

 , Mikhail G. Kolonin 3,*
, Mikhail G. Kolonin 3,* , Dimitris Anastassiou 1,2,4,*
, Dimitris Anastassiou 1,2,4,*1 Department of Systems Biology, Columbia University, New York, NY 10032, USA
2 Department of Electrical Engineering, Columbia University, New York, NY 10027, USA
3 Institute of Molecular Medicine, University of Texas Health Science Center, Houston, TX 77030, USA
4 Herbert Irving Comprehensive Cancer Center, Columbia University, New York, NY 10032, USA
Abstract
Adipose stromal cells (ASCs) are perivascular mesenchymal progenitors of adipose tissue. In cancer patients, ASCs can mobilize and migrate to the tumor, where they subsequently play an important role in cancer progression. This biological process involves the conversion of recruited ASCs into cancer-associated fibroblasts (CAFs). ASC-derived CAFs influence the tumor microenvironment through extracellular matrix remodeling, vascularization, and immunomodulation. These and other processes mediated by secreted paracrine factors also affect gene expression in carcinoma cells to promote the epithelial-mesenchymal transition (EMT), metabolic adaptation, survival, and invasiveness of cancer cells. ASC-derived CAFs can enhance tumor aggressiveness, accounting in part for the link between obesity and mortality observed in many cancer types that are surrounded by adipose tissue. In this review, we highlight recent findings on the characteristics and functions of ASCs in cancer and discuss their potential as therapeutic targets.
Keywords
- adipose stromal cells
- adipose-derived fibroblasts
- cancer-associated fibroblasts
- tumor microenvironment
The cells in adipose tissue can be separated by enzymatic digestion into the stromal vascular fraction (SVF) and the buoyant fraction [1, 2]. The SVF contains a heterogeneous population of cells that includes adipose stromal cells (ASCs), immune cells and endothelial cells, while the buoyant fraction contains adipocytes and some pre-adipocytes [2]. The immunophenotypes of individual ASC sub-populations have been studied previously [1, 3], but are still not completely understood.
Single-cell RNA sequencing (scRNA-seq) has helped to delineate populations
within white adipose tissue (WAT), including ASCs and their subpopulations [4, 5].
ASCs reside in the perivascular niche and support endothelial function, as well
as differentiating into adipocytes or fibroblasts depending on the cues they
receive [5, 6]. A subpopulation of mesenchymal stromal cells (MSCs) is comprised
of preadipocytes with high adipogenic potential and expression of Platelet
Derived Growth Factor Receptor Alpha (PDGFR
One particular ASC subpopulation has a characteristic signature containing several co-expressed genes, such as Apolipoprotein D (APOD), Decorin (DCN), Lumican (LUM), Complement Factor D (CFD), C-X-C Motif Chemokine Ligand 14 (CXCL14), and Prostaglandin D2 Synthase (PTGDS). This signature has been identified in several studies, indicating consistency across different tissues. For example, it was found among adipocyte progenitors in subcutaneous WAT from the dataset used in study [10], as shown in cluster visceral adipose tissue-derived progenitor cells 4 (VP4) of Supplementary Table 20. Moreover, the abundance of this population in the SVF was demonstrated by analyzing the same dataset in Supplementary Fig. 1 of the paper by Cai et al. [11].
Interestingly, a cell population with the same characteristic signature was found among fibro-adipogenic progenitors (FAPs) in skeletal muscle. For example, in the study by Rubenstein et al. [12] they were reported as LUM+ FAPs in Supplementary Table 1, and included the genes APOD, PTGDS, LUM, DCN, Matrix Gla Protein (MGP), CFD, CXCL14, Dermatopontin (DPT), and Serpin Family F Member 1 (SERPINF1). A similar population was found in Supplementary Fig. 3a of the study by Fitzgerald et al. [13], referred to as a Membrane Metalloendopeptidase (MME)+ FAP population, and which expressed the genes APOD, PTGDS, CXCL14, MGP and Secreted Frizzled Related Protein 2 (SFRP2). These cells can differentiate into adipocytes. Cai et al. [11] referred to this subpopulation of ASCs as “the ASC/FAP population”, with its progenitors having both adipogenic and fibroblastic differentiation potential. Consistent with these findings, Gao et al. [14] identified a fibroblast progenitor subtype in a multi-tissue fibroblast atlas. This was characterized by the expression of adipogenic genes such as APOD, and cluster “c03” shown in Supplementary Table 2 of the report by Gao et al. [14], which includes all ASC/FAP genes. Furthermore, the presence of this progenitor population in both adipose tissue and skeletal muscle is consistent with the finding that adipose tissue is a source of FAP-like cells recruited to the skeletal muscle during remodeling [15].
