


 , Si-Yi Wei 1, Xiao-Tong Fu 1, Xi Cheng 2, Xian-Hua Lin 1,3,*
, Si-Yi Wei 1, Xiao-Tong Fu 1, Xi Cheng 2, Xian-Hua Lin 1,3,*
1 Obstetrics and Gynecology Hospital, Institute of Reproduction and Development, Fudan University, 200011 Shanghai, China
2 Department of Obstetrics and Gynecology, Chinese Academy of Medical Sciences and Peking Union Medical College, 100730 Beijing, China
3 Shenzhen Maternity and Child Healthcare Hospital, Southern Medical University, 518028 Shenzhen, Guangdong, China
Abstract
Preeclampsia (PE) is a serious complication of pregnancy characterized by chronic inflammation and immune dysregulation, which significantly increases the risk of neurodevelopmental disorders in offspring, including the autism spectrum disorder (ASD). This review investigated the potential mechanisms linking PE to ASD, with a particular focus on the role of microglial abnormalities. Epidemiological studies have revealed that prenatal exposure to PE raised the risk of ASD, with affected offspring showing increased odds ratios. Microglia, the prime resident immune cells of the central nervous system (CNS), are critical for normal neurodevelopment, influencing processes such as neural stem cell (NSC) proliferation, synaptic pruning, and normal function of the neural circuit. Early-onset preeclampsia (EOPE) and late-onset preeclampsia (LOPE) may have an impact on the microglia abnormality and ASD through not exactly same pathway. Postmortem studies of ASD have further revealed increased microglial density, altered microglial morphology, and upregulated inflammatory markers in key brain regions, including the hippocampus and prefrontal cortex. Understanding the complex processes and potential mechanisms between EOPE, LOPE, microglial abnormalities, and ASD pathogenesis may highlight the importance of early screening and intervention for children born to mothers with PE. Targeting microglia-mediated pathways may offer novel therapeutic strategies to reduce the risk of ASD in these vulnerable populations.
Keywords
- pre-eclampsia
- autism spectrum disorder
- microglia
- prenatal exposure delayed effects
- neurodevelopmental disorders
Intrauterine development marks the earliest stage of life, where normal progression is essential for individual growth and long-term health. The relationship between fetal development and long-term health, known as the Developmental Origins of Health and Disease (DoHAD), has become a critical topic in non-communicable disease research. Investigating the impact of adverse exposures during early development, particularly intrauterine environmental influences on offspring’s near and long-term health outcomes, is a central focus in modern medicine.
Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder characterized by social deficits and repetitive behaviors, and whose incidence is steadily increasing [1]. In the United States, approximately 2.3% of children aged 8 years and 2.2% of adults are affected, and these numbers are rising [2]. ASD is more than four times as prevalent in boys as in girls and individuals with ASD are more likely to experience depression, anxiety, sleep disturbances, epilepsy, and severe intellectual disabilities, as well as language impairments and psychiatric conditions. These challenges impose significant burdens on families, healthcare systems, and society [3]. Despite extensive research, the etiology of ASD remains unresolved, complicating efforts in treatment and prevention. While genetic mutations are considered primary contributors to ASD, growing evidence indicates that non-genetic factors account for approximately 17–50% of cases [4]. Among these, adverse intrauterine environments during pregnancy play a critical role in the pathogenesis of ASD [5].
Maternal conditions such as obesity, chronic inflammation, and preeclampsia (PE) may alter fetal neurodevelopment through acute and chronic immune stress, increasing the risk of ASD [6, 7]. Hypertensive disorders of pregnancy (HDP), particularly PE, are among the most common pregnancy complications, affecting maternal and fetal outcomes globally [8]. PE, the most severe form of HDP, typically arises after 20 weeks of gestation and is a major cause of maternal and perinatal mortality. Its pathology involves impaired trophoblast invasion, endothelial activation, and maternal immune overactivation [9].
Epidemiological studies have increasingly demonstrated that PE exerts long-lasting effects on offspring neurodevelopment, significantly elevating the risk of ASD and other neurodevelopmental disorders (NDDs) independent of preterm birth or other obstetric complications [10, 11, 12]. Microglia, the resident immune cells of the central nervous system (CNS), account for 5–12% of CNS cells and mediate neuroinflammation. Substantial evidence indicates that microglia dysregulation contributed to ASD pathology by impairing neurogenesis, synaptic pruning, and synaptic plasticity, leading to neuronal deficits, structural brain changes, and behavioral abnormalities [13].
This review aims to elucidate the evidence linking PE-induced microglia dysregulation to abnormal neurogenesis and synaptic plasticity, which may underlie the development of ASD in offspring.
