



 , Lijo N Varghese 1, Rajesh Katare 1,*
, Lijo N Varghese 1, Rajesh Katare 1,*
1 Department of Physiology, HeartOtago, School of Biomedical Sciences, University of Otago, 9010 Dunedin, New Zealand
Abstract
Sarcopenia is the progressive loss of skeletal muscle mass, strength, and function, significantly contributing to frailty, disability, and mortality in aging populations. As life expectancy rises, sarcopenia presents a growing public health challenge, increasing healthcare costs, and diminishing quality of life. Despite its prevalence, sarcopenia is often underdiagnosed due to limitations in current diagnostic tools, including the lack of standardized cut-off values and reliance on physical performance tests. The causes of sarcopenia are multifactorial, involving oxidative stress, chronic inflammation, mitochondrial dysfunction, satellite cell depletion, and impaired angiogenesis. Recent research highlights the role of microRNAs (miRs) in regulating these molecular pathways. miRs influence muscle homeostasis by modulating gene expression related to muscle atrophy, apoptosis, inflammation, and insulin resistance. While non-pharmacological interventions such as resistance training and blood flow restriction therapy remain the primary treatment strategies, their effectiveness is often limited in older adults with reduced muscle regenerative capacity. The identification of miRs as biomarkers could enhance early diagnosis and enable more personalized treatment approaches. However, further research is required to validate their clinical utility and therapeutic potential. This review comprehensively analyses the molecular mechanisms underlying sarcopenia, current diagnostic challenges, and emerging miR-based strategies that could transform its management. Future efforts should focus on integrating these molecular insights into clinical practice to improve early detection and intervention strategies.
Keywords
- sarcopenia
- aging
- muscle
- skeletal
- molecular mechanisms
- microRNAs
As a result of considerable human advancement, life expectancy has significantly increased. In the 1900s, the average life expectancy for men and women was 55 and 58 years, respectively [1]. By 2010, the averages had risen to 78 and 83 years [2]. Consequently, the population is shifting towards an aged demographic, with the number of individuals aged 60 years and over expected to triple by 2050 from 894 million as estimated in 2010 [3]. This phenomenon, termed population ageing [3], has unsurprisingly led to an increased incidence of age-related conditions, such as cancer, cardiovascular disease, organ failure, and sarcopenia [4]. Compared to projections for 2025, by 2060, projections estimate the incidence of diabetes will increase by 39%, hypertension by 27% and dyslipidaemia by 28% for the general population [5]. Regarding sarcopenia, in 2010, the incidence of sarcopenia was estimated to be around 50 million individuals, a value expected to rise to more than 200 million by 2050 [6]. Sarcopenia not only impacts the quality of life, but there is a significant economic and social burden [7, 8, 9]. In the United States alone, the estimated healthcare cost attributed to sarcopenia in 2000 was $18.5 billion, approximately 1.5% of the total healthcare expenditure for that year [10]. Furthermore, individuals with sarcopenia often require nursing home admission, develop physical disabilities and depression, and experience increased risks of hospitalisation, adding further strain on public healthcare systems [8]. The global healthcare system incurs $50 billion in costs due to physical inactivity [11]. Thus, understanding its mechanisms, identifying potential treatment targets, and improving diagnostic capabilities are imperative. If left unaddressed, the burden of sarcopenia will continue to grow.
Sarcopenia is the progressive loss of skeletal muscle mass, strength and function with age [12]. Although it does not directly lead to death, sarcopenia significantly increases the risk of all-cause mortality [13]. Furthermore, sarcopenia is strongly associated with frailty, which increases the likelihood of falls, disability, hospitalisations, and a reduced overall quality of life [13]. Although sarcopenia is referred to as an age-related disease affecting muscle function, for decades, the term was generalised to include muscle wasting regardless of muscle function [14].
Due to the inaccuracy of screening tools, many cases of sarcopenia remain undiagnosed. Diagnoses often rely on a case-finding approach, where individuals are evaluated only after presenting symptoms [6]. This approach is most suitable in care settings such as hospitals and nursing homes, where sarcopenia prevalence is higher [14] and symptoms are more apparent. However, this method is less effective for the general population, as it depends on self-reporting, a notoriously unreliable method for diagnoses. While diagnosing sarcopenia is relatively straightforward—using measurements of muscle mass, strength, and performance—a lack of standardised cut-off points limits its application in clinical practice [14].
Diagnosis typically begins with an evaluation of hand-grip strength to identify probable sarcopenia. However, this method does not account for other factors contributing to reduced muscle strength, such as rheumatoid arthritis [15]. Following the grip strength test, muscle mass is assessed using techniques such as dual-energy X-ray absorptiometry (DEXA), computed tomography (CT), magnetic resonance imaging (MRI), or bioimpedance analysis to estimate lean mass [14, 16].
Although each of these methods has its strengths and weaknesses, the primary limitation is the absence of a widely accepted and standardised cut-off point [14]. Despite these challenges, a definitive diagnosis can often be made with muscle mass measurements, though the severity of sarcopenia cannot always be determined. The final diagnostic step is a physical performance test, which evaluates an individual’s ability to perform daily tasks independently. Common physical performance assessments include 400-meter timed walks, gait speed tests, or timed up-and-go tests. These evaluations are crucial in determining sarcopenia severity and guiding appropriate interventions [14]. While current diagnostic methods for sarcopenia lack standardised parameters, extensive cohort studies, such as those conducted by the European Working Group on Sarcopenia in Older People (EWGSOP), have developed evidence-based cut-off recommendations that should be widely adopted [17]. However, the criteria for identifying pre-sarcopenia remain unconvincing. For instance, current guidelines suggest that individuals are considered pre-sarcopenic if diagnosed with a reduction in muscle mass. Ideally, pre-sarcopenia should be diagnosed before the onset of muscle loss, highlighting the need for alternative diagnostic approaches.
One promising avenue is using biomarkers, which are already well-established for other diseases, such as troponins in cardiovascular disease [18]. In sarcopenia, biomarkers could complement current diagnostic methods by increasing accuracy and enabling earlier identification of at-risk individuals [19, 20]. Unfortunately, no clinically validated biomarkers for sarcopenia currently exist. Identifying such biomarkers could revolutionise the diagnostic process, allowing for earlier intervention when it is most effective.
Treating sarcopenia remains challenging due to the lack of approved pharmacological interventions. Although various drugs, including growth hormone, testosterone, and selective androgen receptor modulators, have been tested, their results have been underwhelming [14, 21]. While bimagrumab, an activin type II receptor antagonist, has shown promise in phase II clinical trials by significantly increasing lean muscle mass, no pharmacological treatment has yet been approved [22]. As such, the primary approach to managing sarcopenia currently lies in non-pharmacological interventions such as resistance training, blood flow restriction training (BFRT), suspension-based resistance training (S-RT) and nutritional supplements.
Resistance training is the most effective intervention among non-pharmacological strategies, supported by substantial evidence [11, 23]. High-volume resistance training has increased muscle mass and strength in individuals with sarcopenia [24, 25]. However, its effectiveness diminishes in older adults due to blunted muscle hypertrophic potential, emphasising the importance of early diagnosis and intervention [25, 26, 27]. In addition to improving muscle mass and strength, resistance training enhances muscle functionality, as evidenced by improvements in timed up-and-go tests [28], gait speed [29], and standing balance [29]. Resistance training combats age-related muscle wasting by stimulating muscle protein synthesis and inducing metabolic and morphological adaptations with sustained effects lasting for 24 hours [30]. Furthermore, resistance training activates key molecular pathways implicated in sarcopenia, such as phosphatidylinositol 3kinase protein (PI3K)/Akt [31], mitogen-activated protein kinase (MAPK) [32], mammalian target of rapamycin (mTOR) [33], and forkhead box O3 (FOXO3) [33]. It also increases anabolic hormone production, such as testosterone [34], which activates satellite cells, promotes muscle regeneration, and slows sarcopenia progression [35].