ASCs can be recruited into tumors to enhance the supportive properties of the tumor microenvironment (TME), as indicated by resistance to anti-cancer therapy in mouse models [16, 17, 18, 19]. The recruitment and infiltration of ASCs into tumors is driven by specific molecular and signaling mechanisms that create a favorable environment for cancer progression. One such mechanism involves chemokine signaling, whereby chemokines such as C-X-C Motif Chemokine Ligand 1 (CXCL1) and C-X-C Motif Chemokine Ligand 8 (CXCL8) bind to receptors such as C-X-C Motif Chemokine Receptor 1 (CXCR1) and C-X-C Motif Chemokine Receptor 2 (CXCR2) on ASCs and guide their migration towards the tumor [17]. Mouse models have demonstrated the importance of the CXCL1 chemokine gradient for obesity-dependent tumor ASC recruitment that promotes the progression and metastatic potential of prostate cancer [17, 20].
Once recruited, ASCs infiltrate the tumor and contribute to the stromal compartment by secreting agents that stimulate vascularization, immune modulation, and tissue remodeling [21]. ASC mobilization from WAT is initiated by molecular triggers such as the interaction between adipocyte-secreted proteins and integrins on the surface of ASCs [22]. Signaling pathways such as IL-22 also activate ASCs, thereby increasing their recruitment and integration into the tumor stroma where they produce ECM components and promote cancer cell survival [22, 23]. These processes highlight the important role of chemokine-mediated recruitment and integrin signaling in ASC mobilization. In summary, ASCs are key contributors to the tumor-supportive stroma, and as such represent a potential target for therapeutic intervention [20].
We have previously studied the role of ASC-derived cancer-associated fibroblasts
(CAFs) in epithelial-mesenchymal transition (EMT) induction and cancer
aggressiveness [24, 25, 26]. C-X-C Motif Chemokine Ligand 12 (CXCL12 (alias,
SDF1)) is a paracrine chemokine secreted by ASCs and adipose-derived CAFs and
which signals via CXCR4 and CXCR7 to promote obesity-associated cancer
progression via EMT. The SVF derived from WAT of obese mice shows significantly
higher CXCL12 expression than the SVF from lean mice. Signaling by CXCL12 results
in the activation of Signal Transducer and Activator of Transcription 3 (STAT3),
Nuclear Factor Kappa-light-chain-enhancer of activated B cells (NF-
Single-cell analysis of biopsies from many cancer types has identified computationally-derived clusters that include cells with the same gene signature as the APOD+DCN+LUM+ ASC/FAP population mentioned previously. This indicates they are likely to be recruited ASCs. For example, Chen et al. [28] identified a fibroblastic population in esophageal cancer that was characterized by gene expression for CFD, APOD, Gelsolin (GSN), and Peptidase Inhibitor 16 (PI16). Zhang et al. [29] clustered stromal cells into nine subtypes, including one labeled as “normal activated fibroblasts” that was characterized by expression of APOD, CFD, DPT, CXCL14, PTGDS, and MGP. Other examples of the signature in breast cancer are found in Supplementary Table 9 (inflammatory CAF (iCAF) minor) of the study by Wu et al. [30], and Supplementary Table 3 (iCAF) of the study by Cords et al. [31], both of which feature the ASC markers APOD and CFD at the top.
