown
To elucidate the molecular mechanism underlying AURKA-mediated ferroptosis regulation, we performed Western blot analysis in NCI-H1975 cells following AURKA knockdown. Compared to controls, AURKA knockdown increased the protein expression of NRF2 and HO-1, while concurrently reducing KEAP1 levels (Fig. 6D,E). Co-treatment with the NRF2 inhibitor ML385 reversed these effects, suggesting that AURKA regulates ferroptosis, at least in part, through the NRF2/HO‑1 pathway.
Interestingly, qRT-PCR analysis revealed a decrease in NRF2 mRNA levels following AURKA knockdown (Fig. 6F), despite the observed increase in NRF2 protein level. This discordance implies that AURKA may regulate NRF2 post-transcriptionally through KEAP1, a negative regulator of NRF2 stability, thus supporting a mechanism involving protein-level modulation [38].
To further investigate NRF2 subcellular localization, immunofluorescence staining was performed, with DAPI used to label nuclei. Localization was inferred based on fluorescence patterns: signal peripheral to DAPI was considered membrane-associated, perinuclear distribution suggested cytoplasmic localization, and complete colocalization with DAPI indicated nuclear localization. In control NCI-H1975 cells, NRF2 was primarily cytoplasmic (Fig. 6G). AURKA knockdown promoted nuclear accumulation of NRF2 proteins, a change that was effectively reversed by ML385 (Fig. 6G,H). These findings collectively demonstrate that AURKA knockdown activates the NRF2/HO-1 axis by promoting NRF2 nuclear translocation, thereby inducing ferroptosis in EGFR-mutant LUAD cells.
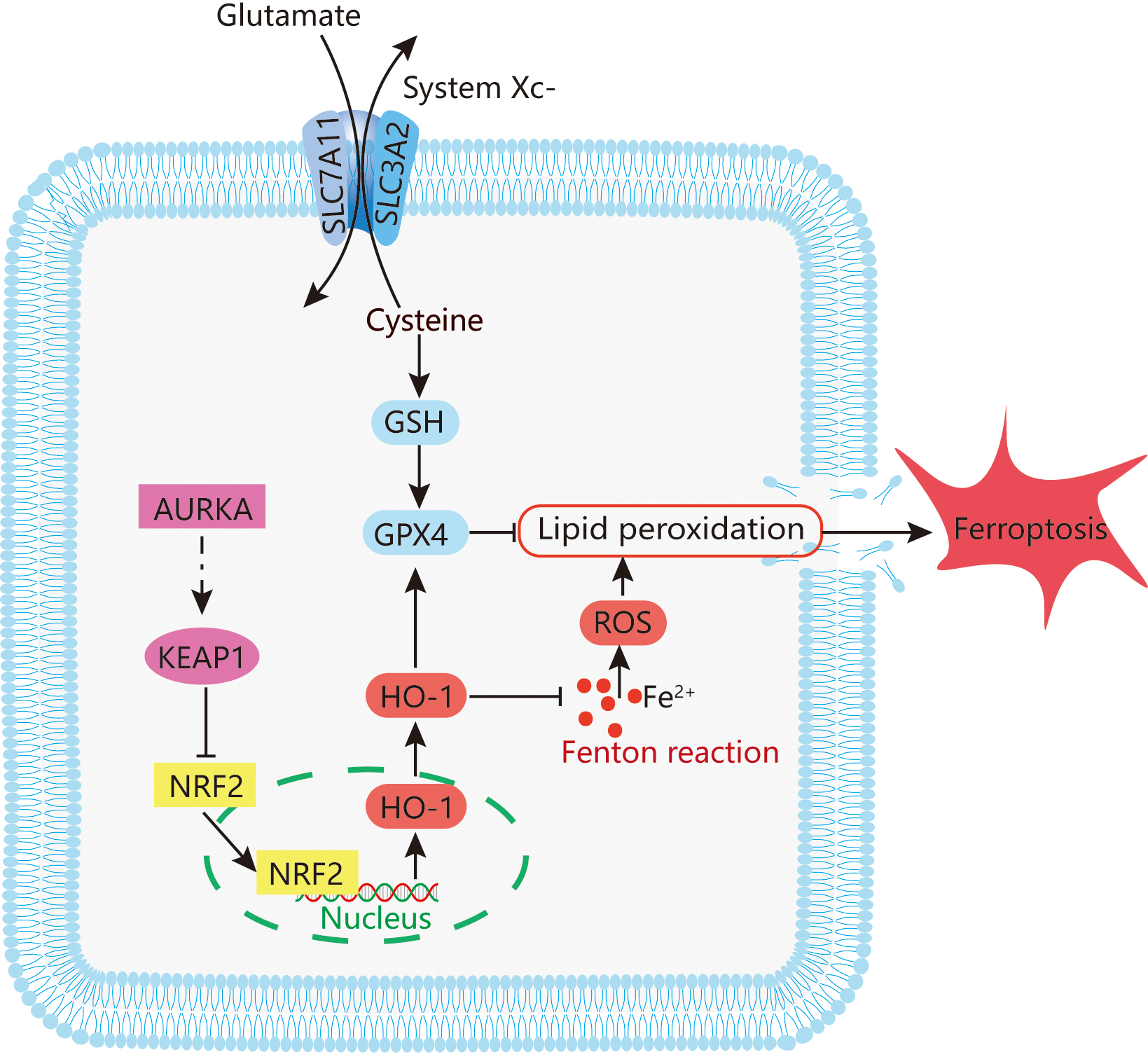
The incidence and mortality of lung cancer in China continue to rise annually [39]. Although EGFR-TKIs have significantly improved clinical outcomes in patients with EGFR-mutant non-small cell lung cancer, the emergence of acquired resistance remains a major therapeutic challenge [6, 7]. In this study, we investigated the role of AURKA in LUAD, with particular emphasis on its regulatory function in ferroptosis via the NRF2/HO-1 signaling pathway (Fig. 7). Our results reveal a strong association between AURKA expression and EGFR mutation status, suggesting a key role for AURKA in the progression of EGFR-mutant LUAD.
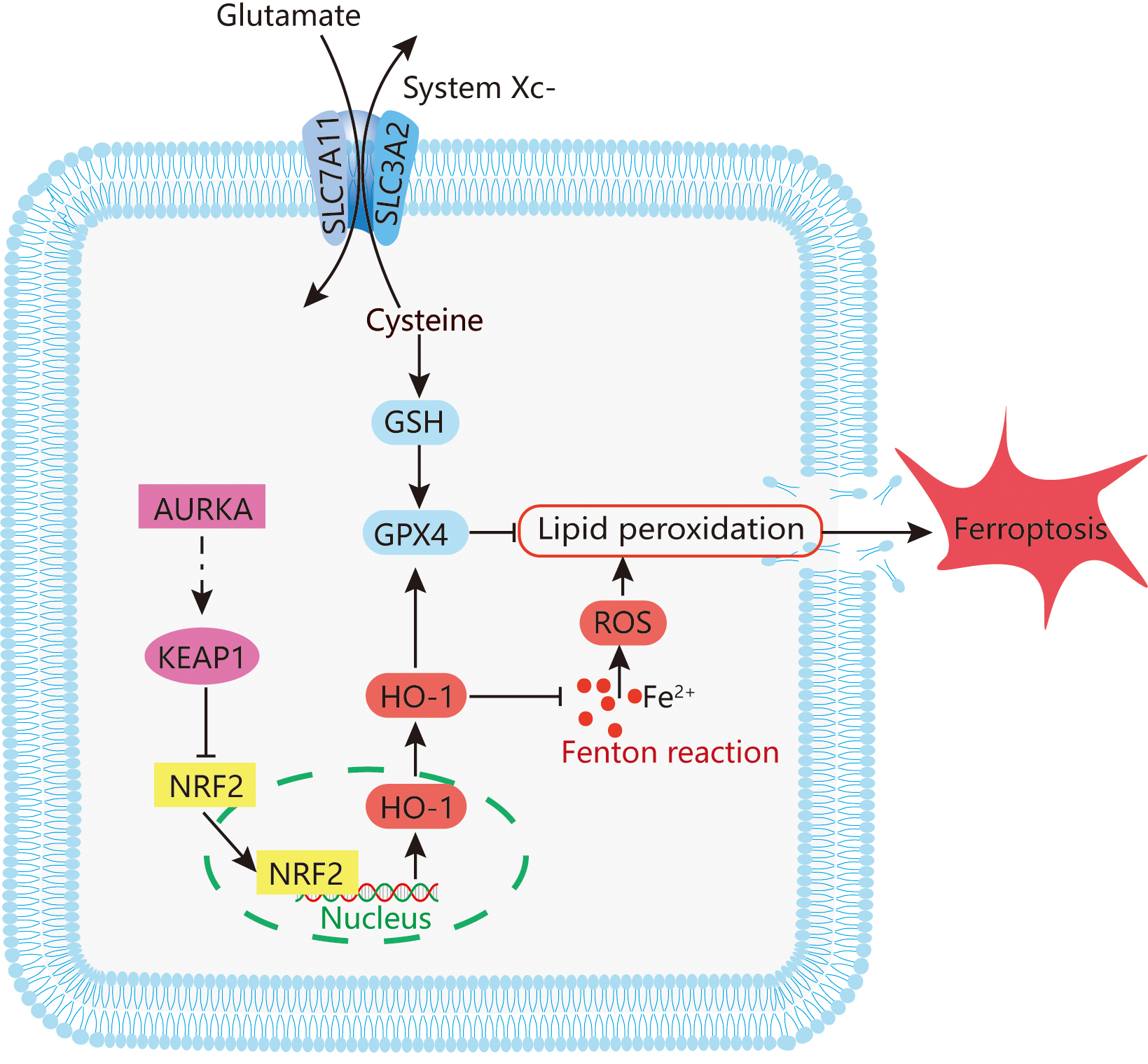 Fig. 7.