BFRT is another potential intervention for sarcopenia. When combined with nutritional supplementation also shows promise, though to a lesser extent [36, 37]. This technique involves performing strength training while restricting venous return but maintaining arterial blood flow [14]. BFRT creates a hypoxic environment that amplifies metabolite accumulation, thereby enhancing muscle hypertrophy with lower loads than traditional resistance training [38]. This makes BFRT more tolerable for older adults or frail individuals [39, 40]. Studies suggest that low-load BFRT can achieve results comparable to high-load resistance training. However, the potential adverse effects of BFRT remain poorly understood, and its application should currently be supervised by trained professionals [14, 41]. Further research into its safety and the development of standardised training protocols could make BFRT a more accessible option for sarcopenic individuals.
S-RT is a novel exercise modality that involves anchoring body segments to suspended straps to perform multi-joint, multi-planar movements in an unstable environment. This approach challenges the musculoskeletal and neuromuscular systems by requiring continual engagement of stabilizing muscles to maintain balance and control during exercises [42].
A study by Aguilera-Castells et al. [43] suggests that S-RT elicits comparable acute responses to traditional resistance training performed in stable environments. In particular, a systematic review reported greater core muscle activation during exercises such as push-ups, inverted rows, prone bridges, and hamstring curls using S-RT compared to conventional weight-based RT. This enhanced activation may offer unique benefits for older adults with sarcopenia, a condition characterized by progressive muscle loss and functional decline [43]. Further, a 12-week randomized controlled trial comparing suspension and traditional resistance training in older men demonstrated that S-RT led to significant improvements in handgrip strength, a key indicator of overall muscle function [44]. Furthermore, suspension training has been associated with favorable changes in body composition and bioimpedance vector patterns, as well as increased serum levels of muscle growth factors, suggesting a potential role in promoting muscle hypertrophy and metabolic health in sarcopenic individuals [45]. However, despite these advantages, there are notable limitations to S-RT in this population. The unstable nature of the training environment may pose a higher risk of falls or injury, particularly among frail older adults with poor balance or comorbid conditions. Additionally, proper technique and supervision are crucial to avoid compensatory movements or joint strain. Accessibility and familiarity with suspension systems may also limit widespread adoption among elderly populations.
For individuals with comorbidities such as cardiovascular disease, diabetes, or chronic obstructive pulmonary disease, resistance training may be impractical or impossible [46]. In such cases, nutritional supplementation might be the only viable option [47]. However, evidence supporting its efficacy in sarcopenia is weak. For example, whey protein supplementation has not significantly improved lean body mass, strength, or physical function [48]. Similarly, multi-nutrient supplementation (a combination of carbohydrates, fats, and proteins) has not demonstrated notable benefits without concurrent exercise [49, 50]. Vitamin D plays a role in muscle regulation and is commonly deficient in older adults [51, 52]. However, another study found no significant improvements in muscle mass or strength following vitamin D supplementation alone [53]. Nutritional supplementation is most effective when combined with exercise and administered early in sarcopenia’s progression [54, 55]. Over the past few decades, a growing body of research has indicated that incorporating creatine supplementation into resistance training regimens may enhance muscle mass and performance outcomes in older adults more effectively than resistance training alone. Nonetheless, the strength of this evidence is constrained by the small sample sizes and limited statistical power of many existing studies [56, 57]. Unfortunately, because sarcopenia is often diagnosed after significant muscle loss has occurred, the potential benefits of supplementation are often diminished. This underscores the need for early diagnostic methods to enable timely interventions.
The lack of effective treatment options for sarcopenia means that, in advanced stages, individuals may become bedridden and require palliative care. This decline is often accompanied by emotional distress, with sarcopenia being linked to an increased risk of depression [58]. As the condition progresses, the quality of life deteriorates significantly. While developing effective treatments is essential, advancements are limited by an incomplete understanding of the disease’s mechanisms. Therefore, further research into sarcopenia’s molecular and physiological underpinnings is critical to identifying new therapeutic targets.
Muscle mass begins to decline at a rate of approximately 1–2% per year after the age of 50, and by old age, muscle strength is approximately 30–40% lower than in individuals in their 20s [12]. At the organ level, sarcopenia is associated with morphological changes, including myosteatosis (fat and connective tissue infiltration), reductions in muscle fibre size (atrophy) and number, and shifts in muscle fibre composition [59]. However, the cellular and molecular mechanisms driving these changes remain under-researched. Current evidence suggests that sarcopenia is a multifaceted disease arising from a complex interplay between multiple dysfunctional processes. The processes range from dysfunction within the regeneration process, with evidence linked to satellite cell senescence and reduced function, to chronic inflammation and oxidative stress (Fig. 1) [12, 60]. Additionally, age-related changes outside of the muscle itself may contribute to the progression of sarcopenia, such as hormonal changes, neural deficits and dysregulated angiogenesis (Fig. 1) [61]. A key step in treating sarcopenia will arise from elucidating the various mechanisms. As such, this review aims to explore the firmly established evidence surrounding the proposed mechanisms of sarcopenia.
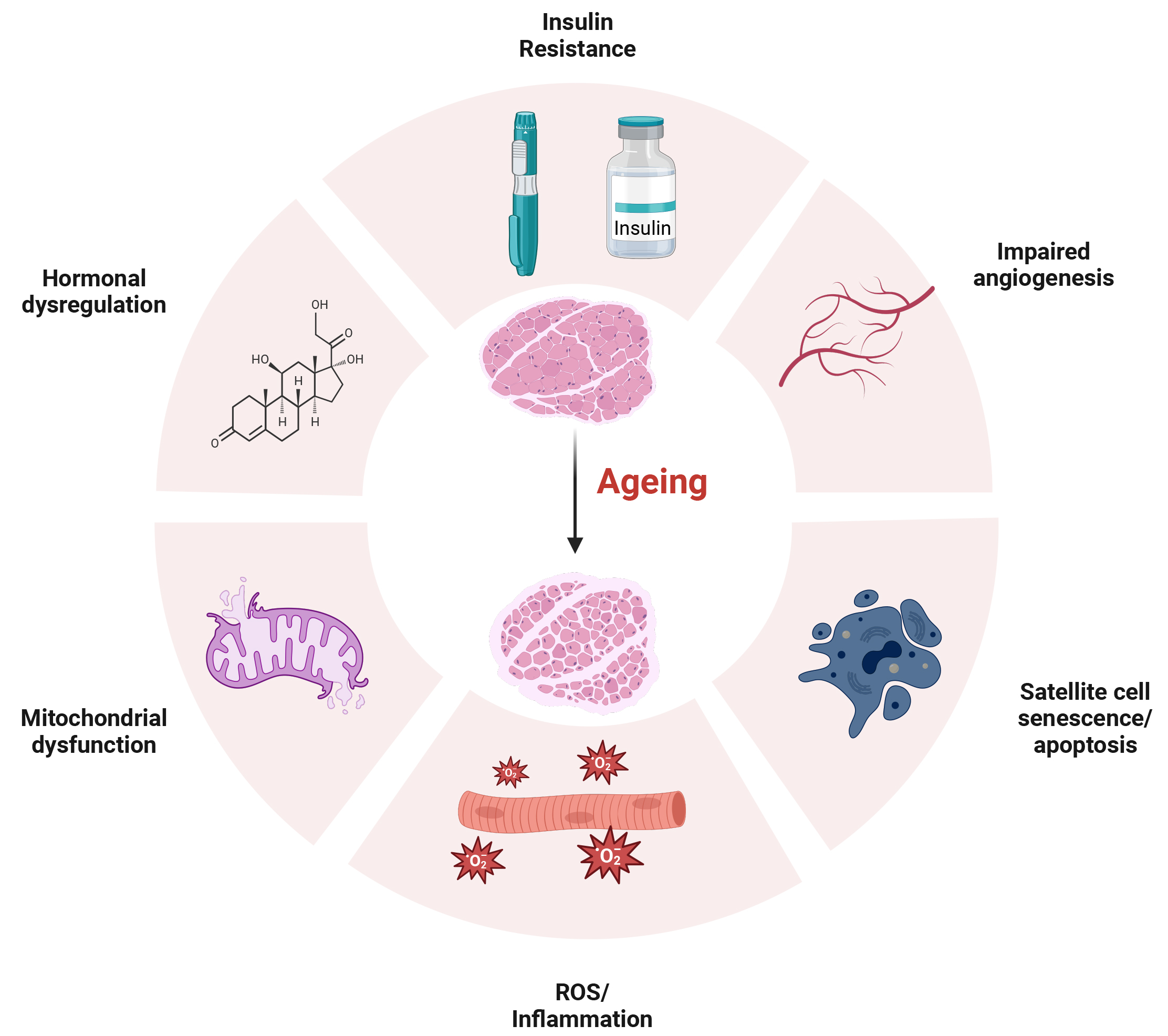 Fig. 1.
