
 , Yongxue Lv 3,†, Kejun Liu 1,2, Zhengquan Li 1,2, Bendong Chen 1,2, Yang Bu 1,2,*
, Yongxue Lv 3,†, Kejun Liu 1,2, Zhengquan Li 1,2, Bendong Chen 1,2, Yang Bu 1,2,*
1 Department of Hepatobiliary Surgery, General Hospital of Ningxia Medical University, 750004 Yinchuan, Ningxia Hui Autonomous Region, China
2 School of Clinical Medicine, Ningxia Medical University, 750004 Yinchuan, Ningxia Hui Autonomous Region, China
3 School of Basic Medicine, Ningxia Medical University, 750004 Yinchuan, Ningxia Hui Autonomous Region, China
†These authors contributed equally.
Abstract
Mediator complex subunit 10 (MED10) serves as a critical regulator of eukaryotic gene expression by facilitating RNA polymerase II activity. Our investigation aims to characterize MED10’s functional contributions and underlying molecular pathways in hepatocellular carcinoma (HCC) development.
MED10 expression patterns in HCC and their correlation with clinicopathological parameters and patient outcomes were examined using bioinformatics databases and immunohistochemistry. Subsequently, we systematically investigated the biological functions of MED10 in the malignant progression of HCC through comprehensive in vitro experiments, including assessments of cell migration (transwell and wound healing assays), proliferative capacity (cell counting kit-8, colony formation, and 5-Ethynyl-2′-deoxyuridine assays), and cell cycle progression (flow cytometry analysis). Furthermore, we elucidated the underlying molecular mechanisms using real-time quantitative PCR (RT-qPCR), western blotting, immunofluorescence staining, and public database analyses. Furthermore, an in vivo subcutaneous xenograft model was employed to validate MED10’s impact on tumor growth.
The results revealed a marked increase in MED10 expression levels within HCC tissues, showing a strong association with unfavorable clinical outcomes. Mechanistically, MED10 induced the epithelial-mesenchymal transition (EMT) and enhanced HCC cell migration. Moreover, MED10 overexpression drives HCC cell cycle progression and proliferation by activating rapidly accelerated fibrosarcoma 1 (RAF1), a process potentially mediated through the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK)/cellular myelocytomatosis oncogene (c-Myc) signaling axis.
MED10 promotes HCC cell migration and EMT but, more importantly, also drives cell cycle progression and proliferation via RAF1 activation, and is related to the MEK/ERK/c-Myc axis.
Keywords
- hepatocellular carcinoma
- mediator complex subunit 10
- cell cycle
- proliferation
- MAPK signaling pathway
- prognosis
Hepatocellular carcinoma (HCC), the predominant type of primary liver cancer, ranks as the sixth most prevalent malignancy globally and accounts for the fourth highest mortality rate among cancer-related deaths [1]. According to projections made by the World Health Organization, HCC is anticipated to claim over one million lives annually by 2030, representing a growing global health crisis [2]. Due to its elusive early symptoms and heterogeneous nature, only a small proportion of patients with HCC are diagnosed sufficiently early to undergo radical resection, which is considered the preferred initial treatment [3]. Molecular-targeted therapies have transformed HCC management, yet their application remains restricted due to incomplete mechanistic insights [4]. This underscores the critical need to unravel HCC’s molecular underpinnings.
Mediator complex subunit 10 (MED10) is a significant component of the mediator complex initially identified in yeast [5]. Functioning as an essential bridge connecting RNA polymerase II with transcription factors, the mediator complex serves as a master regulator of gene expression in eukaryotes [6]. This transcriptional control system governs fundamental biological phenomena ranging from cellular proliferation to organismal aging. Additionally, it critically influences pathological contexts, such as tumor cell metabolic activity and proliferation [7]. Structural or functional perturbations in mediator subunits typically reshape the transcriptional landscape of downstream effector genes, creating permissive conditions for malignant transformation and clinical disease evolution [8]. MED1 aggravates breast cancer by regulating macrophage M2 polarization [9]. MED12 mutations drive leiomyoma malignant progression through aberrant activation of the tryptophan/kynurenine/aryl hydrocarbon receptor signaling cascade [10]. Parallelly, in non-small cell lung cancer, tDR-007333-mediated AS-MED29 activation promotes aggressive tumor phenotypes [11]. In ER-positive breast cancer, MED16 regulates autophagy mediated by the mechanistic target of rapamycin signaling pathway and promotes tumor progression and tamoxifen sensitivity [12]. Accumulating evidence supports MED10’s pivotal contribution to oncogenesis. Aberrant MED10 expression has been shown to promote oncogenic and refractory phenotypes in bladder urothelial carcinoma, primarily through its regulation of hsa-miR-590. A recent study further demonstrated that MED10 enhances cisplatin resistance in HCC by facilitating phosphatase and tensin homolog (PTEN) ubiquitination [5, 13]. These results position MED10 as a promising molecular target for HCC intervention strategies. However, the additional roles MED10 plays in HCC malignant progression and its underlying mechanisms require further exploration and validation, which may offer novel therapeutic targeting strategies for HCC treatment.
This study systematically investigated MED10’s clinical significance and biological roles in HCC to discover innovative prognostic markers and therapeutic targets capable of revolutionizing HCC clinical practice. Our data establish MED10 upregulation in HCC tissues as a predictor of adverse clinical outcomes, mechanistically demonstrating its function as an oncogenic driver that promotes eithelial-mesenchymal transition (EMT) and metastatic competence. Importantly, further investigation indicated that MED10’s influence on tumor cell proliferation and cell cycle progression was partly driven by aberrant activation of the rapidly accelerated fibrosarcoma 1 (RAF1). Together, these findings highlight MED10 as a potential dual-purpose biomarker for prognosis and a molecular target for HCC therapy.
RNA-seq data and associated clinical records for HCC (n = 374) and normal liver tissues (n = 50) were retrieved from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). MED10 expression patterns across multiple tumor types were further evaluated using the TIMER2.0 platform (http://timer.cistrome.org/) [14].
TCGA-LIHC patient data were stratified into high- and low-MED10 expression
groups based on median expression values. GSEA was conducted using the
clusterProfiler R package (version 4.10.0, https://bioconductor.org/packages/clusterProfiler/)
to delineate functionally relevant pathways between
these groups [15]. Statistical significance was defined as p
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using the Metascape platform (https://metascape.org/gp/index.html#/main/step1) [16].
To investigate potential functional associations, we generated a PPI network using the STRING database (https://cn.string-db.org/) by analyzing MED10 alongside cell cycle regulatory genes [17].
Target genes of the transcription factor, c-Myc, were predicted using the TRRUST (https://www.grnpedia.org/trrust/), CHEA (https://maayanlab.cloud/chea3/), ENCODE (https://www.encodeproject.org), and CHIP Atlas (https://chip-atlas.org/) databases [18, 19, 20, 21]. Biological processes involved in c-Myc expression were predicted using the TRRUST database.
Bioinformatic analysis utilizing the JASPAR database (http://jaspar.genereg.net) [22] predicted the presence of c-Myc response elements within the promoter sequences of cyclin-dependent kinase 4 (CDK4), cyclin-dependent kinase 6 (CDK6), and cyclin D1 (CCND1).
This retrospective analysis utilized archived paraffin-embedded tissues and
clinical records from 215 HCC patients who underwent surgical resection at
Ningxia Medical University General Hospital between 2010–2020. The study cohort
was selected based on strict inclusion criteria: (1) histologically confirmed HCC
diagnosis, (2) treatment-naïve status prior to surgery, (3) availability of
complete clinical datasets, and (4) paired tumor and normal liver tissue samples
(collected
Antibodies against the following were used: p-RAF1 (9427, Cell Signaling
Technology, Danvers, MA, USA), MED10 (DF13141, Affinity, Nanjing, Jiangsu,
China), E-cadherin (60335-1-Ig, Proteintech, Wuhan, Hubei, China), p-ERK1/2
(AF1015, Affinity, Nanjing, Jiangsu, China), Cyclin D1 (ab16663, Abcam,
Cambridge, Cambridgeshire, UK), RAF1 (ab181115, Abcam, Cambridge, Cambridgeshire,
UK), MEK1/2 (9122, Cell Signaling Technology, Danvers, MA, USA), Vimentin
(PA1-16759, Invitrogen, Carlsbad, CA, USA), ERK1/2 (11257-1-AP, Proteintech,
Wuhan, Hubei, China), CDK6 (ab222395, Abcam, Cambridge, Cambridgeshire, UK),
matrix metallopeptidase 13 (MMP13, ab39012, Abcam, Cambridge, Cambridgeshire,
UK), c-Myc (5605, Cell Signaling Technology, Danvers, MA, USA), N-cadherin
(66219-1-Ig, Proteintech, Wuhan, Hubei, China), p-MEK1/2 (9154, Cell Signaling
Technology, Danvers, MA, USA), CDK4 (ab108357, Abcam, Cambridge, Cambridgeshire,
UK), marker of proliferation ki-67 (Ki67, 9027, Cell Signaling Technology,
Danvers, MA, USA) and
Following dewaxing of the paraffin-embedded tissue sections in xylene, a graded alcohol series was used for rehydration. To expose target antigens, the sections were microwaved in sodium citrate buffer (C1010, Solarbio, Beijing, China). After cooling to room temperature, the samples were rinsed three times with phosphate-buffered saline (PBS, P1010, Solarbio, Beijing, China) and then treated with a peroxidase-blocking solution (PV-9001, ZSGB-BIO, Beijing, China) for 10 minutes. Subsequent PBS washes were performed before blocking nonspecific binding sites with 5% bovine serum albumin (BSA, SW3015, Solarbio, Beijing, China) for an additional 10 minutes. Primary antibody incubation was carried out overnight at 4 °C, followed by PBS washes and a 1-hour room-temperature incubation with secondary antibodies (PV-9001, ZSGB-BIO, Beijing, China). The primary antibodies used were as follows: MED10 (1:100, DF13141; Affinity, Nanjing, China), Ki67 (1:200, 9027, Cell Signaling Technology, Danvers, MA, USA). Following another PBS wash, the sections were developed using a 3,3’-Diaminobenzidine (DAB) kit (ZLI9018, ZSGB-BIO, Beijing, China), counterstained with hematoxylin (G1120, Solarbio, Beijing, China), dehydrated through an alcohol gradient, cleared in xylene, and finally mounted for microscopic imaging. Two independent pathologists assessed immunohistochemical staining, scoring both the proportion of positive cells (1–3) and staining intensity (0–4). The product of these scores yielded a composite value, with the median score serving as the threshold to classify samples into low- and high-expression groups.
The tissue sections were first dewaxed in xylene and rehydrated using a graded alcohol series. Antigen retrieval was performed by heating the sections in sodium citrate buffer (C1010, Solarbio, Beijing, China). Afterward, the tissue sections were incubated with an endogenous peroxidase blocker (PV-9001, ZSGB-BIO, Beijing, China) for 10 minutes, followed by permeabilization with 0.3% Triton X-100 (T8200, Solarbio, Beijing, China) for 20 minutes. Nonspecific binding sites were then blocked with 5% BSA (SW3015, Solarbio, Beijing, China) for 1 hour. The sections were incubated with the primary antibody at 4 °C overnight, followed by a one-hour room-temperature incubation with a fluorescent secondary antibody. The primary antibodies used were as follows: MED10 (1:150, DF13141, Affinity, Nanjing, Jiangsu, China), CDK6 (1:150, ab222395, Abcam, Cambridge, UK), CDK4 (1:250, ab108357, Abcam, Cambridge, UK), Cyclin D1 (1:50, ab16663, Abcam, Cambridge, UK). Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI, C0065, Solarbio, Beijing, China), and the slides were mounted for imaging using an inverted fluorescence microscope (DMi8, Leica, Wetzlar, Hesse, Germany).