The presence of recruited and infiltrated ASC/FAPs in cancer biopsies is in most cases thought to represent inflammatory CAFs, as described below. This is because computationally-derived clusters are typically labeled with one of several pre-assigned cell subtypes. To date, the ASC/FAP population has not been widely recognized as a stromal population in the TME, and as a consequence this population is usually assumed to be a type of CAF.
The TME reflects the progression to aggressive disease [32, 33]. CAFs are a
heterogeneous and plastic population of non-malignant mesenchymal cells within
this microenvironment that modulate tumor vascularization and growth, EMT,
chemotherapy resistance, ECM remodeling, metastatic dissemination, and
immunosuppression [34]. They are recruited early in cancer development and evolve
as the disease progresses [35, 36]. Several major CAF subtypes are generally
recognized, with the main ones being myofibroblastic CAFs (myCAFs) and
inflammatory CAFs (iCAFs). MyCAFs exhibit a matrix-producing, contractile
phenotype with high alpha-smooth muscle actin (
Tumor infiltrating ASCs in their original, undifferentiated progenitor state should not be classified as one of the three widely recognized CAF subtypes described above. However, they are often included within non-homogeneous, computationally-derived clusters. Since part of the cell population in such clusters expresses inflammatory markers, this has been thought to represent iCAFs in their original state, meaning that even genes such as APOD and CFD have been mischaracterized as iCAF markers. In reality, iCAFs are characterized by the signature previously reported by Elyada et al. [39], which includes the expression of IL6, C-C Motif Chemokine Ligand 2 (CCL2), and Hyaluronan Synthase 1 (HAS1). For example, “cluster 1” in Fig. 5D of the study by Dominguez et al. [41] is marked by the expression of CFD, PTGDS, and C7, all of which are expressed in ASCs. This cluster is distinct from “cluster 2” marked by IL6, HAS1, and CCL2, and which corresponds to the true iCAF population. Similarly, Chen et al. [28] identified iCAFs in esophageal cancer as a population that expressed inflammatory genes such as CXCL1, CXCL8, and Interleukin 24 (IL24). Ho et al. [42] found both iCAF and “adipose-like CAF” in head and neck squamous cell carcinoma (HNSCC), with iCAFs expressing IL6 and CXCL1, while adipose-like CAFs expressed APOD and CFD, and clearly included recruited ASCs. The distinction between ASCs and iCAFs is further supported by the cross-tissue human fibroblast atlas. This identifies inflammatory fibroblasts as expressing IL6, CXCL1 and CXCL2, whereas APOD and PI16 expression is found in progenitor cells that are distinct from IL6-expressing cells [14]. Therefore, recruited ASCs in their original state should not be confused with IL6+ iCAFs, although some may independently transition to that state.
There are many examples in different cancer types where recruited ASCs with a consistent gene signature have been misclassified as iCAFs [43, 44, 45, 46, 47, 48, 49, 50, 51]. For example, transcriptomic analysis shown in Extended Data Fig. 7 of the study by Cui Zhou et al. [43] identified a stromal subpopulation in pancreatic ductal adenocarcinoma (PDAC) with a signature that included the marker genes APOD, PTGDS, CXCL14, CFD, DCN, and LUM. The study also identified 12 genes (APOD, C3, PTGDS, C7, IGFBP3, SFRP4, CXCL14, CFD, SFRP2, DCN, FBLN1, and LUM) that formed a “chemoresistance signature”. Chemoresistant samples of pancreatic cancer showed a three-fold enrichment of this signature. Similarly, Supplementary Table 6-1 in the study by Wang et al. [49] identified the iCAF cluster C0, with high expression levels of APOD, PTGDS, MGP, DCN, LUM, SERPINF1, CFD, CXCL14, and DPT. Additionally, Zhao et al. [44] reported that iCAFs in obesity-associated breast cancer showed upregulation of genes including CFD, APOD, and CXCL14. Chen et al. [45] found a cluster labeled iCAF in bladder cancer that expressed genes such as APOD, DCN, LUM, CFD, CXCL14, PTGDS, MGP, SERPINF1, and DPT. Pu et al. [46] identified a cell population in thyroid carcinoma, labeled iCAF, that was characterized by high expression of ASC genes including APOD, CFD, DCN, and CXCL14. However, these authors noted a lack of key iCAF markers such as IL6 and IL8 (Supplementary Fig. 9e in study [46]). Finally, Thorlacius-Ussing et al. [47] suggested the existence of two separate populations of iCAFs in PDAC, with one being IL6 positive and the other IL6 negative. In fact, the IL6-negative cluster contained mostly the ASC population (Supplementary Fig. 4 in study [47]) without inflammatory gene markers.