Fig. 1.
Ageing-associated changes in skeletal muscle. Ageing leads to various physiological changes that contribute to sarcopenia. Key factors involved include insulin resistance; impaired angiogenesis; satellite cell senescence and apoptosis; reactive oxygen species (ROS) accumulation and inflammation; mitochondrial dysfunction; and hormonal dysregulation. Created with Biorender.com.
Given its heavy functional demands, skeletal muscle possesses a high regenerative capacity [62], primarily mediated by progenitor satellite cells [63]. These muscle stem cells are indispensable for muscle repair, as their elimination completely prevents regenerative myogenesis [64]. Evidence suggests that impaired satellite cell function contributes significantly to sarcopenia. Studies have demonstrated a decline in the number and function of satellite cells with age, leading to reduced muscle regenerative capacity [65, 66, 67]. This has been illustrated by the diminished ability of aged myofiber-derived clones to proliferate compared to those from younger individuals [68]. However, the rate of proliferation and differentiation of muscle precursor cells between young and aged individuals may not differ significantly [69]. Instead, alterations due to senescence have been reported with senescent cells exhibiting impaired expression of differentiation markers, reduced fusion capacity, and the formation of smaller myotubes, suggesting that cellular senescence plays a pivotal role in sarcopenia [69].
Oxidative stress is another critical factor implicated in the pathogenesis of sarcopenia. It has been closely linked to cellular senescence [70] and several other hallmarks of sarcopenia [71]. Oxidative stress markers, such as malondialdehyde (MDA) and 4-hydroxy-2,3-nonenal (HNE), are elevated in sarcopenic individuals compared to non-sarcopenic controls [72]. Similar increases in MDA have been observed in aged populations and animal models, suggesting a strong relationship between oxidative stress and age-related muscle loss [73]. Oxidative stress often leads to a pro-inflammatory state. Elevated levels of inflammatory markers, including C-reactive protein and fibrinogen, have been observed in individuals with sarcopenia [74, 75]. This evidence supports a model in which oxidative stress triggers inflammation, further exacerbating muscle degeneration. Together, these processes create a detrimental feedback loop that accelerates sarcopenia progression.
In addition to inflammation and oxidative stress, evidence suggests that mitochondrial dysfunction may play a role in age-related muscle loss [76]. Mitochondria are highly dynamic organelles that undergo constant remodelling through mitophagy, fission, biogenesis, and fusion in response to fluctuating cellular conditions [77]. Although their primary role is to provide ATP, they participate in signal transduction, cell cycle regulation, and thermogenesis [78]. Given their critical role in maintaining skeletal muscle homeostasis, impairments in mitochondrial biogenesis or function can have severe consequences [77]. Several studies have demonstrated a significant association between mitochondrial dysfunction and an age-related increase in mitochondrial DNA damage [79, 80, 81]. Under normal conditions, when mitochondria become dysregulated, they undergo fission and mitophagy to maintain a healthy mitochondrial population [77]; however, this process becomes impaired with age [77]. In aged mice, mitochondria appear swollen and exhibit a loss of cristae, suggesting dysregulated mitophagy [82]. Furthermore, Goljanek-Whysall et al. [82] found that overexpressing microRNA (miR)-181a, a regulator of mitochondrial dynamics in aged mice, restored a healthy mitochondrial population and improved muscle function, as evidenced by enhanced force production. A recent study showed that recombinant Sestrin 2 treatment reduced DNA damage and improved mitochondrial function in an in vitro model of sarcopenia [83]. These findings indicate that mitochondrial function and regulation are tightly linked to muscle performance and suggest that miR manipulation may have therapeutic potential for treating sarcopenia [82].
Another proposed mechanism contributing to age-related muscle loss is endothelial dysfunction coupled with impaired angiogenesis. Angiogenesis, forming new blood vessels from existing ones, is essential for remodelling the vascular network and may be key in developing sarcopenia. Skeletal muscle requires an adequate blood supply to deliver nutrients and oxygen while removing metabolic waste products, and its microcirculation must be highly adaptable to meet increased demands during exercise or growth. As muscle mass increases, so does the need for enhanced blood flow. The accompanying increase in shear stress on arteriole and capillary walls is a stimulus for angiogenesis [84]. This process involves the migration, proliferation, and differentiation of endothelial cells and pericytes [85]. However, research indicates that the angiogenic response becomes impaired with ageing. For example, studies comparing young and old mice subjected to hindlimb hypoxia/ischemia have found that older mice exhibit reduced capillary density at the injury site, suggesting diminished angiogenic potential [86, 87]. Similarly, when used as a model, endothelial cells derived from older donors or aged human umbilical vein endothelial cells demonstrate reduced proliferation, migration, and tube formation compared to cells from younger individuals [86]. In addition, retinal vessels in aged rats often appear distorted with reduced luminal patency, further supporting the notion that angiogenesis is dysregulated with age [88]. The net effect of impaired angiogenesis is a reduced blood flow to skeletal muscle, which limits oxygen and nutrient delivery and hampers the clearance of metabolic waste. These changes can lead to reduced protein synthesis and contribute to the muscle atrophy observed in sarcopenia [89, 90]. These findings suggest that dysregulation of angiogenesis, likely initiated by endothelial dysfunction, plays an important role in the progression of age-related muscle loss.
The pathogenesis of sarcopenia is multifactorial, with several dysregulated processes, including oxidative stress, chronic inflammation, cellular senescence, and impairments in mitochondrial and angiogenic function playing significant roles. Although these mechanisms are not exhaustive, their combined effects contribute to the decline in muscle mass, regenerative capacity, and overall muscle function characteristic of sarcopenia. While our current understanding of these processes provides valuable insights into the aetiology of sarcopenia, the disease’s complexity and the absence of effective interventions underscore the need for further research to elucidate its underlying molecular pathways fully.