Fig. 7.
Potential mechanism mediating ferroptosis by AURKA in LUAD. The figure was originally created by the authors using Adobe Illustrator (version 26.0, San Jose, CA, USA).
Ferroptosis is a newly characterized form of regulated cell death, mechanistically distinct from apoptosis and autophagy [10]. It is driven by iron-dependent lipid peroxidation, and has been implicated in organ damage as well as targeted cancer therapy [40]. The KEAP1/NRF2/HO-1 axis is a central regulatory pathway that modulates ferroptosis by controlling antioxidant responses and intracellular redox homeostasis [41]. Accumulating evidence implicates ferroptosis in diverse disease processes, including cancers [42], Alzheimer’s disease [43], Parkinson’s disease [44], ischemia-reperfusion injury [45], and stroke [46] — and it has increasingly been recognized as a relevant mechanism in lung cancer pathophysiology [47].
Our data demonstrated that AURKA knockdown induces ferroptosis in LUAD cells. This was confirmed by reversal of lipid peroxidation and ferroptosis markers upon treatment with Fer‑1, a lipid ROS scavenger and Fe2+ chelator. Moreover, inhibition of the NRF2 using ML385 suppressed HO-1 expression and attenuated the ferroptosis phenotype, highlighting the central role of NRF2/HO‑1 pathway in mediating ferroptosis triggered by AURKA suppression. Together, these results suggest that AURKA negatively regulates ferroptosis, at least in part, through modulation of the KEAP1/NRF2/HO‑1 signaling axis.
AURKA, a serine/threonine kinase that belongs to the Aurora family [48], is a critical regulator of mitosis, particularly through its roles in centrosome maturation, spindle assembly, and chromosome segregation [49]. Aberrant overexpression of AURKA has been documented in a wide range of malignancies [50, 51, 52], where it contributes to tumorigenesis and resistance to anticancer therapies [53]. In addition to its classical role in cell division, AURKA has been shown to regulate mitochondrial dynamics and support metabolic reprogramming via enhanced mitochondrial interconnectivity [54], providing further mechanistic insight into its role in ferroptosis resistance. Although Aurora kinases, including AURKA, have been extensively studied in LUAD and their roles in tumor growth and cell cycle regulation are well characterized, most previous studies have focused on mitotic functions and therapeutic targeting through kinase inhibition [48, 49, 50, 51, 52]. Several AURKA inhibitors have entered clinical trials, underscoring its translational significance. However, the ferroptosis-related role of AURKA, especially via the KEAP1/NRF2/HO-1 axis in the context of EGFR-mutant LUAD, remains poorly defined. Our study thus extends the functional repertoire of AURKA beyond canonical cell cycle control and highlights a novel connection between this mitotic kinase and ferroptosis resistance. However, our data uniquely demonstrate that AURKA directly modulates the KEAP1/NRF2/HO-1 signaling axis in EGFR-mutant LUAD cells, thereby regulating ferroptosis sensitivity. This contrasts with prior work where AURKA was primarily associated with metabolic adaptation rather than ferroptotic regulation [55]. Furthermore, our finding that AURKA inhibition leads to increased NRF2 nuclear translocation and upregulation of HO-1 highlights a novel regulatory link between a mitotic kinase and ferroptosis-related antioxidant defenses. Notably, although we assessed total AURKA expression in this study, we did not examine its phosphorylation status, such as phosphorylation at Thr288, which is known to be essential for its activation and substrate recognition. Since phosphorylation may influence AURKA’s capacity to regulate downstream signaling, including KEAP1/NRF2/HO-1, future studies employing phospho-specific antibodies and kinase activity assays will be necessary to determine whether AURKA’s ferroptosis-related effects are dependent on its kinase activity.