Recruited ASCs have been found to undergo fibroblastic differentiation as part of a multi-faceted, cancer invasiveness-related biological mechanism [11] (Fig. 1). By analyzing datasets from multiple cancer types, including a particularly rich dataset from pancreatic cancer [52], Zhu et al. [53] identified a cell transition starting from APOD-expressing ASCs. The endpoint of this transition is an aggressive type of CAF with prominent expression of Collagen Type XI Alpha 1 Chain (COL11A1), referred to as “CAFs in aggressive tumors (aCAFs)”. These also have a well-defined and characteristic gene expression signature that includes THBS2, INHBA, POSTN, COL10A1, and MMP11. First identified by Kim et al. [54], the aCAF gene signature was strongly associated with cancer invasiveness and poor prognosis. It was also found to be associated with tumor stage, consistent with the presence of adjacent adipose tissue. For example, in ovarian cancer the aCAF signature only occurs after the tumor cells have reached the omentum in stage III disease, whereas in breast cancer it occurs in stage I already. Following analysis of six cancer types, Ma et al. [51] reported a potential transition from an APOD+CFD+MGP+CXCL14+ population labeled as iCAF (Fig. 2b in study [51]), to a COL11A1+COL10A1+MMP11+POSTN+ population labeled mCAF (Fig. 2i in study [51]). In pleomorphic sarcoma, Lu et al. [55] identified APOD as a stem cell-related gene involved in cell transition, with decreased APOD expression accompanied by increased COL11A1 expression.
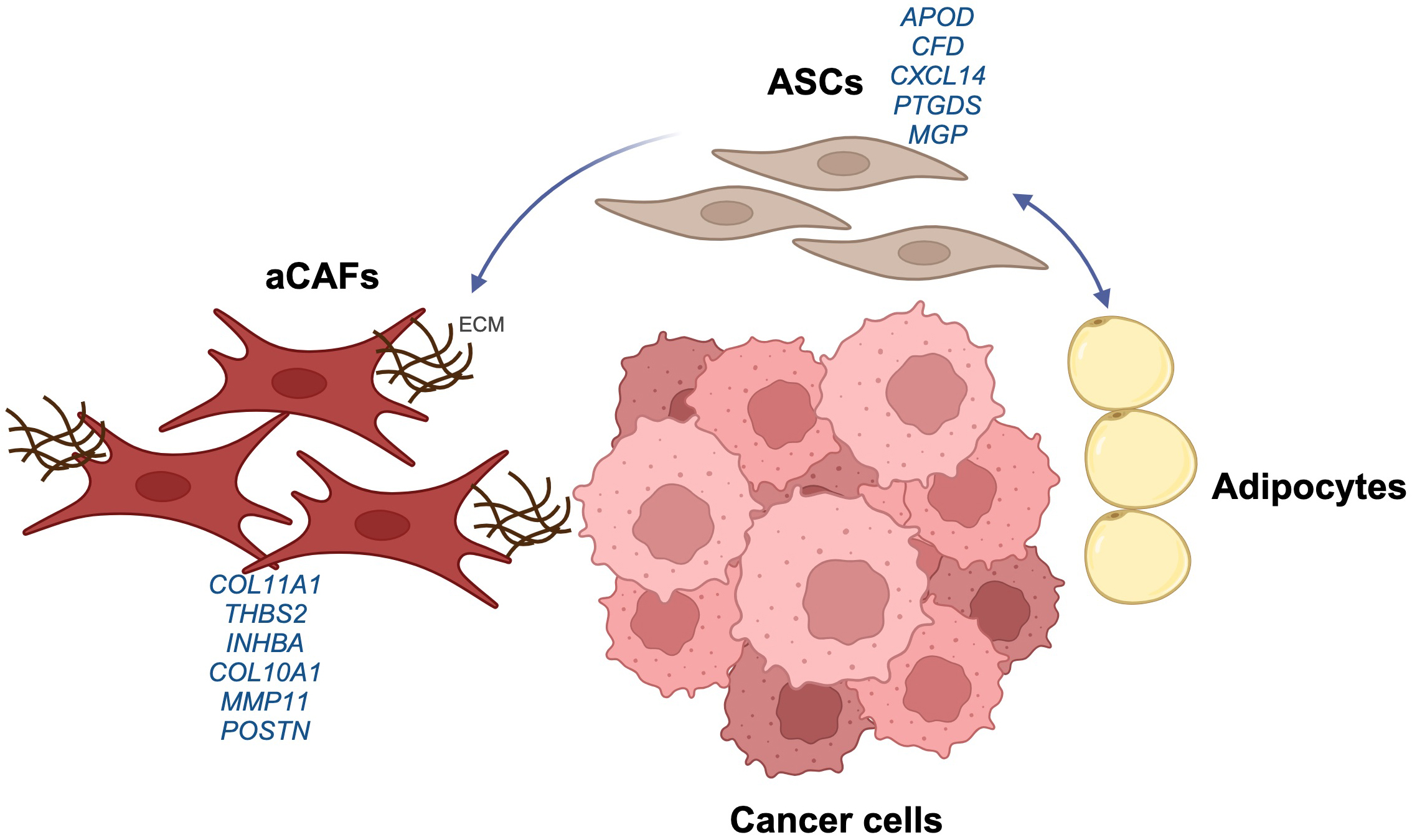 Fig. 1.
Fig. 1.
Adipose stromal cells (ASCs) as a source of aggressive cancer-associated fibroblasts (aCAFs) in tumors. The diagram summarizes the transition of ASCs to aCAFs within the tumor microenvironment, showing some of the associated gene markers. The ASCs have both adipogenic and fibroblastic differentiation potential, and differentiate into