An emerging promising area of research is miR. miRs are short, noncoding RNA molecules, typically 20–22 long nucleotides. Although evidence exists for multiple miR biogenesis pathways, the canonical pathway is the most common. In this pathway, miR genes in the DNA are transcribed by RNA polymerase II into a primary miR (pri-miR) [91]. This immature transcript is then processed in the nucleus by the DROSHA/Di-George syndrome critical region 8 (DGCR8) complex into a precursor miR (pre-miR) [91]. Following export from the nucleus via exportin-5, the pre-miR is further processed in the cytoplasm by DICER into a miR duplex [91]. Finally, this duplex is incorporated into the RNA-induced silencing complex (RISC) with Argonaute (AGO) proteins, during which the duplex unwinds and the passenger strand is degraded [91]. RISC then targets the 3′ untranslated regions (UTR) of specific mRNAs; complete complementary binding typically results in mRNA cleavage, while imperfect binding leads to translational suppression (Fig. 2). Many miRs have been evaluated for their therapeutic potential, with several miR-therapeutics currently undergoing clinical trials [92]. For example, miR-122 has completed a phase 2 clinical trial for the treatment of hepatitis virus infection, while miR-16 has completed a phase 1 trial for malignant pleural mesothelioma. Other microRNAs currently under investigation include miR-92 (for wound healing and heart failure), miR-132 (for heart failure), and miR-29 (for keloid disorder). While miRs exert effects within the cells that produce them, they can also be released into the extracellular environment. Once released, they may be taken up by neighboring cells or enter the systemic circulation. In circulation, miRs remain stable and are protected from degradation by binding to carrier proteins or by being encapsulated within microvesicles or nanovesicles. This stability enables miRs to exert regulatory effects on distant cells and tissues. Owing to these properties, miRs are being explored not only for their therapeutic potential but also as diagnostic biomarkers, as their expression is often dysregulated in various disease states [93]. Due to this property, in addition to their therapeutic applications, miRs are also being investigated as diagnostic biomarkers, as many exhibit dysregulated expression in disease states [94].
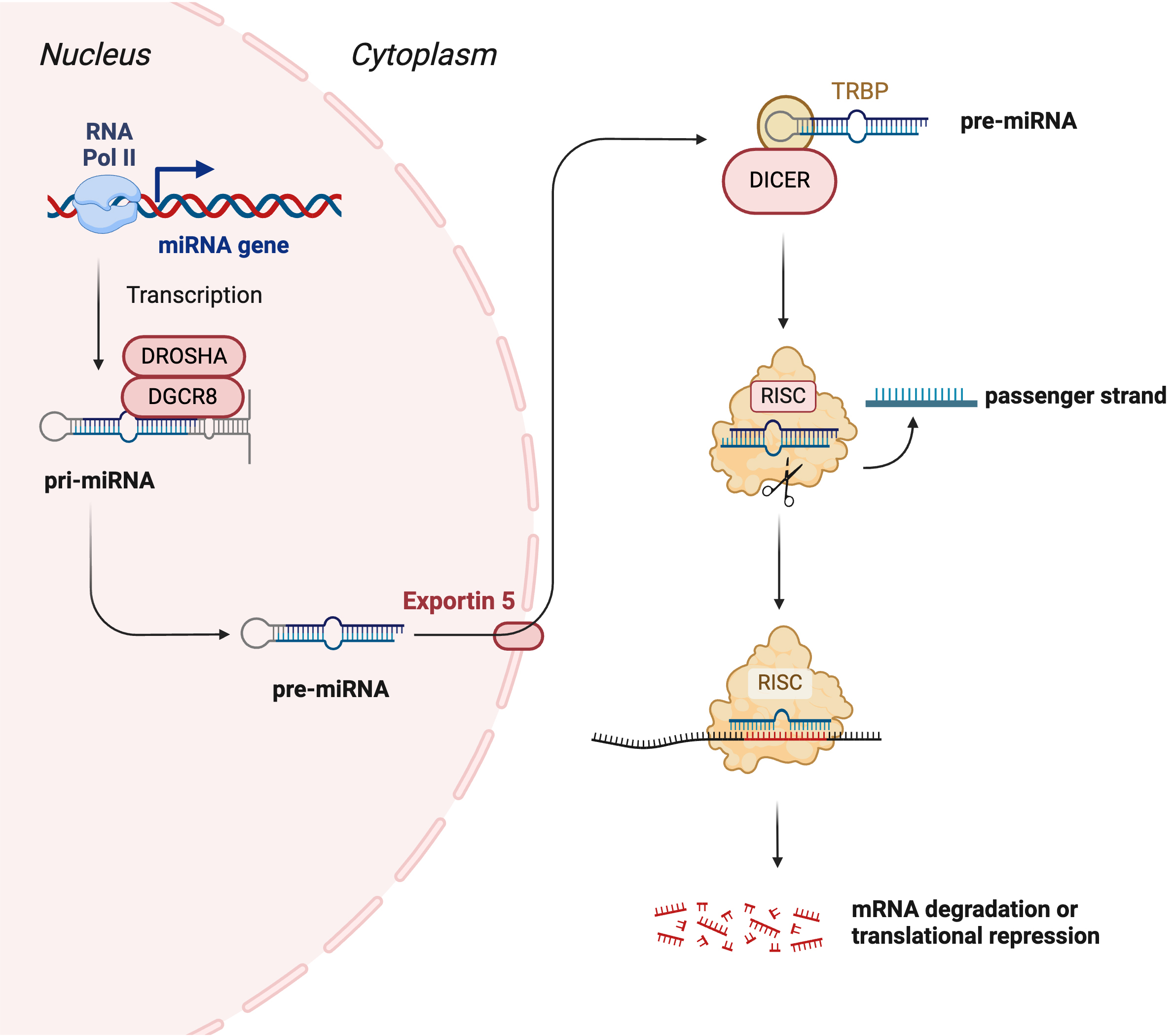 Fig. 2.
Fig. 2.
Canonical pathway of microRNA biogenesis. In the canonical pathway, the miRNA gene is transcribed by RNA polymerase II to form a primary miRNA (pri-miRNA). The pri-miRNA is cleaved by DROSHA and Di-George syndrome critical region 8 (DGCR8) to form the precursor miRNA (pre-miRNA). Pre-miRNA is transported to the cytoplasm by exportin-5. In the cytoplasm, pre-miRNA is cleaved by DICER to form a duplex mature miRNA. The mature miRNA is loaded to RNA-induced silencing complex (RISC), leading to the cleavage of the passenger strand. miR-RISC binds to the target mRNA leading to its degradation or translational repression depending on the binding complementarity. Created with Biorender.com.
Recent studies have begun to elucidate the role of miRs in age-related muscle loss. Differential expression analyses comparing young and aged samples have identified many miRs whose levels change with age [95, 96, 97]. The following section will summarize miRs associated with pathological features in sarcopenia. Fig. 3 summarizes known miRs associated with sarcopenia.