The human hepatoma cell lines MHCC97L, MHCC97H, HCCLM3, Hep3B, Huh-7, and SK-Hep-1, along with the immortalized hepatocyte line LO2, were kindly provided by the Institute of Liver Diseases at Fudan University (Shanghai, China). All cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM; VivaCell, Shanghai, China) supplemented with 10% fetal bovine serum (FBS, ExCell Bio, Shanghai, China) and 1% penicillin-streptomycin (P1400, Solarbio, Beijing, China) under standard culture conditions (37 °C, 5% CO2). Prior to experimentation, cell line authenticity was confirmed through short tandem repeat (STR) profiling, and mycoplasma contamination testing yielded negative results.
For MED10 overexpression (ov-MED10 group), HCCLM3 cells were transduced with the lentiviral construct PDS279-PL-CMV-GFP-CC-DB-Puro-MED10, while control cells received the empty vector PDS279-pL-CMV-GFP-cc-dB-puro. MED10 knockdown (shMED10) was achieved using the lentiviral vector VP013-U6-MCS-CMV-ZsGreen-PGK-PURO-MED10-SHRNA, with the VP013-U6-MCS-CMV-ZsGreen-PGK-PURO vector serving as the shcontrol. All lentiviruses (Tsingke Biotech, Beijing, China) were transfected at 30–50% cell confluence using Lipo3000 (Invitrogen, Carlsbad, CA, USA) per manufacturer protocols. After 14 h, the viral medium was replaced with fresh complete medium. Transduced cells were selected with 2 µg/mL puromycin (P8230, Solarbio, Beijing, China) for 4–5 days, with transduction efficiency verified via green fluorescence microscopy. Where indicated, ov-MED10 cells were treated with 10 µM LY3009120 (S7842, Selleck Chemicals, Houston, TX, USA). MED10 targeting sequences are provided in Supplementary Table 1.
Total RNA was isolated from cells using the MolarPure Cell/Tissue Total RNA Kit (19221ES, Yeasen Biotechnology, Shanghai, China). cDNA synthesis was performed with PrimeScript RT Master Mix (RR036A, Takara Bio, Kyoto, Japan), followed by RT-qPCR analysis using TB Green Premix Ex Taq II (RR820A, Takara Bio, Kyoto, Japan) on a validated thermal cycler. Gene expression was calculated using the 2-ΔΔCt method. The primers used to amplify these genes (Table 1) were designed and synthesized by Tsingke Biotech (Beijing, China).
| Gene | Primer sequences (5′-3′) | |
| MED10 | Forward | GAGAAGTTTGACCACCTAGAGGA |
| Reverse | TGGGGATTTCGACCTTGATCT | |
| CDH1 | Forward | ATTTTTCCCTCGACACCCGAT |
| Reverse | TCCCAGGCGTAGACCA AGA | |
| CDH2 | Forward | AGCCAACCTTAACTGAGGAGT |
| Reverse | GGCAAGTTGATTGGAGGGATG | |
| VIM | Forward | AGTCCACTGAGTACCGGAGAC |
| Reverse | CATTTCACGCATCTGGCGTTC | |
| GAPDH | Forward | GGCAAATTCCATGGCACCGTCAAGG |
| Reverse | GCCAGCATCGCCCCACTTGATTTTG |
MED10, Mediator complex subunit 10; CDH1, Cadherin 1; CDH2, Cadherin 2; VIM, Vimentin; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase.
The total protein content was isolated from HCC tissues and cultured cells using
radioimmunoprecipitation (RIPA) lysis buffer (P0013B, Beyotime Biotechnology,
Shanghai, China) supplemented with protease inhibitors, phosphatase inhibitors,
and phenylmethylsulfonyl fluoride (KGB5303-100, KeyGEN BioTECH, Nanjing, Jiangsu,
China). After 20 min ice incubation, lysates were centrifuged (12,000 g, 10 min, 4
°C) to remove debris. Protein concentrations were quantified via bicinchoninic
acid (BCA) assay (KGB2101-100, KeyGEN BioTECH, Nanjing, Jiangsu, China), followed
by denaturation with loading buffer at 100 °C for 10 min. Samples were
separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were
blocked for 1 h at room temperature with 5% BSA or skim milk, then incubated
with primary antibodies overnight at 4 °C. The primary antibodies used were as
follows: p-RAF1 (1:1000, 9427, Cell Signaling Technology, Danvers, MA, USA),
MED10 (1:1500, DF13141, Affinity, Nanjing, Jiangsu, China), E-cadherin (1:2000,
60335-1-Ig, Proteintech, Wuhan, Hubei, China), p-ERK1/2 (1:2000, AF1015, Affinity,
Nanjing, Jiangsu, China), Cyclin D1 (1:1000, ab16663; Abcam, Cambridge,
Cambridgeshire, UK), RAF1 (1:1000, ab181115; Abcam, Cambridge, Cambridgeshire,
UK), MEK1/2 (1:1000, 9122, Cell Signaling Technology, Danvers, MA, USA), Vimentin
(1:1500, PA1-16759, Invitrogen, Carlsbad, CA, USA), ERK1/2 (1:3000, 11257-1-AP,
Proteintech, Wuhan, Hubei, China), CDK6 (1:1000, ab222395, Abcam, Cambridge,
Cambridgeshire, UK), MMP13 (1:2000, ab39012, Abcam, Cambridge, Cambridgeshire,
UK), c-Myc (1:1000, 5605, Cell Signaling Technology, Danvers, MA, USA),
N-cadherin (1:2000, 66219-1-Ig, Proteintech, Wuhan, Hubei, China), p-MEK1/2
(1:1000, 9154, Cell Signaling Technology, Danvers, MA, USA), CDK4 (1:1500,
ab108357, Abcam, Cambridge, Cambridgeshire, UK), and
500 µL of DMEM supplemented with 10% FBS was placed in the lower
chamber of a Costar Transwell system (Corning, New York, NY, USA), while the upper chamber
received 200 µL of serum-free DMEM containing 8
Confluent cell monolayers in 6-well plates were wounded using a sterile 200-µL pipette tip. After gentle PBS washing to remove debris, cells were maintained in serum-free medium. Wound closure was documented at 0 and 48 hours usingan inverted fluorescence microscope (DMi8, Leica, Wetzlar, Hesse, Germany).
Cell proliferation was assessed using the Cell Counting Kit-8 (CCK-8) assay (B34304; Biomake, Beijing, China). HCC cells (2000 cells/well) were seeded in 96-well plates and cultured at 37 °C. At designated timepoints (12, 24, 48, 72, 96 h), 10 µL CCK-8 reagent in 100 µL DMEM was added per well. Following 2 h incubation, absorbance at 450 nm was measured using a microplate reader, with background subtraction from control wells.
Cells were seeded in 6-well plates at 500 cells/well and cultured for 14 days. Resulting colonies were fixed with 4% paraformaldehyde (15 min), PBS-washed, and stained with 0.1% crystal violet (30 min). Colony quantification was performed using ImageJ software (version 1.8.0, National Institutes of Health, Bethesda, MD, USA).
Cell proliferation was assessed using the BeyoClick™ EdU-555 Kit (C0075S; Beyotime, Shanghai, China). HCC cells grown in 24-well plates for 24 h were pulse-labeled with 10 µM EdU at 37 °C for 2 h. After fixation with 4% PFA (15 min) and permeabilization with 0.3% Triton X-100 (10 min), Click reaction solution was applied for 30 min in darkness. Nuclei were counterstained with Hoechst 33342 (5 min), and images acquired using an inverted fluorescence microscope (DMi8, Leica, Wetzlar, Hesse, Germany).
HCC cells were harvested, washed in cold PBS, and centrifuged at 1000
Female BALB/c-nu nude mice (4–5-week-old) were sourced from Beijing Weitong
Lihua Co., Ltd. (Beijing, China) and raised under standard conditions in a
specific pathogen-free environment. Twenty-four nude mice were randomly divided
into four experimental groups (6 mice/group): (1) control (injected with
HCCLM3control cells), (2) ov-MED10 (injected with HCCLM3ov-MED10), (3)
shcontrol (injected with Hep3Bshcontrol), and (4) shMED10 (injected with
Hep3BshMED10) groups. Subcutaneous xenograft tumors were established by
injecting 150 µL of a cell suspension containing 5
Statistical analyses were performed using SPSS (version 26.0, IBM, Armonk, NY,
USA) and GraphPad Prism (version 9.4.0, San Diego, Boston, MA, USA). Normally
distributed continuous variables were compared between two groups with Student’s
t-test and among multiple groups with ANOVA. Associations between MED10
expression and clinicopathological features were evaluated using
t-tests, Wilcoxon rank-sum, chi-square, or Fisher’s exact tests.
Survival curves were generated by Kaplan-Meier analysis with log-rank testing.
MED10’s diagnostic utility was assessed through receiver operating characteristic
(ROC) curve analysis, while Cox proportional hazards regression identified
survival predictors. A MED10-based prognostic nomogram was developed and
validated using calibration curves. Statistical significance was defined as
p
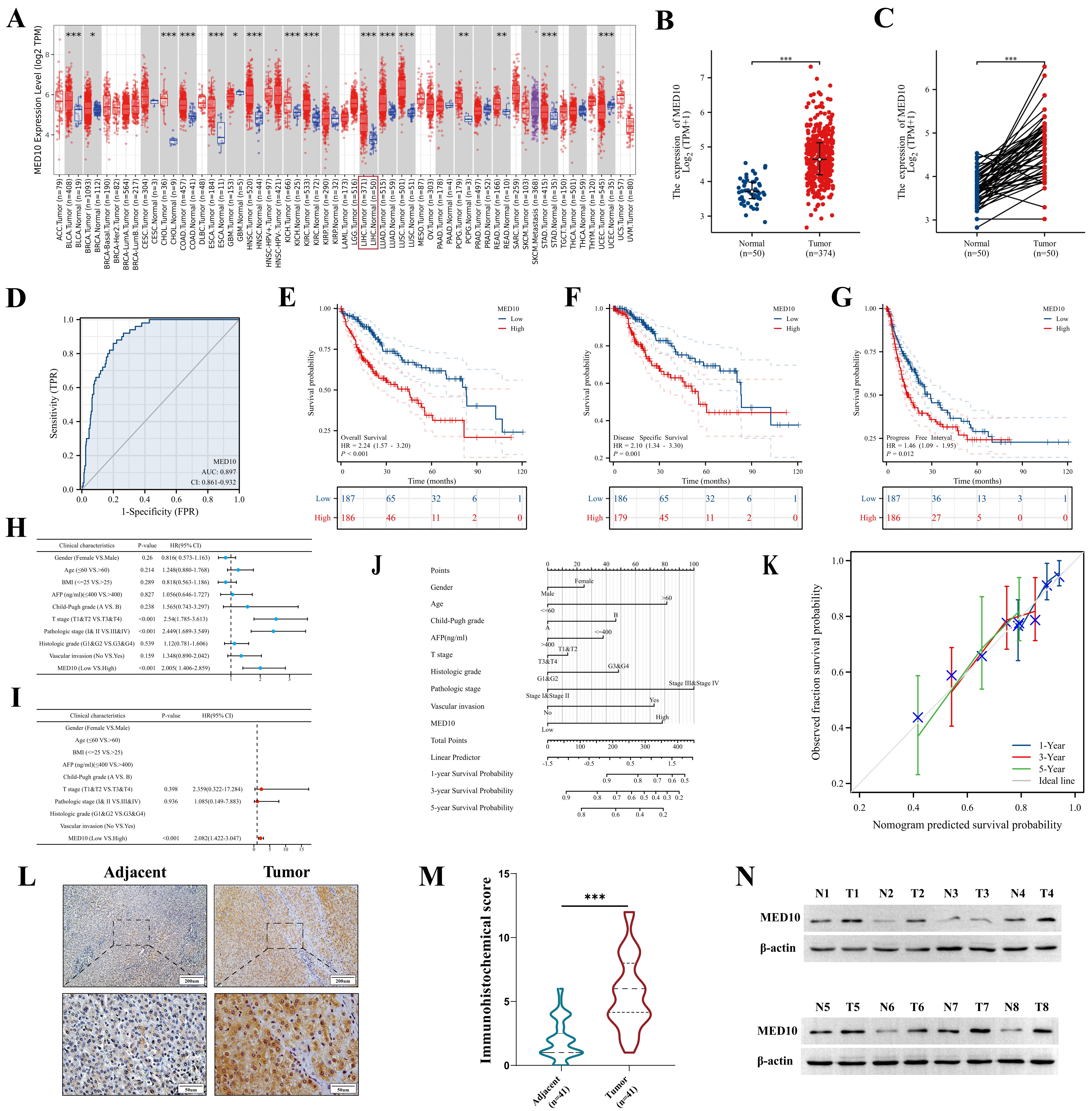
Initial analysis of MED10 expression across multiple cancers using TIMER2.0 revealed significant upregulation in tumor tissues, particularly hepatocellular carcinoma (HCC), versus normal counterparts (Fig. 1A). TCGA database analysis further confirmed elevated MED10 expression in HCC tissues relative to both paired and unpaired adjacent normal liver samples (Fig. 1B,C). ROC curve assessment demonstrated strong diagnostic potential for HCC (area under the curve (AUC) = 0.897, Fig. 1D). Analysis of MED10’s clinical relevance in HCC demonstrated significant correlations between its expression and key clinicopathological features, including body mass index, alpha-fetoprotein (AFP) levels, T stage, histologic grade, and vascular invasion (Table 2). Kaplan-Meier analysis demonstrated markedly inferior overall survival (OS), disease-specific survival (DSS), and progression-free interval (PFI) in HCC patients with elevated MED10 expression compared to low-expression cohorts (Fig. 1E–G). Univariate and multivariate Cox regression analyses consistently established MED10 as an independent prognostic determinant (Fig. 1H,I). We subsequently developed a clinical nomogram incorporating MED10 expression with eight clinicopathological parameters—gender, age, Child-Pugh grade, AFP level, T stage, histologic grade, pathologic stage, and vascular invasion—to predict 1-, 3-, and 5-year OS probabilities (Fig. 1J). The model’s precision was validated through calibration curves showing exceptional concordance between predicted and observed survival rates across all timepoints (Fig. 1K), collectively confirming MED10’s clinical utility as a prognostic biomarker.