Targeted inhibition of AURKA has demonstrated therapeutic potential in clinical
trials. In a phase I clinical trial enrolling 40 individuals with late-stage
cancers, AT9283—a dual inhibitor of AURKA and AURKB—was found to reduce tumor
burden, although treatment was associated with reversible toxicities such as
dose-dependent myelosuppression, gastrointestinal symptoms, fatigue, and alopecia
[56]. A subsequent phase II trial of 219 patients with treatment-refractory
cancers tested the pan-Aurora kinase inhibitor PHA-739358, which showed limited
antitumor efficacy [57]. In another phase II study of
249 patients, the AURKA-selective inhibitor Alisertib achieved a modest response
rate of 11.2%, but 43% of participants experiencing severe drug-related adverse
events [58]. In a randomized, double-blind phase II study, the addition of
Alisertib to paclitaxel significantly prolonged median survival in patients with
small cell lung cancer (7.20 months vs. 4.47 months). However, this combination
was associated with a higher rate of grade
Based on this rationale, we explored the combined effects of AURKA inhibition and Sorafenib, a multi-kinase agent known to induce ferroptosis. Our in vitro results revealed that AURKA suppression enhances sorafenib-induced lipid peroxidation and ferroptosis, suggesting a synergistic interaction. These findings provide a strong rationale for further investigation of AURKA–ferroptosis-targeting combinations. However, as these observations are limited to in vitro models, additional validation in animal models is required to substantiate their therapeutic potential and assess safety profiles.
Additionally, comprehensive bioinformatics analyses using TCGA-LUAD datasets revealed that elevated AURKA expression correlates with poor survival outcomes and increased immune cells infiltration. Through the application of a ferroptosis-focused random forest classifier, we further uncovered a mechanistic link between AURKA expression and EGFR mutation status. These findings are in line with previous studies that have implicated AURKA in tumor progression and therapy resistance [8, 9]. GSEA also confirmed that AURKA is involved in multiple oncogenic and ferroptosis-related signaling pathways, supporting its candidacy as a therapeutic target in LUAD.
RNA sequencing of AURKA-silenced NCI-H1975 cells revealed significant transcriptomic changes associated with ferroptosis. Enrichment analyses using GO, KEGG, Reactome, and WikiPathways consistently highlighted the involvement of the NRF2 signaling pathway and other ferroptosis-related processes following AURKA knockdown. Functional assays confirmed hallmark features of ferroptosis, including elevated intracellular Fe2+, MDA and lipid peroxides, accompanied by reduced GSH levels and diminished mitochondrial membrane potential. Transmission electron microscopy further validated these findings by revealing classical ferroptotic morphological changes, such as shrinkage and cristae loss.
Interestingly, AURKA expression was lower in EGFR-mutant LUAD tumors compared to their wild-type counterparts. This seemingly paradoxical observation may reflect the dependency of EGFR-mutant tumors on EGFR-driven signaling, which could suppress alternative oncogenic pathways such as those involving AURKA [61, 62]. Additionally, feedback inhibition from EGFR signaling may downregulate growth-promoting factors like AURKA to conserve cellular resources and minimize pathway redundancy [8, 63].
As resistance to EGFR inhibitor develops, tumors often shift their reliance to alternative oncogenic drivers such as mesenchymal–epithelial transition (MET) amplification [64]. In this context, AURKA may emerge as a compensatory pathway, contributing to drug resistance. Ongoing studies are investigating the potential role of AURKA in mediating gefitinib resistance, which may further clarify its mechanistic involvement in EGFR-targeted therapy failure.
We also observed that protein levels of GPX4 and SLC7A11 were relatively higher in the AURKA knockdown group compared to cells treated with AURKA inhibitors. This discrepancy could be attributed to residual AURKA activity following siRNA-mediated suppression, as siRNA does not completely abolish protein expression. In contrast, pharmacologic inhibitors more effectively block AURKA kinase activity, leading to stronger downstream suppression.