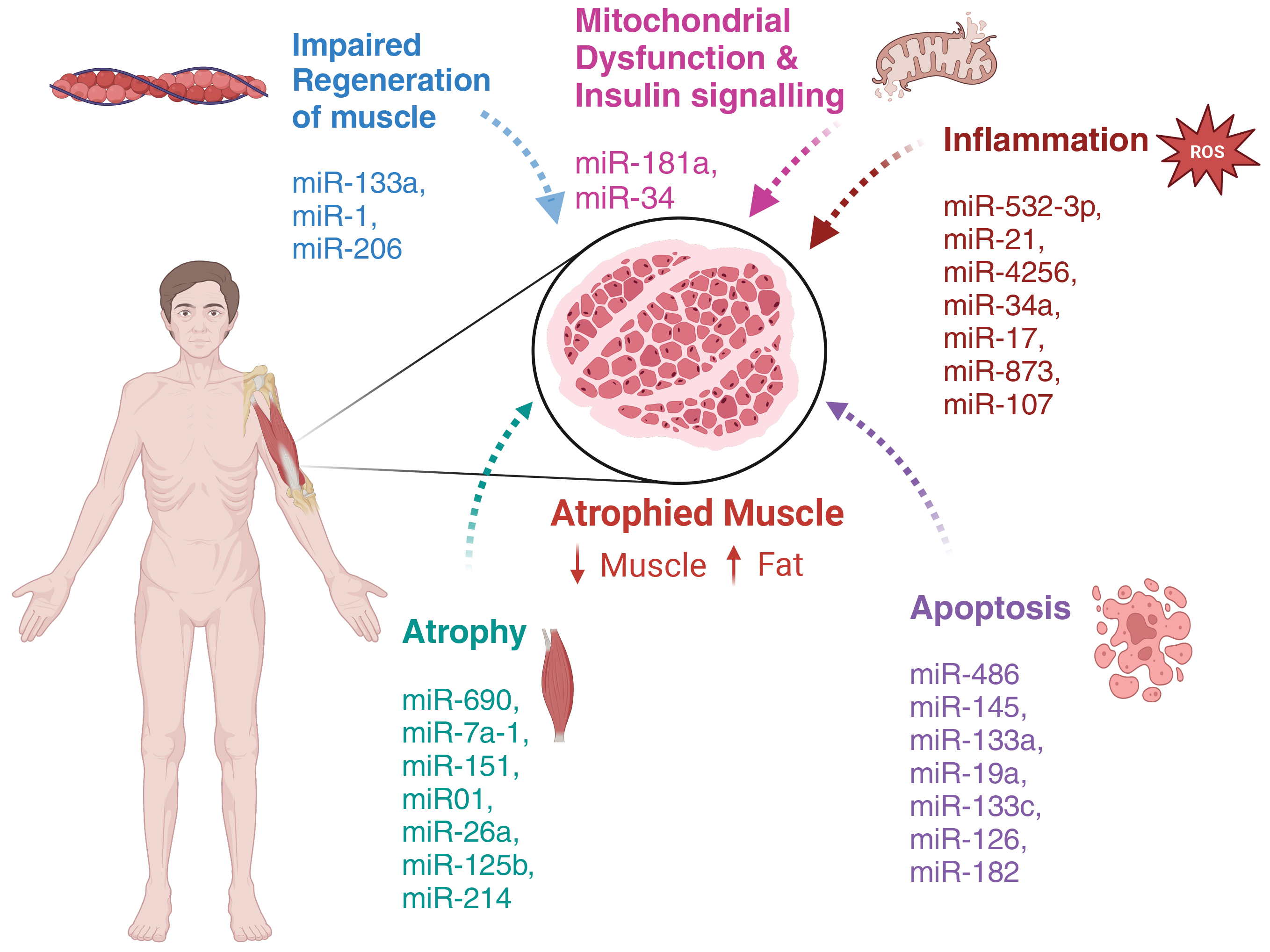 Fig. 3.
Fig. 3.
microRNAs involved in age-associated progression of sarcopenia. Ageing leads to skeletal muscle atrophy, characterized by a reduction of muscle mass and an increase in deposition of adipose tissue. Age-associated dysregulation of microRNA expression leads to impaired regeneration of muscle, mitochondrial dysfunction and insulin signaling, increased inflammation, apoptosis, and atrophy. These processes drive muscle atrophy and functional decline with age. Created with Biorender.com.
A systematic search strategy was developed to identify relevant literature without bias. Keyword-based searches were conducted in both PubMed and Scopus databases using Boolean operators (“AND”, “OR”) to combine search terms. In PubMed, the keywords used were: “age-related muscle loss” OR “age-associated muscle loss” OR “sarcopenia” AND “microRNA” OR “miRNA” OR “miR”. In Scopus, the author keyword function was used with the terms: skeletal muscle AND ageing OR aging OR deterioration AND microRNA OR miRNA OR miR. To ensure the inclusion of recent and relevant primary research, studies published before 2020, review articles, and those lacking a direct comparison between young and aged groups with statistically significant results were excluded. Both search strategies were applied to both databases to ensure comprehensive coverage and minimize the risk of omitting eligible studies. As this review aims to identify skeletal muscle miRNAs dysregulated with aging, studies were excluded if they were not directly related to skeletal muscle aging or if their methodologies did not include a young/adult group compared to an aged group. In total, 243 publications were initially identified across both databases. After excluding studies published before 2020, 142 publications remained. Further screening to eliminate studies lacking comparisons between young and old groups yielded a final selection of 12 relevant studies (summarized in Tables 1,2,3, Ref. [82, 96, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107]).
| Xhuti et al. (2023) [100] | Yu et al. (2022) [96] | Jia et al. (2020) [101] | Shao et al. (2022) [98] | |
| Samples & controls | Human: young group averaging 21.8 & aged group averaging 74.9. The number of participants varied depending on the analysis. | Mice: 3-month-old (young) and aged 17-month-old (aged) (n = 2; male mice). | Sika Deer: 1, 3, 5 and 10 years old (n = 3 for each group). | Mice: 3-month-old & 24-month-old (n = 6; male). |
| Human: Adult group averaging 31 years (n = 9) & aged group averaging 71 years (n = 10). | ||||
| Methods | Exosome isolation & characterisation. Skeletal muscle biopsy, histology, nanoparticle tracking, TEM & cell culture. | Quantitative reverse transcription polymerase chain reaction (qRT-PCR), miR target prediction and protein gene network mapping. | qRT-PCR, miR-mRNA interaction network construction, gene ontology & Kyoto encyclopedia of genes and genomes (KEGG) Analyses. | Histology, force generation, histology, qRT-PCR, western blot (WB) & luciferase. |
| miRs; targets (analysed) | miR-1-3p, -23a-3p, -23b-5p, -92a, -146a-5p, -181a-3p, -199a-3p, -206, -422a (↓). miR-34a (↑). | 130 miRs exhibited a 2-fold change with ageing. Raising the threshold to 5-fold identified 14 miRs. | miR-378b & miR-17-5p (↑); WWP1, DEK, DHCR24, PTTG, & HSPA9. | miR-690 (↑); Mef2c. |
| miR-7 & miR-199a-5p (↓); UCP3, FUS, CAV1, RAB6A, & TOM1. | ||||
| Results | Loss of skeletal muscle mass and reduced cross sectional area (CSA) of Type IIA fibers. Exercise ameliorated some miRs and protein changes. | Gene mapping identified Notch1, Egr2 and MyoD as significant nodes. | Alteration in calcium signalling pathway was the major gene signalling pathway during ageing. Enrichment analysis identified changes in metabolism and energy. | Ageing caused a reduction in CSA, maximal tetanic force & elevated levels of atrophy genes. |
Up and down arrows indicate if miR was upregulated or downregulated
respectively. WWP1, WW domain-containing E3 ubiquitin protein ligase 1;
DHCR24, 3
| Liang et al. (2023) [99] | Fukuoka et al. (2021) [102] | Shin et al. (2020) [103] | Gao et al. (2021) [104] | |
| Samples & Controls | Rats: 6 & 21-months-old (n = 3; male). | Mice: 6-weeks & 23-months-old (n = 5; male). | Human: age varying from 25–80 years (n = 20). | Rats: 8 and 18 months old. Sedentary or exercise starting at 8 or 18 months of age (n = 12; female). |
| Mice: 3 (young) & 24 months (aged) (n = 6). | ||||
| Methods | Exercise training, muscle weight, histology, qRT-PCR, miR target prediction & western blotting analysis. | Cell culture, target prediction, qRT-PCR, miR administration, histology, strength test, reporter assay. | Cell culture, luciferase assay, qRT-PCR, western blotting analysis, morphometric analysis, force & fatigue analysis. | Exercise, qRT-PCR, in-silico analysis of signalling pathways, target prediction & western blotting analysis. |
| miRs; targets (analysed) | miR-7a-1-3p (↑); Ar & IGF-1. | miR-199-3p (↓); Lin28 & Suz12. | 42 miRs in the Dlk1-Dio3 cluster were analysed. | miR-486, -145, -133a, -19a & -133c (↓). |
| miR-135a-5p (↓); FAK. | miR-126, -23a, -182, -206, -199a & -208b (↑). | |||
| miR-151-5p (↑); BDNF. | ||||
| Results | Exercise ameliorated dysregulation of atrophy-associated miRs & proteins in aged rats. | Reduced expression of muscle differentiation markers with age. miR overexpression improved muscle function. | miR-376-3c reduced muscle atrophy. | Anti-apoptotic, mitochondrial function & autophagy gene markers downregulated with age. miR-486 target PTEN. |
Up and down arrows indicate if miR was upregulated or downregulated respectively. Ar, androgen receipt; IGF-1, insulin growth factor-1; FAK, focal adhesion kinase; BDNF, Brain-derived neurotrophic factor; Dlk1-Dio3, delta-like homolog 1 gene and the type III iodothyronine deiodinase; PTEN, Phosphatase and tensin homolog deleted on chromosome 10.