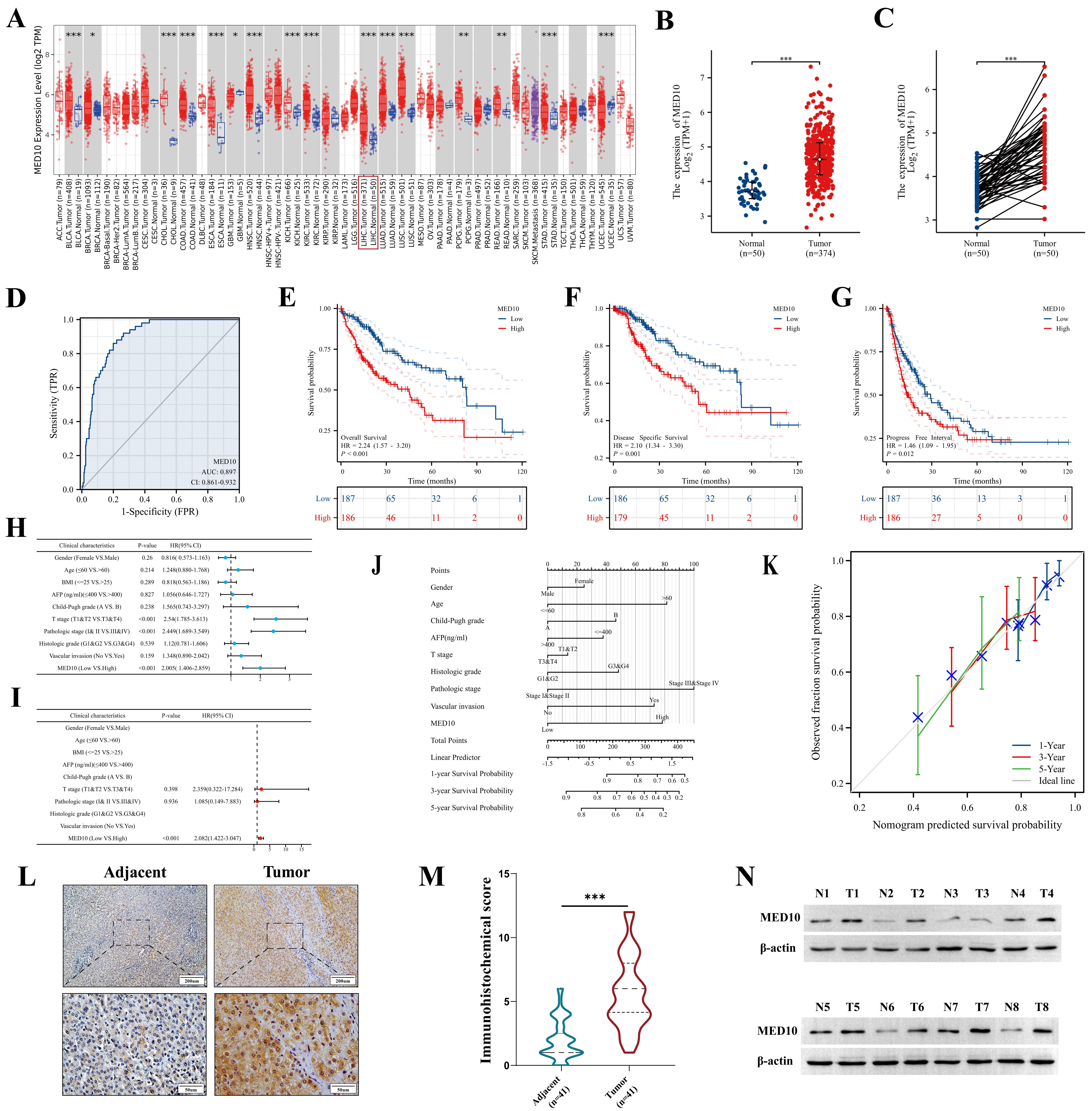 Fig. 1.
Fig. 1.
MED10 overexpression in HCC correlates with poor prognosis. (A)
Expression levels of MED10 across pan-cancer. The red box indicates higher expression of MED10 in liver hepatocellular carcinoma (LIHC) compared to adjacent normal tissues. (B,C) Elevated MED10 in HCC versus
adjacent normal liver tissues. (D) Diagnostic receiver operating characteristic
(ROC) curve for MED10 in HCC (area under the curve (AUC) = 0.897). (E–G)
Survival analyses showing reduced overall survival (OS), disease-specific
survival (DSS), and progression-free interval (PFI) in high-MED10 expressors.
(H,I) Univariate and multivariate Cox regressions identifying MED10 as
independent prognostic factor. (J) Prognostic nomogram predicting 1-/3-/5-year
OS. (K) Nomogram calibration curves. (L) Representative Immunohistochemistry
(IHC) of MED10 in HCC and normal tissues. Scale bars: 200 μm (top) and 50 μm (bottom). (M) Quantitative IHC scoring in 41
paired samples. (N) Western blotting confirmation of MED10 overexpression.
*p
| Characteristics | Expression of MED10 | Statistic | p-value | Method | ||
| Low (n = 187) | High (n = 187) | |||||
| Gender, n (%) | 0.110 | 0.740 | Chisq test | |||
| Female | 62 (16.6%) | 59 (15.8%) | ||||
| Male | 125 (33.4%) | 128 (34.2%) | ||||
| Age, n (%) | 1.298 | 0.255 | Chisq test | |||
| 83 (22.2%) | 94 (25.1%) | |||||
| 104 (27.8%) | 93 (24.9%) | |||||
| BMI, n (%) | 5.567 | 0.018 | Chisq test | |||
| 79 (23.4%) | 98 (29.1%) | |||||
| 92 (27.3%) | 68 (20.2%) | |||||
| AFP (ng/mL), n (%) | 17.129 | Chisq test | ||||
| 129 (46.1%) | 86 (30.7%) | |||||
| 20 (7.1%) | 45 (16.1%) | |||||
| Child-Pugh grade, n (%) | 0.904 | Fisher test | ||||
| A | 116 (48.1%) | 103 (42.7%) | ||||
| B | 12 (5.0%) | 9 (3.7%) | ||||
| C | 1 (0.4%) | 0 (0.0%) | ||||
| Fibrosis ishak score, n (%) | 6.076 | 0.108 | Chisq test | |||
| 0 | 49 (22.8%) | 26 (12.1%) | ||||
| 1/2 | 16 (7.4%) | 15 (7.0%) | ||||
| 3/4 | 11 (5.1%) | 17 (7.9%) | ||||
| 5/6 | 45 (20.9%) | 36 (16.7%) | ||||
| Pathologic T stage, n (%) | 7.942 | 0.047 | Chisq test | |||
| T1 | 104 (28%) | 79 (21.3%) | ||||
| T2 | 42 (11.3%) | 53 (14.3%) | ||||
| T3 | 32 (8.6%) | 48 (12.9%) | ||||
| T4 | 6 (1.6%) | 7 (1.9%) | ||||
| Histologic grade, n (%) | 15.254 | 0.002 | Chisq test | |||
| G1 | 37 (10.0%) | 18 (4.9%) | ||||
| G2 | 96 (26.0%) | 82 (22.2%) | ||||
| G3 | 47 (12.7%) | 77 (20.9%) | ||||
| G4 | 5 (1.4%) | 7 (1.9%) | ||||
| Pathologic stage, n (%) | 7.210 | 0.066 | Yates’ correction | |||
| Stage I | 98 (28%) | 75 (21.4%) | ||||
| Stage II | 40 (11.4%) | 47 (13.4%) | ||||
| Stage III | 34 (9.7%) | 51 (14.6%) | ||||
| Stage IV | 2 (0.6%) | 3 (0.9%) | ||||
| Vascular invasion, n (%) | 4.284 | 0.038 | Chisq test | |||
| No | 118 (37.1%) | 90 (28.3%) | ||||
| Yes | 49 (15.4%) | 61 (19.2%) | ||||
TCGA, The Cancer Genome Atlas; HCC, hepatocellular carcinoma; BMI, body mass index; AFP, alpha-fetoprotein.