aCAFs by interacting with cancer cells. Created with https://www.biorender.com. ECM, extracellular matrix; APOD, Apolipoprotein D; CFD, Complement Factor D; CXCL14, C-X-C Motif Chemokine Ligand 14; PTGDS, Prostaglandin D2 Synthase; MGP, Matrix Gla Protein; COL11A1, Collagen Type XI Alpha 1 Chain; THBS2, Thrombospondin 2; INHBA, Inhibin Subunit Beta A; COL10A1, Collagen Type X Alpha 1 Chain; MMP11, Matrix Metallopeptidase 11; POSTN, Periostin.
Many other studies have also suggested that ASCs serve as precursors to CAFs
within tumors, with some demonstrating their ability to transition into
myofibroblast-like cells that contribute to tumor progression. For example, Kidd
et al. [56] showed that adipose-derived cells could differentiate into
Strong et al. [59] proposed that obesity alters adipose-derived
stromal/stem cells, making them more likely to convert into CAFs in the presence
of tumor-derived factors. This in turn promotes the proliferation, invasiveness,
and expression of pro-tumorigenic factors by breast cancer cells [59]. Okumura
et al. [60] demonstrated that ASCs contribute to pancreatic tumor
progression by being recruited to extra-pancreatic invasion sites, where they
produce dense collagen matrices that enhance tumor growth. In ovarian cancer,
Tang et al. [61] suggested that adipose-derived mesenchymal stem cells
(ADSCs) from the omentum can differentiate into CAF-like cells via the
TGF-
Importantly, both in vitro and in vivo experiments conducted by Miyazaki et al. [62] confirmed that adipose-derived stromal cells have the ability to differentiate into CAFs. Specifically, direct co-culture of ASCs with tumor cells resulted in the expression of genes such as COL10A1, COMP, and INHBA [62]. Using mouse xenografts derived from human tumor cell lines, the same authors subsequently demonstrated the ability of ASCs to differentiate into CAFs with strongly upregulated expression of COL11A1 [63].