| Chen et al. (2020) [105] | Borja-Gonzalez et al. (2020) [106] | Goljanek-Whysall et al. (2020) [82] | Kukreti et al. (2020) [107] | |
| Samples & Controls | Human: young group averaged 26.3 years old, and the aged group averaged 64.3 (aged) (n = 24; male). | Mice: 6-month-old (young) and 24-month-old (aged) (n = 3; male). | Mice: 6-month-old (young), and 22–24-month-old (aged) (n = 3; male). | Mice: 3 months (young) old and 2 years (aged) old (n = 3; male). |
| Cells: Human myoblasts. | ||||
| Methods | RNA isolation, microarray, qRT-PCR, western blotting analysis, serum cytokine measurement, cell culture & chromatin immunoprecipitation. | Satellite cell isolation, transfection & myogenesis, myoblast cell culture, qRT-PCR & MTT assay. | qRT-PCR, western blotting analysis, functional analysis, histology & liquid chromatography-mass spectrometry. | Cell culture, target prediction, qRT-PCR, western blotting analysis, reporter assay, glucose uptake assay, ceramide immunocytochemistry & histology. |
| miRs; targets (analysed) | 20 miRs changed with age with 6 of interest. | miR-21 (↑); FOXO3 or IL6R. | miR-181a (↓); p62/SQSTM1, parkin, DJ-1. | miR-34a (↑); CERK. |
| miR-532-3p (↓) as the top dysregulated miR; BAK1. | ||||
| Results | miR-532-3p associated with ageing induced apoptosis and inflammation. | miR-21 inhibition increased PTEN, IL6R & FOXO3. miR-21 may reduce regenerative potential. | miR-181a mediate the tuning of mitochondrial dynamics through several autophagy genes (upregulated in ageing). | Downregulated CERK caused dysregulation of the insulin signalling pathway. Reduced glucose uptake & insulin resistance. Antagonising miR ameliorated sensitivity. |
Up and down arrows indicate if miR was upregulated or downregulated respectively. BAK1, Bcl-2 homologous antagonist/killer 1; FOXO3, Forkhead box O3; IL6R, Interleukin 6 receptor; SQSTM1, Sequestosome-1; DJ-1, Parkinson disease protein 7; CERK, ceramide kinase.
Skeletal muscle atrophy is a key component of sarcopenia. Several studies have identified miRs involved in atrophy (Tables 1,2). Shao et al. (2022) [98] (Table 1) identified miR-690 as upregulated in ageing male mice extracellular vesicles (EVs) and human muscle. Initially detected in atrophy- and ageing-induced cell models, miR-690 was validated in human and murine samples. In vitro and in vivo experiments confirmed that miR-690 targets myocyte enhancer factor 2C (Mef2c), a transcription factor involved in skeletal muscle regeneration. Inhibition of miR-690 in aged mice reduced atrophy markers (muscle-specific ring finger protein-1 and atrogin-1), concurrently increasing muscle cross-sectional area (CSA) and maximal tetanic force, suggesting that miR-690 promotes skeletal muscle atrophy. Additionally, overexpression of miR-690 reduced Mef2c expression, impairing satellite cell myogenic potential and differentiation. These findings indicate a dual role of miR-690: promoting skeletal muscle atrophy and inhibiting differentiation. Similarly, Liang et al. (2023) [99] investigated miRs during ageing in male rat muscle (Table 2). While primarily assessing the effects of exercise on ageing, the study also explored skeletal muscle ageing mechanisms. Results showed that miR-7a-1-3p and miR-151-5p were significantly upregulated, targeting androgen receptor (Ar), insulin-like growth factor 1 (IGF-1), and brain-derived neurotrophic factor (BDNF), respectively. Conversely, miR-135a-5p, which targets focal adhesion kinase (FAK), was downregulated. Ar, IGF-1, and BDNF promote muscle cell proliferation, differentiation, and regeneration, while IGF-1 has an added role in inhibiting atrophy [98]. FAK plays a role in muscle morphology, metabolism, and insulin sensitivity, and its impaired signalling has been linked to age-related muscle regeneration decline [108]. However, Liang et al. (2023) [99] only validated four out of 764 identified miRs, potentially overlooking additional key targets.
Fukuoka et al. (2021) [102] (Table 2) analyzed atrophy-associated miRs in male mice serum using DNA microarrays. They identified miR-1, miR-26a, let-7e, miR-125b, miR-133a, miR-133b, miR-214, miR-199-3p, miR-351, and miR-6412 as significantly downregulated. In vitro experiments showed that serum from young mice induced stronger myogenic differentiation than aged serum, suggesting reduced myogenic capacity with ageing. Using luciferase assays, they confirmed that Lin28b and Suz12 were key targets. Lin28b, an RNA-binding protein, regulates miR processing and modulates miR-1 expression [102]. Suz12, a component of the polycomb repressive complex 2 (PRC2), represses chromatin and suppresses myogenic differentiation [109]. Therefore, Lin28b and Suz12 could be targeted as novel therapeutic targets for age-related muscle loss. Notably, administering a miR-199-3p mimic in aged mice mitigated CSA loss and increased differentiation markers in injured muscle, suggesting its role in hypertrophy and muscle regeneration. Additionally, the mimic downregulated atrogin-1, an E3 ligase crucial for protein degradation and muscle atrophy [110]. This result showcases the potential use of miR-199 mimics as a therapeutic tool for treating age-related muscle loss. Shin et al. (2020) [103] (Table 2) focused on miR-376c-3p, identified within a cluster of 42 miRs from the delta-like homolog 1 gene and the type III iodothyronine deiodinase (Dlk1-Dio3) region in murine muscle. Unlike Liang et al. (2023) [99], they confirmed seven miRs directly targeting atrogin-1 using luciferase assays. In vitro and in vivo findings showed that miR-376c-3p increased myotube diameter and had anti-apoptotic effects in transfected mice. Moreover, they demonstrated downregulation of miR-337-3p, miR-379-5p, miR-431-5p, miR-485-5p, miR-1197-3p, miR-409-5p, miR-411-3p, miR-134-3p, miR-1193-5p, miR-377-3p, and miR-127-3p in human muscle samples from individuals aged 25 to 50 years. Correlation analysis confirmed that these miRs negatively correlated with age. However, no significant link was drawn between miR-376c-3p and ageing, possibly due to stage-specific expression fluctuations. A more consistent age-stratified sample size could have clarified this discrepancy.