This retrospective study validated public database findings through experimental analysis. IHC confirmed significantly elevated MED10 expression in HCC versus adjacent liver tissues (Fig. 1L–N). Western blotting corroborated these results, demonstrating increased MED10 levels in HCC specimens consistent with TCGA data (Fig. 1L–N). Among 215 HCC cases stratified by median immunohistochemistry scores, high MED10 expression correlated significantly with elevated aspartate aminotransferase levels, larger tumor size, microvascular tumor thrombi, and advanced T stage (Table 3). These findings establish MED10 as a key pathogenic driver in HCC, where overexpression predicts aggressive disease and poor prognosis.
| Characteristics | Expression of MED10 | Statistic | p-value | Method | ||
| High (n = 107) | Low (n = 108) | |||||
| Age (years), mean |
54.5 |
55.9 |
–0.911 | 0.363 | T test | |
| Gender, n (%) | 0.015 | 0.903 | Chisq test | |||
| Male | 82 (50.0%) | 82 (50.0%) | ||||
| Female | 25 (49.0%) | 26 (51.0%) | ||||
| HBsAg, n (%) | 0.030 | 0.863 | Chisq test | |||
| Positive | 93 (50.0%) | 93 (50.0%) | ||||
| Negative | 14 (48.3%) | 15 (51.7%) | ||||
| WBC ( |
5.2 (4.3, 6.3) | 5.1 (4.2, 6.4) | 0.992 | Wilcoxon | ||
| NE ( |
56.4 (49.3, 64.7) | 54.4 (48.6, 63.4) | 0.308 | Wilcoxon | ||
| Lym ( |
31.1 |
31.9 |
–0.589 | 0.557 | T test | |
| Hb (g/L), median (IQR) | 144 (133.5, 157.0) | 149 (134.5, 158.0) | 0.338 | Wilcoxon | ||
| PLT ( |
166 (119.0, 210.0) | 151 (120.3, 193.0) | 0.425 | Wilcoxon | ||
| AFP (ng/mL), n (%) | 2.235 | 0.135 | Chisq test | |||
| 72 (53.7%) | 62 (46.3%) | |||||
| 35 (43.2%) | 46 (56.8%) | |||||
| ALB (ng/mL), median (IQR) | 38.2 (36.0, 41.3) | 38.6 (35.0, 41.6) | 0.755 | Wilcoxon | ||
| AST (ng/mL), median (IQR) | 36.2 (28.7, 54.1) | 29 (23.5, 45.8) | 0.024 | Wilcoxon | ||
| PT (S), median (IQR) | 12.1 (11.4, 13.2) | 12.2 (11.6, 13.4) | 0.562 | Wilcoxon | ||
| Tumor size (cm), n (%) | 25.213 | Chisq test | ||||
| 64 (69.6%) | 28 (30.4%) | |||||
| 43 (35.0%) | 80 (65.0%) | |||||
| Microvascular tumor thrombus, n (%) | 35.747 | Chisq test | ||||
| Yes | 68 (73.1%) | 25 (26.9%) | ||||
| No | 39 (32.0%) | 83 (68.0%) | ||||
| Degree of differentiation, n (%) | 1.118 | 0.572 | Chisq test | |||
| Low | 40 (54.8%) | 33 (45.2%) | ||||
| Middle | 49 (46.8%) | 55 (53.6%) | ||||
| High | 18 (47.4%) | 20 (52.6%) | ||||
| T stage, n (%) | 20.452 | Yates’ correction | ||||
| T1 | 8 (22.2%) | 28 (77.8%) | ||||
| T2 | 53 (48.2%) | 57 (51.8%) | ||||
| T3 | 44 (68.8%) | 20 (31.2%) | ||||
| T4 | 2 (40.0%) | 3 (60.0%) | ||||
HBsAg, Hepatitis B Surface Antigen; WBC, White Blood Cell; NE, Neutrophil; Lym, Lymphocyte; Hb, Hemoglobin; PLT, Platelet; ALB, Albumin; AST, Aspartate Aminotransferase; PT, Prothrombin Time.
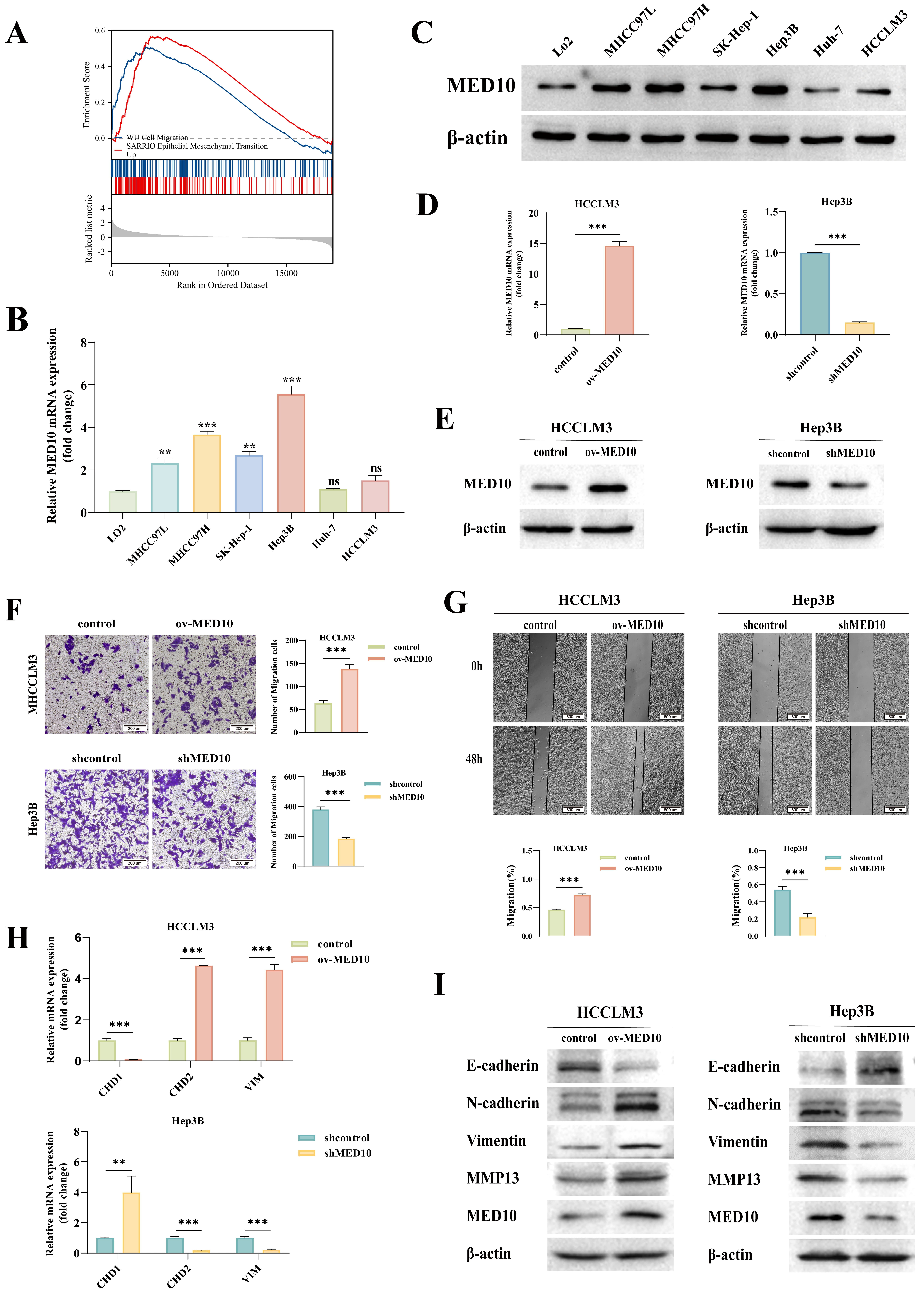
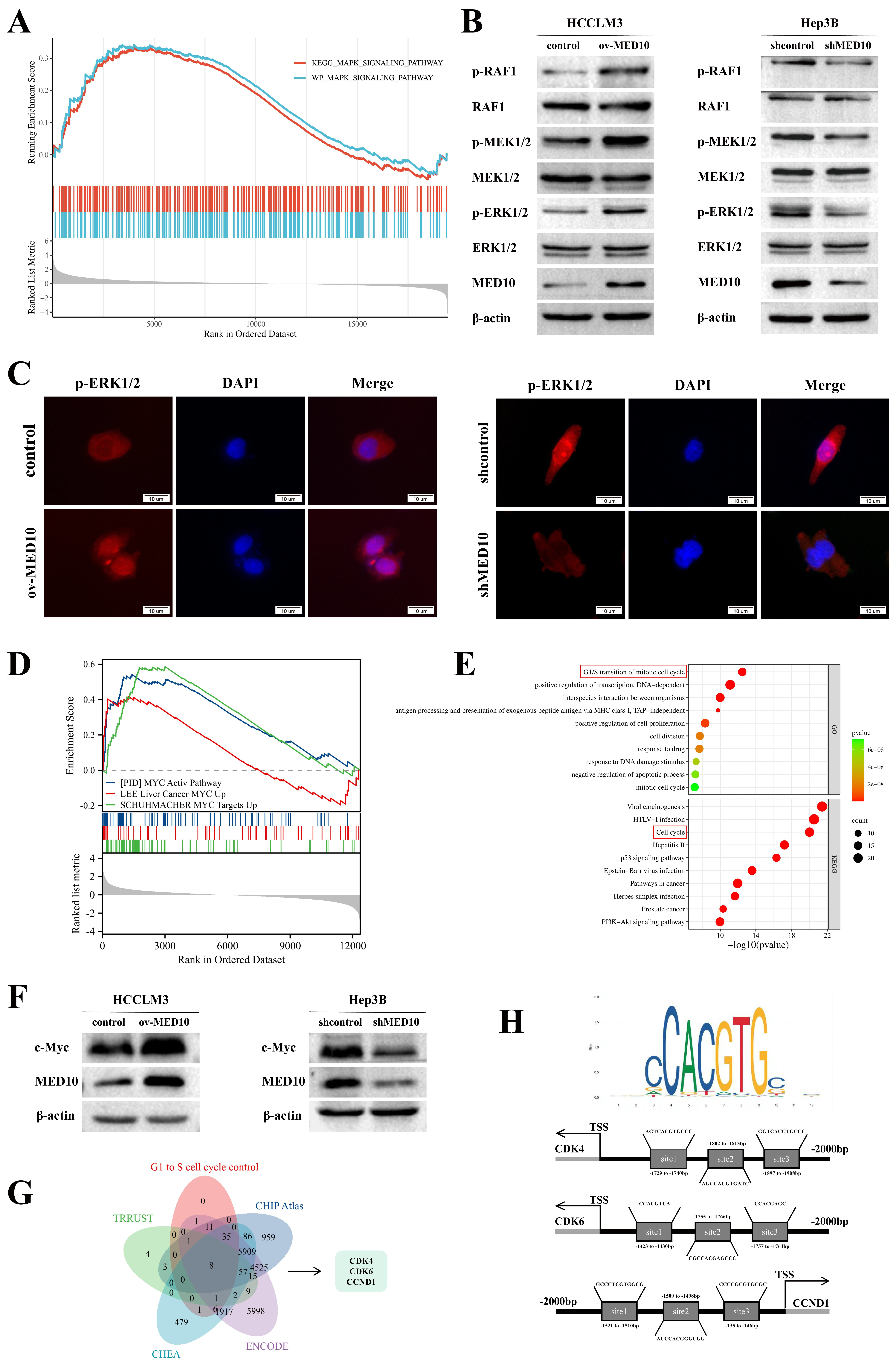
Analysis of TCGA data and clinical cohorts identified significant associations between elevated MED10 expression and microvascular invasion/tumor thrombi in HCC patients (Tables 2,3). GSEA revealed marked enrichment of cell migration and EMT pathways in high-MED10 expressors (Fig. 2A), implicating MED10 in metastatic progression. Supporting this observation, both transcriptional and translational analyses revealed MED10 dysregulation in HCC cell lines (MHCC97L, MHCC97H, SK-Hep-1, Hep3B), showing significantly elevated mRNA and protein expression relative to normal hepatocytes (LO2) (Fig. 2B,C). To functionally characterize MED10, we engineered stable HCC cell lines with modulated expression: MED10 was overexpressed in low-expressing HCCLM3 cells and knocked down in high-expressing Hep3B cells. Successful overexpression (ov-MED10) and knockdown (shMED10) were validated by RT-qPCR and Western blotting (Fig. 2D,E). Functional assessments via Transwell and scratch assays established MED10’s pro-migratory role in HCC: forced expression accelerated cellular motility, whereas genetic ablation attenuated migration (Fig. 2F,G). To investigate MED10’s role in EMT, we assessed EMT marker expression via RT-qPCR and Western blotting. MED10 overexpression elevated mesenchymal markers (N-cadherin, vimentin) while suppressing the epithelial marker E-cadherin. Conversely, MED10 knockdown increased E-cadherin and reduced mesenchymal markers (Fig. 2H,I). Protein levels of the metastatic regulator MMP13 mirrored these changes, increasing with MED10 overexpression and decreasing upon knockdown.