Pan-cancer single-cell transcriptomic analyses further support the above conclusions, as evidenced by the fact that clusters which include genes such as APOD and CFD are typically located adjacent to clusters that include COL11A1. For example, Fig. 4 in the study by Hornburg et al. [64] shows two clusters in ovarian cancer, with each being defined by characteristic genes for the two populations (APOD, CFD, MGP, DPT, CXCL14 and COL11A1 for the first cluster; MMP11, POSTN, and INHBA for the second cluster). Similarly, Fig. 2 in the study by Wang et al. [49] on pancreatic cancer shows adjacent clusters C0 and C3, characterized by marker genes including APOD, PTGDS, MGP, DCN, and LUM for C0, and MMP11, POSTN, COL10A1, COL11A1, INHBA, THBS2, SULF1 for C3. Other workers have also demonstrated adjacent clusters characterized by ASC gene markers and CAFs expressing genes such as COL11A1, COL10A1 and MMP11. In PDAC, these include cluster 1 and clusters 3, 4 and 6 [47], “iCAF” and “myCAF” in study [48], and C1 and C0 in study [41]. In breast cancer, similar marker profiles were observed for the adjacent CFD-expressing “iCAF1” and COL11A1-expressing “iCAF2” clusters [44]. In gastric cancer, Zhao et al. [65] observed a similar pattern in the “poor prognosis” CAF_0 cluster, wherein POSTN appears alongside APOD, CFD, and CXCL14 with expansion of the cluster, consistent with a transition.
Gao et al. [14] analyzed 517 samples spanning 11 different tissues. Their RNA velocity results revealed a transition from cluster c03 expressing the ASC/FAP signature, to cluster c16 with SFRP4 as the top differentially expressed gene, identified as marking the start of the transition in another study [53]. The final transition to c04 included the top marker genes COL11A1, THBS2, INHBA, COL10A1, MMP11, POSTN, and SPARC. It remains to be determined whether any of these genes play a role in the pro-carcinogenic properties of ASC-derived CAFs, and whether their targeting could have therapeutic value.
CAFs have often been considered to represent a potential therapeutic target
[36, 66, 67, 68, 69]. According to some studies, selective depletion of certain CAF
subtypes can impede tumor growth and improve treatment outcomes. For example,
conditional depletion of FAP+ stromal cells in transgenic mice led to slower PDAC
growth [70]. Similarly, depletion of Leucine Rich Repeat Containing 15 (LRRC15)+
CAFs in pancreatic tumors reduced their overall CAF content, decreased tumor
burden, and boosted the responsiveness to anti-programmed cell death ligand-1
(anti-PD-L1) immunotherapy [71]. Collectively, these findings indicate there may
be therapeutic potential in modulating the aCAF population to improve cancer
treatment. However, the general approach of targeting CAFs remains a subject of
ongoing debate. Depletion of
It remains to be determined whether selective inactivation of ASC-derived CAFs
can provide a unique therapeutic benefit. A few studies have investigated the
effects of ablating CAFs that express ASC markers [25, 73, 74]. Our group used
the hunter-killer peptide D-CAN, which specifically targets ASCs and their
non-glycanated decorin- and PDGFR
The recruitment of ASCs in cancer and their differentiation into CAFs is associated with invasiveness, poor prognosis, and resistance to therapy. Although the identification of viable therapeutic targets that play driver roles in the clinical setting show considerable promise, several challenges remain. Accumulating evidence suggests that blocking ASC-derived CAFs can help to suppress tumor growth and metastatic dissemination, as well as overcoming therapy resistance. The identification of specific markers expressed by adipose-derived CAFs, together with a better understanding of their function, may lead to targeted treatments against these cells that could transform cancer management with adjuvant therapies.
MGK and DA designed the study; LC and MGK contributed to the literature review; LC drew the figure; LC, MGK, and DA wrote, reviewed, and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
M.G.K is supported by the Levy-Longenbaugh Fund and the Bovay Foundation.
The authors declare no conflict of interest. Given his role as the Guest Editor, Mikhail G. Kolonin had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Sung Eun Kim.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.