Collectively, these studies highlight a complex regulatory network of miRNAs involved in age-related skeletal muscle atrophy, with both pro-atrophic (e.g., miR-690, miR-7a-1-3p) and anti-atrophic (e.g., miR-199-3p, miR-376c-3p) roles identified. While several miRs have been functionally validated in vitro and in vivo, discrepancies remain in their reported expression patterns across models, time points, and sample types. For instance, differences in findings between tissue-derived and circulating miRs underscore the need for more tissue-specific investigations. Furthermore, the limited temporal and histological data hinder our understanding of the causal relationship between miR dysregulation and muscle degeneration. Nevertheless, identifying recurrent targets such as atrogin-1, IGF-1, and Mef2c suggests conserved molecular pathways through which miRs may modulate muscle atrophy, presenting promising avenues for therapeutic intervention.
Apoptosis, often accompanying atrophy, is another key mechanism associated with sarcopenia [111]. Gao et al. (2021) [104] (Table 2) investigated the role of apoptosis in age-related muscle loss and evaluated the timing of exercise interventions for its prevention. They analyzed miR expression in female rat skeletal muscle, identifying miR-486, miR-145, miR-133a, miR-19a, and miR-133c as downregulated, while miR-126, miR-23a, miR-182, miR-206, miR-199a, and miR-208b were upregulated. In addition to miR expression changes, they observed downregulation of anti-apoptotic, mitochondrial function, and autophagy markers alongside increased atrophy markers. This study specifically focused on miR-486, as it was significantly upregulated in their exercise intervention group. Using a luciferase assay, they confirmed phosphatase and tensin homolog (PTEN)—a negative regulator of the PI3K/Akt pathway—as a direct target of miR-486. In vitro, applying a miR-486 mimic reduced PTEN mRNA expression while increasing phosphorylated Akt levels. These molecular changes were accompanied by improved mitochondrial function, enhanced autophagy, and inhibition of atrophy and apoptosis, suggesting that miR-486 is critical in mediating these processes. In addition to regulating apoptosis, miR-486 is also extensively studied for its role in muscle regeneration in sarcopenia. This is through the direct action of miR-486 on insulin growth factor 1 to activate the PI3K/Akt pathway that promotes muscle growth [112]. Chronic exercise has been shown to positively impact muscle growth in sarcopenia by altering miR-486 expression [113]. Interestingly, the findings related to miR-23a, miR-206, and miR-199a contrast with those of Xhuti et al. (2023) [100], who reported downregulation of these miRs. One possible explanation for this discrepancy is the small sample size in Xhuti et al. (2023) [100] which may have led to chance findings. Additionally, differences in species and sex composition could contribute to the variation, as Xhuti et al. (2023) [100] analyzed both male and female human samples, while Gao et al. (2021) [104] examined only female rats.
The studies reviewed suggest that apoptosis is closely intertwined with miRNA regulation in the progression of sarcopenia, with several miRs playing dual roles in modulating both apoptotic and atrophic pathways. Notably, miR-486 emerges as a central regulator, promoting mitochondrial integrity and autophagy while inhibiting PTEN-mediated apoptosis. However, inconsistencies in the expression patterns of miR-23a, miR-206, and miR-199a between studies underscore the influence of experimental variables such as species, sex, and sample size. These differences highlight the need for standardized models and sex-specific analyses in future research. Despite these limitations, the convergence on pathways like PI3K/Akt signaling reinforces the importance of miRs as upstream modulators of apoptosis and points to their potential as therapeutic targets in mitigating sarcopenia-associated muscle degeneration.
Chen et al. (2020) [105] (Table 3) investigated the role of
inflammation and apoptosis in male sarcopenic patients. Their study confirmed the
involvement of inflammation in age-related muscle loss by demonstrating increased
levels of pro-inflammatory cytokines in sarcopenic individuals. miR expression
analysis identified miR-532-3p, miR-4256, miR-34a-3p, miR-663a, miR-922,
miR-126-5p, miR-328-3p, miR-22-3p, and miR-370 as downregulated, while miR-17-3p,
miR-873-5p, miR-1225, miR-192-3p, miR-2052, miR-590-3p, miR-30a-3p, miR-208,
miR-107, and miR-361-5p were upregulated. The three most upregulated and
downregulated miRs were validated in human samples, with miR-532-3p emerging as a
key candidate. Further analysis confirmed that Bcl-2 homologous antagonist/killer
1 (BAK1) was a direct target of miR-532-3p. Through in vitro analysis,
they identified transcription factors capable of binding to the promoter region
of miR-532p. Specifically, binding sites for nuclear factor
kappa-light-chain-enhancer of activated B cells (NF-
These studies highlight the role of specific miRNAs, such as miR-532-3p and
miR-21, in linking inflammation to muscle deterioration in sarcopenia. Both
appear to regulate key targets involved in apoptosis and inflammatory signaling,
including BAK1 and IL6R. Upstream regulation by factors like NF-
As discussed above, mitochondrial dysfunction plays a key role in sarcopenia [76]. Recent studies have investigated the role of miRs in mitochondrial dynamics and insulin signaling in age-related muscle loss [82, 107] (Table 3). Goljanek-Whysall et al. (2020) [82] examined mitochondrial function in male mice skeletal muscle and identified a significant downregulation of key mitochondrial proteins with ageing, including p62/sequestosome 1 (SQSTM1), Parkin, and Parkinson disease protein 7 (DJ-1). Interestingly, while histological analysis showed malformed mitochondria characterised by swelling, contractile and autophagy proteins were upregulated. This suggests that although autophagy proteins are present, the process of mitophagy and biogenesis is impaired. Using target prediction analysis, the miR-181 family was identified as a regulator of mitochondrial dynamics, with experimental validation showing downregulation of miR-181a in aged mice by directly inhibiting p62 and Park2 (parkin). In vitro experiments further confirmed that miR-181a overexpression increased mitophagy, with a concurrent increase in ATP production. However, cell viability remained unaffected, suggesting that miR-181a fine-tunes mitochondrial dynamics rather than directly influencing cell survival [82]. Functionally, miR-181 upregulation ameliorated mitochondrial morphology and improved muscle function in a mouse model, suggesting a minor but significant role in age-related muscle loss through modulation of mitochondrial homeostasis. Kukreti et al. (2020) [107] investigated the interplay between miR-34a and ceramide kinase (CERK) in male mice muscle. While sirtuin-1 (SIRT1) has been reported as a target of miR-34a, this study failed to demonstrate a significant downregulation of SIRT1in their model. Instead, using target prediction analysis and luciferase assays, CERK was identified as a direct target of miR-34a. Further analysis confirmed that upregulation of protein phosphatase 2A (PP2A) and c-Jun N-terminal kinases (JNK) which are associated with insulin signalling following CERK inhibition. Furthermore, miR-34a overexpression exhibited similar molecular changes, while miR-34a inhibition restored insulin signalling. These findings suggest that miR-34a plays a role in insulin resistance in ageing muscle by suppressing CERK expression and disrupting insulin signalling cascades.