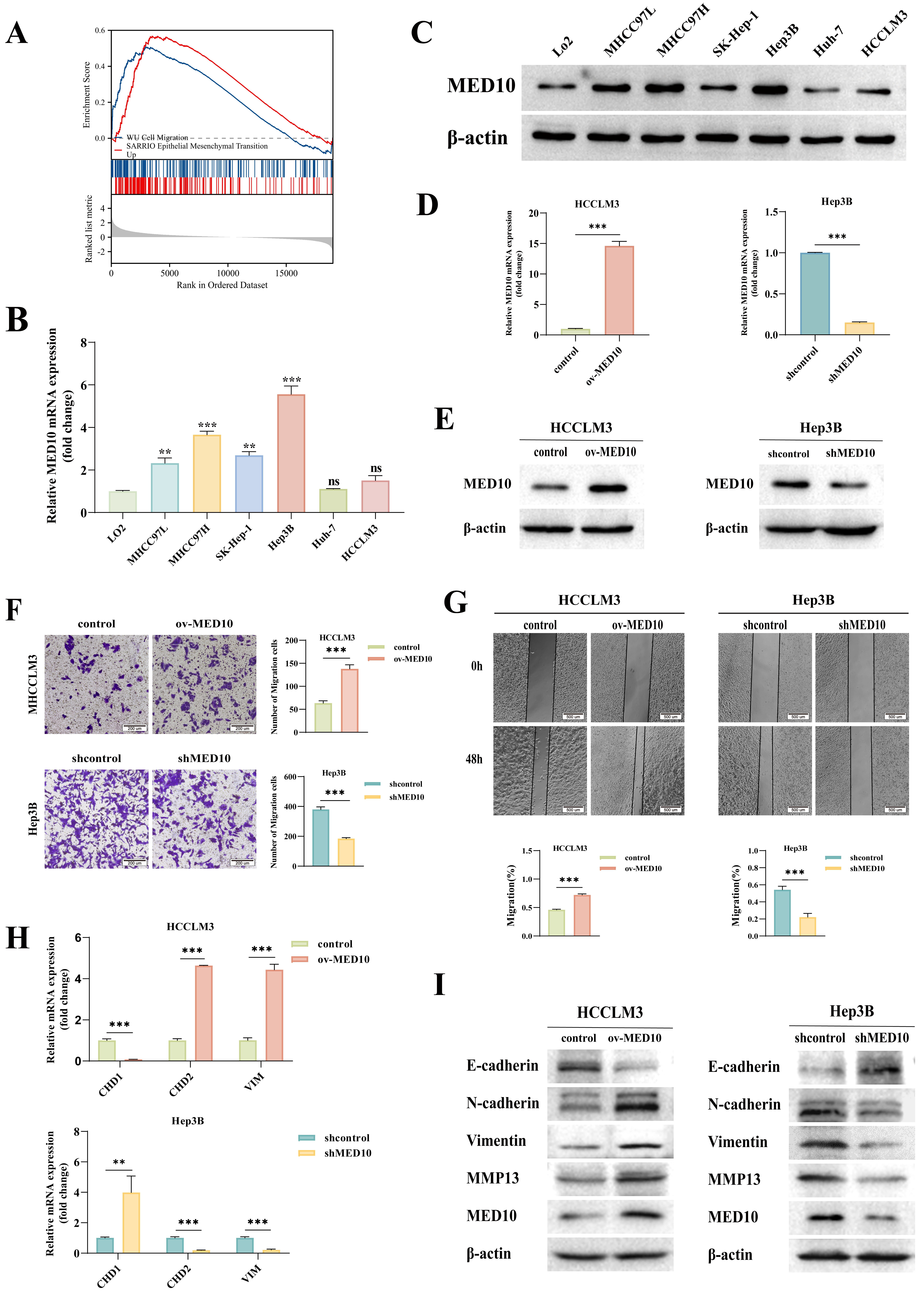 Fig. 2.
Fig. 2.
MED10 promotes migration and EMT in HCC. (A) Gene Set
Enrichment Analysis (GSEA) showing significant enrichment of cell migration and
EMT pathways. (B,C) MED10 mRNA/protein expression in normal hepatocytes (LO2)
versus HCC cell lines. (D,E) Validation of MED10 (over)expression (ov-MED10) and
knockdown (shMED10) models. (F,G) Enhanced migration capacity after MED10
overexpression versus suppression in knockdown models (Transwell/wound-healing). Scale bars: 200 μm (F) and 500 μm (G).
(H,I) EMT marker shifts: MED10 overexpression downregulated E-cadherin while
upregulating N-cadherin, vimentin, and MMP13; knockdown reversed these effects.
ns: p
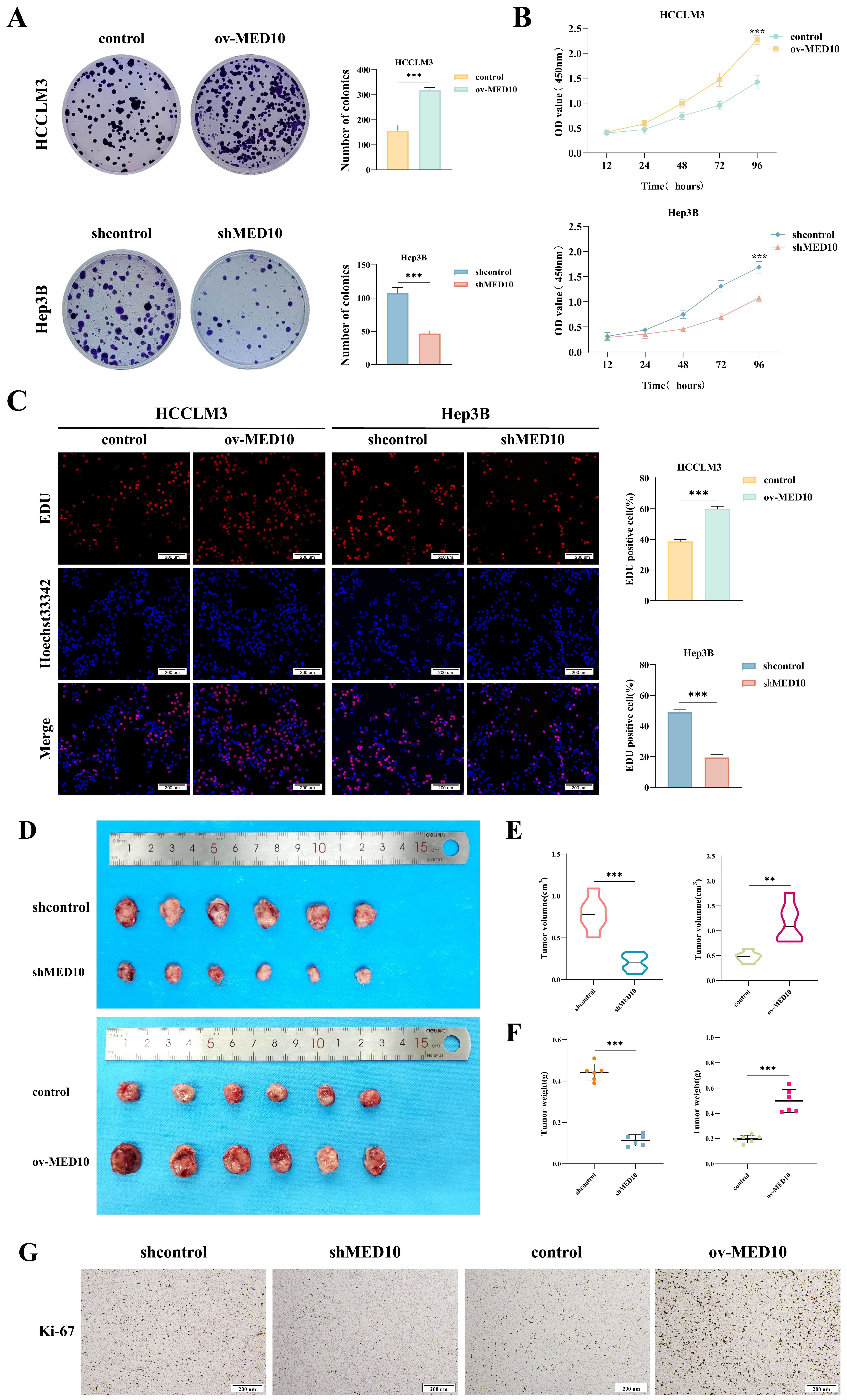
Analysis of TCGA data and HCC clinical cohorts revealed significant correlations between MED10 expression and advanced T stage/tumor size (Tables 2,3). Building on these clinical associations, we investigated MED10’s functional role in proliferation. Colony formation, CCK-8, and EdU assays consistently demonstrated enhanced proliferative capacity upon MED10 overexpression, whereas MED10 knockdown markedly suppressed HCC cell growth (Fig. 3A–C). To validate these findings in vivo, nude mouse xenograft models showed significantly reduced tumor burden in shMED10 groups versus controls, while ov-MED10 xenografts exhibited increased tumor volume and mass (Fig. 3D–F). IHC analysis of proliferation marker Ki-67 further confirmed these effects: diminished staining in shMED10 tumors and intensified staining in ov-MED10 tumors compared to respective controls (Fig. 3G).
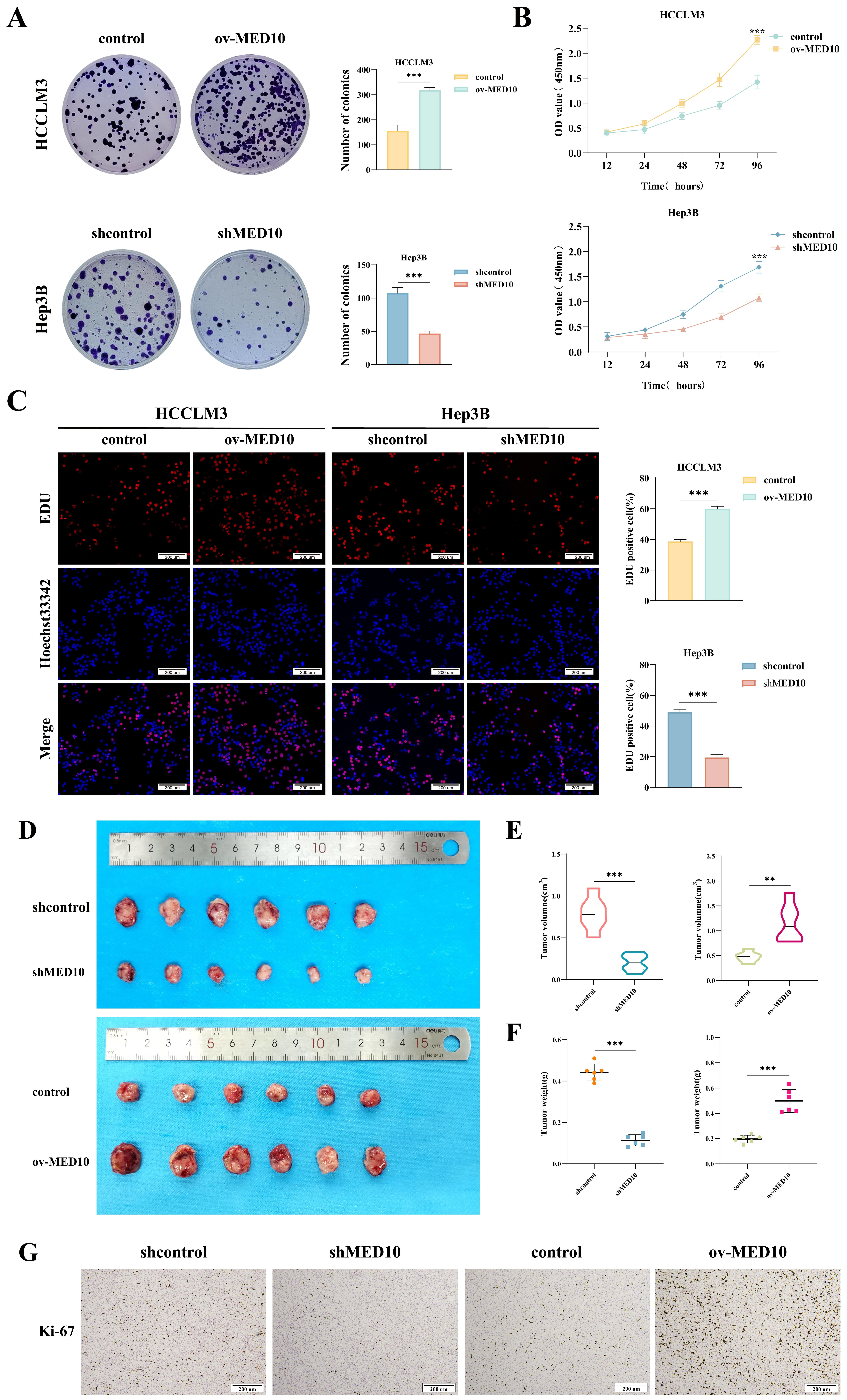 Fig. 3.