Despite the promising findings, our study has several limitations. First, the mechanistic investigations were primarily based on in vitro siRNA-mediated knockdown in a single EGFR-mutant LUAD cell line (NCI-H1975), without in vivo validation. Notably, NCI-H1975 harbors both L858R and T790M mutations, making it intrinsically resistant to first-generation EGFR inhibitors such as gefitinib. Although our data strongly suggest that AURKA inhibition promotes ferroptosis via the KEAP1/NRF2/HO-1 axis, further validation using xenograft or genetically engineered mouse models will be essential to confirm the physiological relevance and translational potential of this pathway. Second, while our results implicate the KEAP1/NRF2/HO-1 pathway as a key downstream mediator, the precise molecular mechanism by which AURKA regulates this axis remains to be clarified. It is unclear whether AURKA directly modulates NRF2 or KEAP1 at the transcriptional or post-translational level. Although we observed changes in KEAP1 and NRF2 protein levels following AURKA knockdown, additional experiments are needed to determine whether AURKA directly phosphorylates these proteins or affects their stability through intermediate signaling cascades. Future studies employing co-immunoprecipitation, chromatin immunoprecipitation, or kinase-substrate interaction analyses will help elucidate these regulatory relationships. Third, the TCGA transcriptomic dataset primarily includes Western populations, whereas our experimental validation was based on Asian patient tissues, which may introduce ethnic and genetic variability. Moreover, the clinical relevance of AURKA expression in EGFR-mutant LUAD remains to be further explored in larger and more diverse patient cohorts. Our current IHC validation was limited to six matched tumor-normal LUAD specimens; while these preliminary data support our conclusions, future investigations involving expanded clinical samples with detailed prognostic and therapeutic annotations are warranted. Lastly, to improve the generalizability of our findings, future investigations will incorporate multiple LUAD cell lines and expand into in vivo models to strengthen the mechanistic and therapeutic conclusions.
Importantly, the LUAD cell line used in this study, NCI-H1975, harbors the EGFR L858R/T790M mutation, rendering it intrinsically resistant to first-generation EGFR inhibitors such as gefitinib. This resistance poses a major clinical challenge in LUAD management. Our findings that AURKA knockdown induces ferroptosis in this TKI-resistant background highlight a potential alternative strategy to overcome EGFR-TKI resistance. By targeting the AURKA/KEAP1/NRF2/HO-1 axis, it may be possible to bypass EGFR-dependence and sensitize resistant tumors to ferroptotic cell death, offering a novel therapeutic avenue for patients with EGFR-mutant, drug-refractory LUAD.
In conclusion, this study provides the first evidence that AURKA knockdown induces ferroptosis in EGFR-mutant LUAD. Despite the typical dependence of these tumors on the EGFR signaling, AURKA suppression effectively triggered ferroptotic cell death. These results highlight AURKA as a promising intervention point and propose a viable approach to counteract EGFR inhibitor resistance. The synergistic potential of combining AURKA inhibition with ferroptosis-inducing agents such as sorafenib warrants further preclinical and clinical exploration to optimize treatment strategies for LUAD.
In summary, our study demonstrates that AURKA knockdown induces ferroptosis and impairs proliferation and invasion in EGFR-mutant LUAD cells, primarily through modulation of the KEAP1/NRF2/HO-1 signaling pathway. Transcriptomic analysis and functional assays confirmed that AURKA inhibition leads to increased lipid peroxidation, iron accumulation, and mitochondrial dysfunction—hallmarks of ferroptosis. Furthermore, bioinformatic data revealed that AURKA expression correlates with poor prognosis and immune infiltration. These findings collectively suggest that AURKA acts as a ferroptosis suppressor and potential therapeutic target in EGFR-TKI–resistant LUAD. Future studies incorporating in vivo models and multi-lineage validation will help further establish the translational relevance of targeting AURKA in this context.
The transcriptomics data from this study are available in the NCBI repository (https://www.ncbi.nlm.nih.gov/) under accession number PRJNA1113218. All original contributions are included within the article and supplementary material, and any further inquiries can be directed to the corresponding author.
YFH drafted the manuscript, contributed to the study design, data collection and organization, and analysis. GQL performed the cell and tissue experiments. ZL and JHZ made substantial contributions to the conception and design of the study, provided critical guidance on data interpretation, and supervised all stages of the research to ensure scientific rigor and accuracy. They also resolved key methodological and analytical issues that arose during the work. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was conducted in accordance with the ethical guidelines outlined in the Declaration of Helsinki. It received approval from the Institutional Ethics Committee at Sun Yat-sen University’s Seventh Affiliated Hospital. The approval number for this study is 2024-152, and approval was granted on April 11, 2024. Informed consent was obtained from all participants prior to the collection of human tissue samples. The privacy and confidentiality of all participants were strictly maintained throughout the study.
Not applicable.
This project was supported by Guangdong Traditional Chinese Medicine Project (20222107).
The authors declare no conflict of interest.
During the preparation of this work, the authors utilized ChatGPT (OpenAI) exclusively to assist with proofreading tasks, including identification of spelling errors and grammatical inconsistencies in the final draft. No AI tool was applied to generate or interpret scientific content, data analysis, or conclusions. Following AI-assisted proofreading, all modifications were critically reviewed and approved by the authors, who assume full responsibility for the accuracy and integrity of the published work.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL41293.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.