These studies reveal a role for specific miRNAs, such as miR-181a and miR-34a, in regulating mitochondrial dynamics and insulin signaling in aged muscle. miR-181a appears to promote mitophagy and improve mitochondrial function, while miR-34a contributes to insulin resistance by targeting CERK and disrupting downstream signaling. Although the effects on overall cell viability and SIRT1 expression were limited, the findings highlight how miRs can fine-tune key metabolic pathways linked to sarcopenia. Further research is needed to clarify their broader physiological relevance and therapeutic potential.
Skeletal muscle regeneration is comprised of two distinct yet necessary
processes: satellite cell proliferation and differentiation. Satellite cells,
located beneath the basal lamina surrounding each myofiber, function as stem
cells for muscle growth and repair [114]. Under normal conditions, these cells
remain quiescent under active transcriptional control [115]. Upon receiving
extrinsic signals, satellite cells become activated, migrate to the injury site,
and begin proliferating; a subset of these cells subsequently commits to
differentiate into myofibers [62]. Among the proteins involved in this process,
serum response factor (SRF) is of note as it is the direct target of miR-133a, a
critical skeletal muscle developmental miR [116]. SRF is vital in mediating
satellite cell proliferation and fusion by regulating actin cytoskeleton genes
[117, 118]. Deletion of SRF reduces actin expression and disrupts the actin
cytoskeleton [119], leading to diminished satellite cell motility and fusion. SRF
modulates the actin cytoskeleton by activating the ras homolog gene family,
member A (RhoA), stimulating skeletal muscle
Despite the established role of miR-1, miR-133a and their respective targets in skeletal muscle biology, their precise contribution to age-related muscle loss remains unclear. Although differential expression of these miRs with ageing has been reported, findings are inconsistent. For example, Yu et al. (2022) [96] observed upregulation of miR-1 and miR-133a in the serum of aged rats, while Xhuti et al. (2023) [100] found that miR-1 was downregulated with no significant change in miR-133a expression in human aged skeletal muscles biopsies. Interestingly, Mytidou et al. [126] reported sustained increase in miR-1 and miR-133a until the early adolescence in mice, before reaching a plateau phase and showing no difference at least until 52 weeks of age. In contrast, Ling et al. [127] reported a decrease in miR-1 and miR-133 with maturity in goat skeletal muscle. These studies therefore suggest that the age associated changes in the expression pattern of miR-1 and miR-133 can be different in different species, warranting further research to elucidate their specific roles in the development of sarcopenia.
Considering the plasticity of skeletal muscle, it is key that the supporting microvasculature is healthy and adaptable. The body relies on angiogenesis to maintain a healthyted, the reduced angiogenic response to hypoxia in aged animals suggests dysregulation in angiogenesis [86, 87]. The losses to angiogenic potential with age could be linked to the miR-126 axis. Despite identifying a change in miR-126, the findings vary between studies. For example, Gao et al. (2021) [104] demonstrated an increase in expression, while Chen et al. (2020) [105] reported downregulation of miR-126-5p. Further, the changes to miR expression could translate to a protein level with SPRED-1 identified as downregulated in an aged human umbilical vein endothelial cell model [128].
The miR-126–SPRED-1 axis plays a central role in regulating angiogenesis by promoting endothelial cell proliferation and migration through the Raf-1/ERK and PI3K/Akt pathways. While its importance in vascular integrity is well established, especially in developmental models, its role in ageing skeletal muscle remains ambiguous. Conflicting reports on miR-126 expression, with some studies showing upregulation and others downregulation, highlight the need for further investigation. These discrepancies may reflect differences in tissue type, model systems, or stages of ageing. Although SPRED-1 downregulation has been observed in aged endothelial cells, a clear mechanistic link between miR-126 dysregulation and impaired angiogenesis in sarcopenia has yet to be established.
Sarcopenia is a complex and multifaceted condition characterised by the progressive loss of skeletal muscle mass, strength, and function with age. As global life expectancy continues to rise, sarcopenia poses a growing public health challenge, leading to significant increases in healthcare costs, disability, and reduced quality of life. Despite its prevalence and detrimental consequences, sarcopenia remains underdiagnosed due to limitations in current diagnostic methodologies, including a lack of standardised cut-off values and the reliance on physical performance tests that may not capture early-stage muscle deterioration. At the molecular level, sarcopenia arises from an interplay of multiple mechanisms, including oxidative stress, inflammation, mitochondrial dysfunction, satellite cell exhaustion, and impaired angiogenesis. While these pathways contribute to muscle degeneration, their interactions remain incompletely understood. Recent advances in molecular biology have highlighted the role of miRs as key regulators of these pathogenic processes. Emerging evidence suggests that miRs such as miR-1, miR-133a, and miR-126 play crucial roles in skeletal muscle homeostasis, influencing satellite cell proliferation, differentiation, and vascular integrity. However, their precise contributions to sarcopenia remain elusive, with conflicting reports on their differential expression in aged muscle tissue. Given the complexity of sarcopenia, a multifaceted approach to diagnosis and treatment is necessary. Non-pharmacological interventions such as resistance training and blood flow restriction therapy remain the most effective strategies for mitigating muscle loss. However, their efficacy is limited in older adults with diminished muscle regenerative potential. While pharmacological approaches such as selective androgen receptor modulators and myostatin inhibitors have shown promise, no approved drug therapies exist. Identifying miRs as potential biomarkers and therapeutic targets presents an exciting opportunity to enhance early diagnosis and develop novel treatments tailored to the molecular underpinnings of sarcopenia. However, care should be taken when using miRs as a biomarker due to the factor that miRs associated with sarcopenia may also be influenced by other mechanisms. Hence, future research should elucidate the precise molecular pathways linking miR dysregulation to age-related muscle loss. Large-scale, longitudinal studies are needed to validate the diagnostic utility of miRs and determine their suitability as therapeutic targets. Additionally, efforts should be directed towards integrating miR profiling with current diagnostic tools to enable early detection and intervention. The ultimate goal is to translate these discoveries into clinically effective strategies that preserve muscle function, enhance longevity, and improve the overall quality of life for ageing populations. By advancing our understanding of the molecular mechanisms driving sarcopenia and harnessing the potential of miRs, we can move closer to developing targeted therapies that address this growing health crisis. A concerted effort from researchers, clinicians, and policymakers are required to ensure that sarcopenia is recognised as a critical health priority and that innovative solutions are implemented to combat its far-reaching consequences.
JJK, LNV and RK were responsible for designing and completing the overall study. JJK wrote the first draft of the manuscript, LNV prepared the figures and edited the manuscript, RK designed the idea and wrote the manuscript along with JJK. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
All the figures are original, created using BioRender with appropriate license to publish.
This study is funded by the funding from the University of Otago.
Given his role as the Guest Editor and Editorial Board member, Rajesh Katare had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Elisa Belluzzi.
AI (ChatGPT and Grammarly) was used to check the spelling, grammar and accuracy of the language in the manuscript.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.