Fig. 3.
MED10 promotes HCC cells proliferation. (A–C) Enhanced
proliferative capacity in ov-MED10 cells versus suppression in shMED10 models
across colony formation, CCK-8, and EdU assays. Scale bar = 200 μm. (D) Representative xenograft
tumors from experimental groups post-excision (n = 6). (E,F) Significant
reduction in tumor volume/weight in shMED10 cohort versus increased burden in
ov-MED10 group relative to controls. (G) Immunohistochemical validation of
proliferation marker Ki-67: diminished expression in shMED10 tumors versus
intensified staining in ov-MED10 xenografts. Scale bar = 200 μm. **p
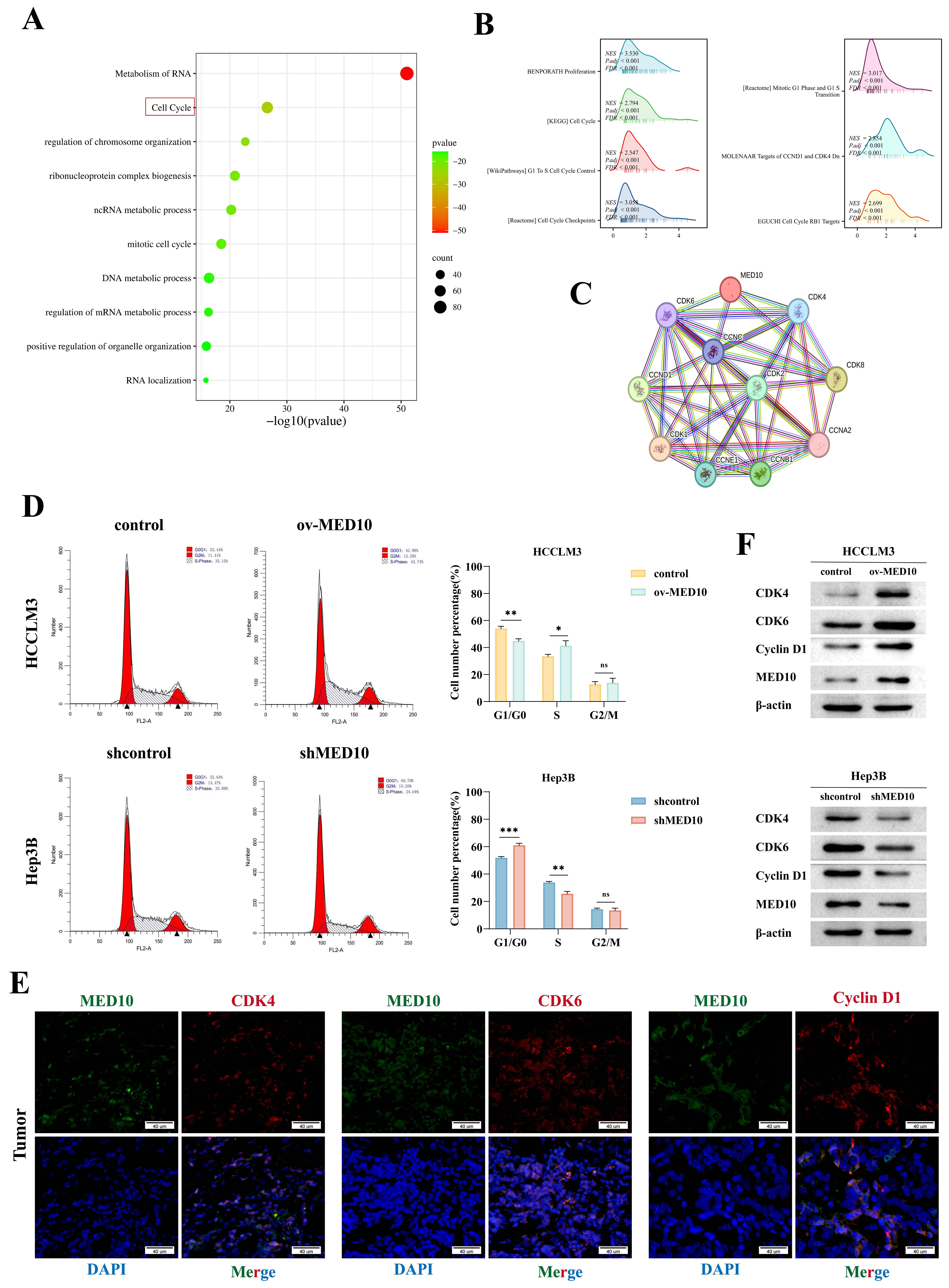
To investigate MED10’s role in proliferation, we analyzed TCGA data through functional enrichment. GO/KEGG pathway analysis revealed pronounced enrichment of MED10-correlated DEGs in cell cycle regulatory pathways (Fig. 4A). GSEA further implicated MED10 in proliferation and cell cycle processes including checkpoint control, G1/S transition, cyclin D1/CDK4 targets, cyclin D-associated G1 events, and RB1-mediated pathways (Fig. 4B). Protein-protein interaction network analysis (STRING) identified predicted binding partners CDK4, CDK6, CDK8, and CCNC (Fig. 4C). These findings establish MED10 as a regulator of cell cycle progression in HCC. Flow cytometry analysis revealed that MED10 overexpression in HCCLM3 cells significantly reduced G0/G1 phase occupancy while increasing S-phase fraction. Conversely, MED10 knockdown in Hep3B cells increased G0/G1 phase cells and decreased S-phase populations, with no significant G2/M phase alterations (Fig. 4D), indicating MED10’s critical role in G1/S transition. Immunofluorescence of HCC tissues demonstrated co-localization of MED10 with key G1/S regulators CDK4, CDK6, and cyclin D1 (Fig. 4E). Western blotting confirmed MED10-dependent regulation: overexpression upregulated while knockdown suppressed CDK4, CDK6, and cyclin D1 expression (Fig. 4F). Taken together, MED10 enhances HCC proliferation by driving the G1/S cell cycle transition, possibly via regulation of CDK4, CDK6, and Cyclin D1.
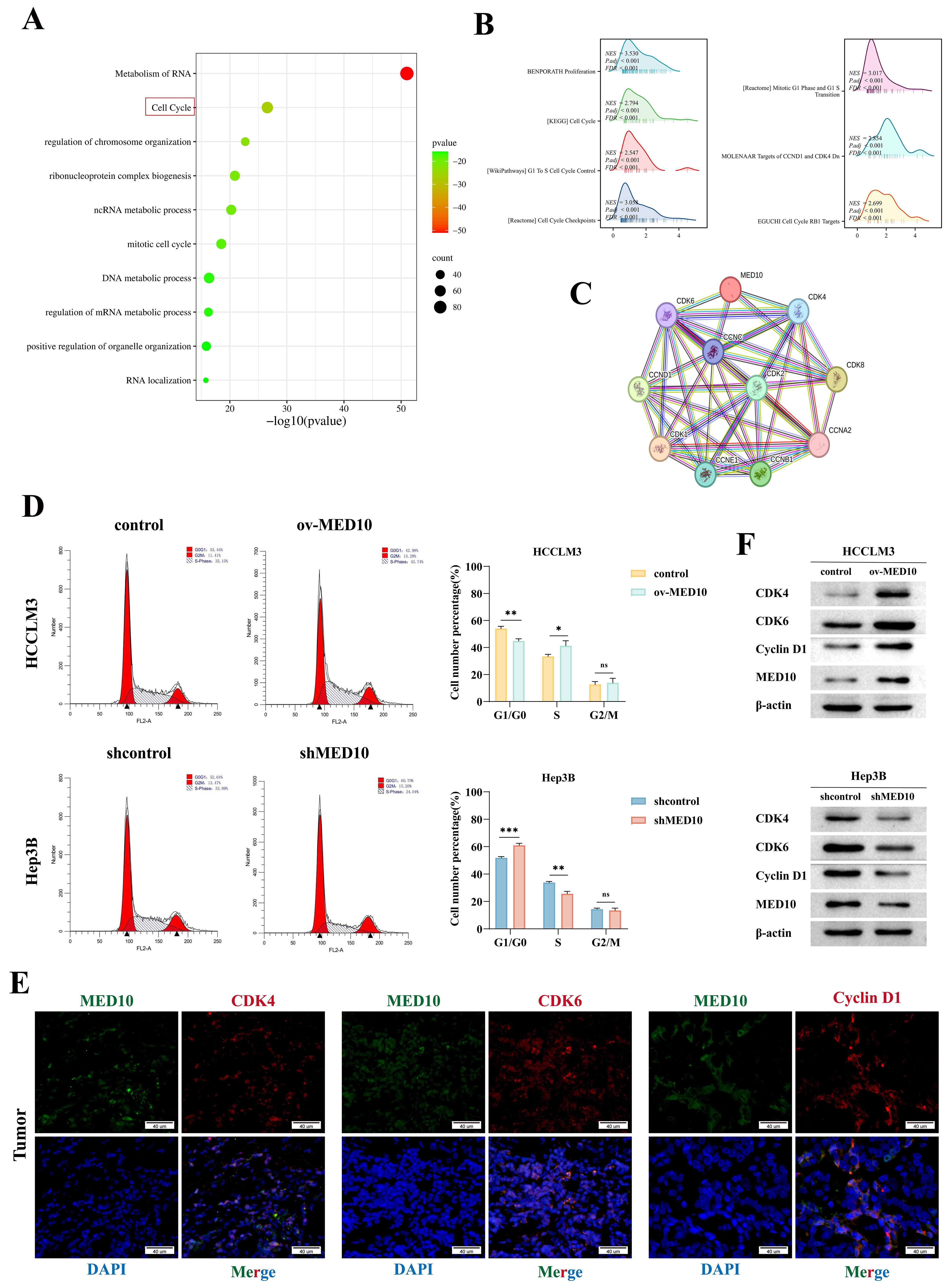 Fig. 4.
Fig. 4.
MED10 orchestrates cell cycle progression in HCC. (A)
Functional enrichment of MED10-associated genes (Gene Ontology (GO)/Kyoto Encyclopedia of Genes and Genomes (KEGG)) in TCGA-LIHC, such as cell cycle, etc. (B) GSEA of
differentially expressed mRNAs stratified by MED10 expression. (C)
Protein-protein interaction network linking MED10 to cell cycle regulators. (D)
Flow cytometry quantification of cell cycle phase distribution following MED10
modulation. (E) Immunofluorescence co-localization of MED10 (green) with CDK4,
CDK6, and cyclin D1 (red) in HCC tissues. Scale bar = 40 μm. (F) Western blotting analysis of CDK4/6
and cyclin D1 expression under MED10 modulation. ns: not significant (p
To elucidate MED10’s cell cycle regulatory mechanism, GSEA identified pronounced mitogen-activated protein Kinase (MAPK) pathway enrichment in high-expressing cohorts (Fig. 5A). Building on established MAPK/ERK functions in proliferation control [23], we interrogated pathway activity. Western blotting confirmed MED10 gain-of-function selectively enhanced RAF1-MEK-ERK cascade phosphorylation without affecting total protein abundance. Conversely, MED10 knockdown suppressed phosphorylation of these kinases (Fig. 5B), indicating MED10-dependent MAPK pathway activation. Since nuclear translocation of p-ERK1/2 is essential for transcriptional activation, immunofluorescence confirmed MED10’s regulation of this process: nuclear p-ERK1/2 accumulation increased with MED10 overexpression and decreased upon knockdown (Fig. 5C). The transcription factor c-Myc, a key MAPK/ERK downstream effector, regulates cyclin and CDK transcription (e.g., CDK4, CDK6, cyclin D1) to control cell cycle progression [24, 25]. GSEA linked MED10 to Myc pathway activation (“Myc active”, “Myc targets up”, “HCC up”; Fig. 5D), while TRRUST analysis confirmed c-Myc’s enrichment in mitotic G1/S transition (Fig. 5E). Western blotting demonstrated MED10-dependent c-Myc regulation: overexpression upregulated and knockdown downregulated c-Myc expression (Fig. 5F). Integrated analysis of TRUST, CHEA, ENCODE, and ChIP Atlas databases predicted eight c-Myc target genes involved in G1/S control, including CDK4, CDK6, and CCND1 (Fig. 5G). JASPAR identified high-score c-Myc binding motifs in these genes’ promoters (Fig. 5H). Collectively, these findings demonstrate that MED10 modulates both the RAF/MEK/ERK signaling pathway activation and c-Myc expression, potentially representing a key mechanism through which MED10 regulates cell cycle progression and proliferation in HCC cells.
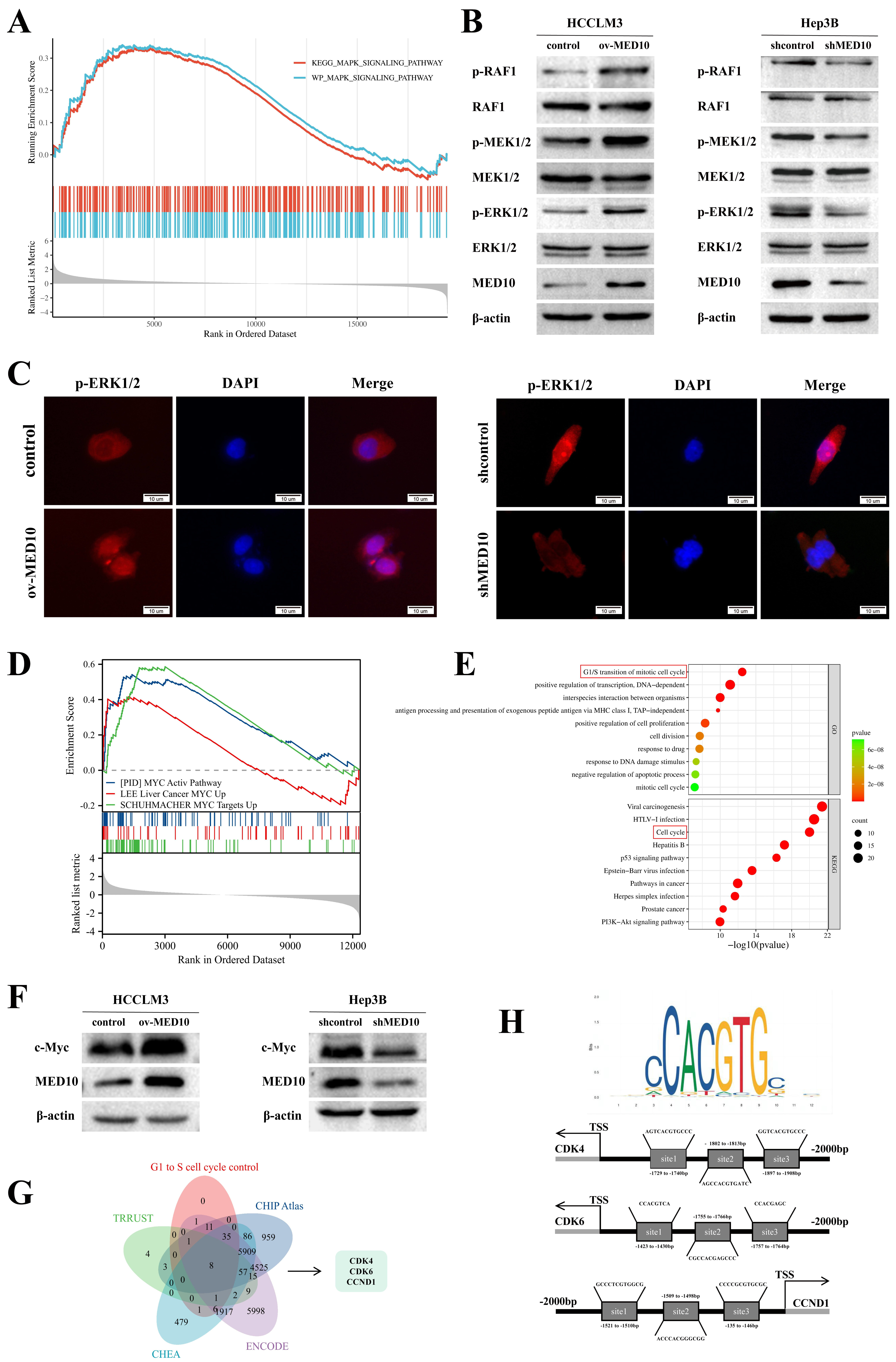 Fig. 5.
Fig. 5.
MED10 regulates activation of the MAPK signaling axis. (A) GSEA linking MED10 to MAPK signaling. (B) Western blotting analysis of MAPK pathway components under MED10 modulation. (C) Subcellular localization of p-ERK1/2 by immunofluorescence. Scale bar = 10 μm. (D) GSEA enrichment of Myc-related pathways. (E) TRRUST-predicted c-Myc involvement in G1/S transition. (F) MED10-dependent c-Myc regulation via Western blotting. (G) Venn intersection of c-Myc target genes (TRUST/CHEA/ENCODE/ChIP Atlas) and G1/S regulators. (H) JASPAR-predicted c-Myc binding motifs in CDK4/CDK6/CCND1 promoters.
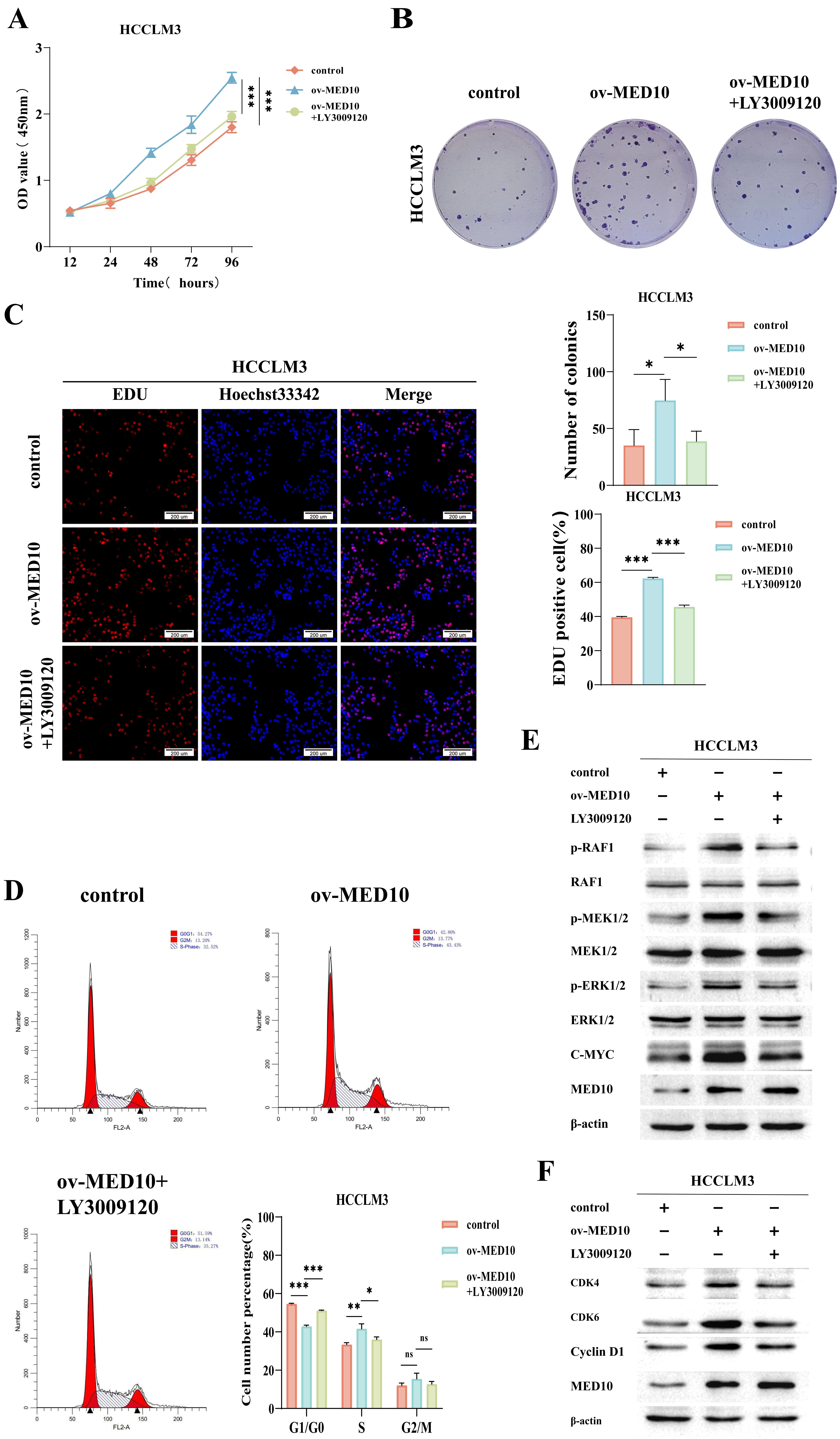
To determine if MED10’s pro-proliferative and cell cycle effects require MAPK signaling, we treated HCC cells with RAF inhibitor LY3009120. Proliferation assays (CCK-8, colony formation, EdU) demonstrated that MED10 overexpression-enhanced growth was partially reversed by LY3009120 (Fig. 6A–C). Flow cytometry confirmed MED10-mediated G0/G1 reduction and S-phase increase were similarly attenuated by RAF inhibition (Fig. 6D). Western blotting analysis revealed LY3009120 partially reversed MED10-driven phosphorylation of RAF1, MEK1/2, ERK1/2, and c-Myc (Fig. 6E), and correspondingly blunted MED10-induced upregulation of CDK4, CDK6, and cyclin D1 (Fig. 6F). Thus, MED10 regulates proliferation and cell cycle progression primarily through RAF, and related to MEK/ERK/c-Myc signaling. Conversely, MED10-promoted migration and EMT remained unaffected by RAF inhibition (Supplementary Fig. 1A–C), indicating these effects occur independently of MAPK pathway activation.
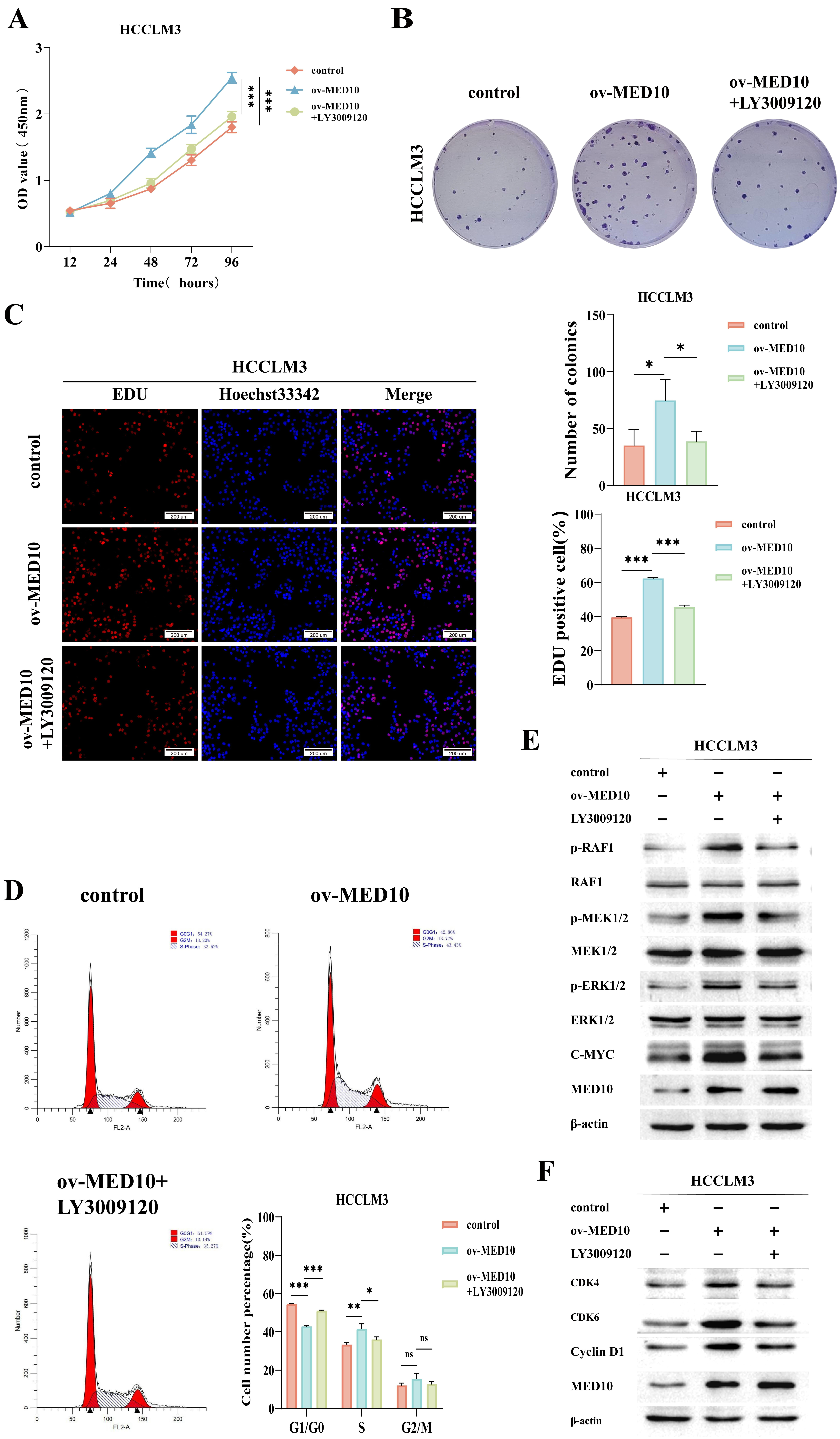 Fig. 6.
Fig. 6.
RAF inhibition reverses MED10-driven proliferation and cell
cycle progression in HCC. (A–C) CCK-8, colony formation, and EdU assays
collectively demonstrated that the RAF inhibitor LY3009120 blocks MED10-driven
proliferation. Scale bar = 200 μm. (D) Furthermore, flow cytometry analysis revealed that LY3009120
rescues the promoting effect of MED10 overexpression on cell cycle progression.
(E) Western blotting assessed MAPK pathway protein expression following RAF1
phosphorylation inhibition in MED10-overexpressing HCCLM3 cells. (F)
Additionally, Western blotting evaluated levels of cell cycle regulators CDK4,
CDK6, and cyclin D1 in these cells after LY3009120 treatment. “ns” denotes
p
Mediator complexes regulate gene expression at multiple stages of transcription, including preinitiation complex formation, transcriptional elongation, and mRNA splicing [26]. Growing evidence implicates dysregulation of Mediator complex subunits in cancer progression, where they play key roles in critical oncogenic processes such as migration, cell cycle, apoptosis, immune escape, tumor cell proliferation, and drug resistance [27, 28, 29, 30, 31]. As a critical subunit of the mediator complex, MED10 expression is markedly elevated in uterine fibroids, and facilitates tumor progression by enhancing proliferative capacity and metastatic potential through hsa-miR-590 regulation in bladder urothelial carcinoma [5, 32]. Notably, recent studies have revealed that MED10 enhances cisplatin resistance in HCC by promoting PTEN ubiquitination, highlighting its significant potential as a therapeutic target. Our current study further uncovers a novel role of MED10 in HCC malignant progression and elucidates its underlying molecular mechanism [13]. In the present investigation, we observed that MED10 exhibits high expression levels in HCC and correlates with unfavorable patient prognosis. Moreover, MED10 was demonstrated to exert critical roles in HCC cell migration and the EMT, and modulated HCC cell cycle progression and proliferation through modulating RAF1 activation. Collectively, this study establishes a theoretical foundation for developing novel diagnostic and therapeutic strategies in HCC.
Elevated MED10 expression was observed in HCC tissues and correlated with poorer patient prognosis. Identified as an independent prognostic risk factor, MED10 formed the basis of a predictive nomogram demonstrating strong performance for HCC patient outcomes. These findings linking MED10 expression to survival and prognosis highlight its significant potential as both a prognostic biomarker and therapeutic target. Furthermore, MED10 levels showed significant associations with key clinical indicators of aggressiveness—including microvascular invasion, tumor thrombosis, larger tumor size, and advanced T stage—suggesting its involvement in HCC metastasis and proliferation.
The EMT represents a pivotal step in tumor invasion and early metastatic dissemination. Within tumor microenvironments, EMT activation drives tumor cell invasion and metastasis. GSEA findings revealed that cell migration and the EMT were significantly enriched in the patient cohort exhibiting high MED10 expression. Our findings reveal that elevated MED10 expression stimulates HCC cell migration and induces EMT, as evidenced by suppressed E-cadherin alongside enhanced N-cadherin and vimentin expression. Conversely, MED10 inhibition reversed this phenotypic shift. Furthermore, we observed a direct correlation between MED10 levels and MMP13 expression, a type I collagenase [33] implicated in extracellular matrix degradation and tumor metastasis. Collectively, these findings demonstrate that MED10 serves as a key regulator in HCC metastasis.
The capacity for uncontrolled proliferation represents a cardinal malignant trait of tumors, which is frequently linked to dysregulated cell cycle progression. GO, KEGG, and GSEA results revealed that MED10 is implicated in biological processes that include cell proliferation, cell cycle regulation, and control of the G1-to-S cell cycle transition. Notably, MED10 overexpression was shown to enhance HCC cell proliferation and facilitate the G1-to-S cell cycle phase transition. The knockdown of MED10 showed the opposite result. Cyclin D1 is the dominant cell cycle protein in the G1 phase. Upon complex formation with CDK4 or CDK6, cyclin D induces phosphorylation of the retinoblastoma protein (Rb), thereby enhancing E2F transcriptional activity. This cascade drives progression through the G1/S phase checkpoint, facilitating the rapid transition from G1 to S phase [34]. We found that the expression of CDK4, CDK6, and CCND1 increased after MED10 overexpression, and that the expression of these three genes was inhibited after MED10 knockdown.
The MAPK/ERK cascade represents one of the canonical pathways within the broader MAPK signaling network. Initiation of the RAS-to-ERK signaling cascade triggers downstream effector molecules, including transcription factors and kinases, which subsequently modulate gene expression patterns [23]. Ultimately, this process drives fundamental biological mechanisms in hepatocellular carcinoma (HCC), such as cell migration, proliferation, cell cycle progression, apoptosis, and drug resistance [35, 36, 37]. Enrichment analysis further established a significant association between MED10 and the MAPK signaling pathway. Notably, we demonstrated that MED10 regulates phosphorylation of the RAF1-MEK1/2-ERK1/2 cascade and facilitates nuclear translocation of p-ERK1/2. The nuclear translocation of activated ERK1/2 and the subsequent stimulation of downstream effector transcription factors are critical events enabling the MAPK signaling cascade to exert its functional outputs. We found that MED10 modulates c-Myc expression. As a critical transcription factor downstream of MAPK/ERK signaling, c-Myc drives expression of key cell cycle regulators including CDK6, CDK4, and cyclin D, ultimately facilitating G1/S phase transition [25, 38]. Using a public database, we found that c-Myc is involved in regulation of the G1/S transition and is a common transcription factor for cyclin D, CDK4, and CDK6. Following RAF inhibitor LY3009120 treatment, phosphorylation of the RAF1-MEK-ERK cascade and c-Myc expression were suppressed, concomitantly reversing MED10-driven proliferation and cell cycle phenotypes.
Although widely utilized as a HCC biomarker for screening and clinical diagnosis, AFP exhibits undetectable elevation in nearly 30% of HCC cases, leading to suboptimal diagnostic sensitivity and specificity [39]. This diagnostic limitation underscores the need for novel biomarkers. Our findings identify dysregulated MED10 expression in HCC tissues that correlates with adverse clinical outcomes. Further, MED10 as a driver of HCC progression, accelerating tumor growth in both cellular models and living organisms, and interference with MED10 significantly inhibited tumor growth, which is mainly related to MED10 mediating activation of the MAPK/ERK axis to regulate the cell cycle process. Collectively, these data position MED10 as a dual-function target with diagnostic and therapeutic potential, establishing a conceptual framework for developing MED10-directed theranostic strategies. However, a paucity of drug development targeting MED10 as a therapeutic target and relevant clinical trials currently exist, necessitating further research to deeply explore the role and underlying mechanisms of MED10 in the malignant progression of HCC.
This work acknowledges certain limitations: while establishing MED10’s association with adverse HCC outcomes, our conclusions derive primarily from retrospective analyses of public datasets and clinical cohorts. Furthermore, information regarding the real-world follow-up of patients was lacking. Future studies should aim to enroll more subjects and conduct prospective clinical studies. Subjects should also be categorized according to population heterogeneity to further validate the potential of MED10 as a prognostic biomarker and its role in patients with HCC that have different genetic backgrounds and environmental factors. Secondly, our preliminary findings suggest that the regulatory effect The regulatory effects of MED10 on cell proliferation and cell cycle progression are dependent on its control of RAF1 activation. Nevertheless, we have yet to gain further insights into the mechanisms by which MED10 activates MAPK signaling axis. Therefore, future studies should continue to comprehensively explore the specific molecular mechanisms involved through additional experiments.
Collectively, this study furnishes multidimensional evidence that highlights the functional significance of MED10 in HCC pathogenesis, as well as its potential utility as a prognostic biomarker for HCC disease progression. Our findings demonstrated that MED10 exerts critical functional roles in key biological processes, including HCC cell migration, the EMT, cell proliferation, and cell cycle regulation. Mechanistically, MED10 primarily regulates proliferation and cell cycle progression by promoting RAF1 activation, a process possibly associated with MEK/ERK signaling and c-Myc expression. These findings collectively offer promising molecular targets for the development of HCC-directed anticancer therapeutic strategies.
All data reported in this paper will be shared by the corresponding author upon reasonable request.
JHL and YXL: validation, formal analysis, investigation, visualization, writing - original Draft, writing - review & editing. KJL: validation, investigation, methodology. ZQL: validation, investigation. BDC: investigation, resources, formal analysis. YB: conceptualization, methodology, supervision, project administration, funding, acquisition, writing - review & editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The clinical research protocol was conducted in full compliance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from all human participants or their legally authorized representatives prior to study enrollment. All animal experiments were performed in strict compliance with the guidelines established by Animal Welfare Act and Ningxia Medical University. The studies involving human participants and animal experimental protocol were reviewed and approved by the Medical Ethics Review Committee of Ningxia Medical University (Approval No. 2025-N092).
Not applicable.
This work was supported by the National Natural Science Foundation of China [grant numbers 81960533], Ningxia natural science foundation [grant numbers 2023AAC03606], Ningxia natural science foundation [grant numbers 2023AAC03507], School-level scientific research project of Ningxia Medical University [grant numbers XM2022041], 2024 First-class Discipline Funding of Ningxia Medical University (Third Clinical College of Medicine) [grant numbers 4301240006], Key R&D Program Talent Introduction Special Project of Ningxia Hui Autonomous Region (2024BEH04154).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL39944.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.