PE, the most severe form of HDP, has highly variable clinical manifestations and
is a major cause of maternal and perinatal mortality [9]. According to the
International Society for the Study of Hypertension in Pregnancy (ISSHP), PE is
defined as new onset hypertension (systolic
EOPE is related to more severe neonatal mortality and is less commonly compared with LOPE [17], which is considered to be typically associated with placental dysfunction, preterm birth and restriction of fetal growth [15]. LOPE comprises about 80% to 90% of PE cases, and is associated with maternal cardiometabolic syndrome or diseases and is mainly the result of cardiovascular susceptibility and maternal endothelial dysfunction [18, 19]. These two phenotypes have different etiologies and time lines, but both can trigger maternal inflammatory and immune responses of varying degrees that leads to adverse maternal and fetal outcomes [18, 20]. In EOPE, this is triggered by insufficient perfusion of the placenta which results in a pro-inflammatory placental state. LOPE is more associated with systemic maternal inflammation due abnormal vessel response due to endothelial cell activation from the stress of pregnancy such as gestational diabetes mellitus and obesity, and inflammatory conditions found in pregnant females [20, 21, 22]. Although EOPE is more associated with poor placental function, LOPE can also lead to uteroplacental malperfusion as a result of compression of placental terminal villi when the placenta outgrows uterine capacity [18, 23].
NDDs are a group of diverse conditions characterized by disruptions in brain development. They present with a wide range of clinical symptoms and severity and are linked to varying levels of cognitive, adaptive functioning, and disability [24]. The etiology of NDDs is multifactorial, and both genetic and environmental factors play important roles in guiding and shaping the dynamically developing process and increasingly functional complexity of brain, as well as the development of NDDs [25]. ASD is recognized as one of canonical NDDs that are especially sensitive to early disturbance to the CNS [25]. According to family and population studies, ASD heritability is about 50% that of mental illness [26], while the genetic cause of the presence of ASD is clear in approximately only 20% of cases [27]. There were several proteins, including Phosphatase and tensin homolog (PTEN), Fragile X mental retardation protein (FMRP), Neurexins (NRXNs), the chromodomain helicase DNA-binding protein 8 (CHD8), SH3 and multiple ankyrin repeat domains protein 3 (SHANK3), have been reported related to ASD. PTEN is a tumor suppressor gene that regulate neuron positioning, dendritic development and synapse formation, and PTEN deficiency was associated with communication disorders, repetitive behaviors [28]. CHD8 gene has been identified as a high-risk gene for ASD, which regulated the transcription of ASD risk factors in human neural progenitor cells, as well as neuronal differentiation, synaptic development, cell adhesion [29]. In addition, a lot of epidemiological researches revealed that early environmental factors, including toxicants, infections, medications, and maternal complications have been familiar to contribute to autism susceptibility [3]. Valproic acid (VAP) is a short-chain fatty acid. Clinical studies, along with other animal researches have reported an increased risk of neural tube defects, developmental delays, cognitive impairment and autism when VPA is taken during pregnancy [30, 31]. Other maternal complications such as PE are risks factors of ASD as well [6].
Several epidemiological studies had found that maternal PE increases the risk of
offspring neurodevelopmental and psychiatric disorders [4, 11, 32, 33, 34], as shown
in Table 1 (Ref. [4, 11, 33, 34, 35, 36, 37, 38]). Systematic review and meta-analysis
revealed a 1.5-fold increased risk of ASD exposure to maternal PE [4]. A
population-based case-control study of 517 children with ASD and 350 controls
with typical development (TD) from 20 California counties, found that children
with ASD were twice as likely to have been exposed to PE in utero compared to
controls (adjusted OR, 2.36; 95% CI, 1.18–4.68) [34]. Another study has also
conducted cohort analyses to assess the risk of developing ASD in relation to PE
exposure, and found approximately 1- to 3-fold increased risk [35]. However, the
specific mechanisms as to how preeclampsia results in an increased risk of autism
are unclear. Animal models mechanistically bridge PE and ASD phenotypes. Liu
et al. [39] established a preeclampsia mouse model by
Nω-nitro-L-arginine methyl ester (L-NAME), and found there were
increased anxiety repetitive behavior and defects of social behavior of adult
offspring, as well as neuro morphologic abnormalities and changed expression
genes related to ASD. High level of serum TNF-
| Reference | Type of study | Participants number | Exposure/case | Risk of ASD |
| Wang et al. 2021 [38] | Population-based cohort study | 52,171 live-born singletons | Preeclampsia | Adjusted HR, 1.36 (95% CI, 1.30–1.42) |
| Sun et al. 2020 [11] | Prospective population-based cohort study | 980,560 children born at term | Preeclampsia (Subtype unexplored) | Adjusted OR, 1.29 (95% CI, 1.08–1.54) |
| Chien et al. 2019 [37] | Observational study | 323 probands with ASD, and 1504 of TDs | Preeclampsia | Significant factor to predict socio-communication deficits, stereotyped behaviors, and total scores of SRS; |
| No risk reported. | ||||
| Maher et al. 2018 [4] | Meta-analysis | 11 studies (including 777,518 participants) | Hypertensive disorders of pregnancy | Pooled adjusted OR of 1.35 (95% CI, 1.11–1.64) |
| 6 studies (including 378, 991 participants) | preeclampsia | Pooled adjusted OR of 1.50 (95% CI, 1.26–1.78), test for subgroup differences, p = 0.33 | ||
| Dachew et al. 2018 [33] | Meta-analysis | 10 studies | Preeclampsia | Pooled RR of 1.32 (95% CI, 1.20–1.45) |
| Walker et al. 2015 [34] | Population-based case-control | Children with ASD (517) vs Controls with TD (350) | Children with ASD | Adjusted OR of 2.36 (95% CI, 1.18–4.68) |
| Mann et al. 2010 [35] | Retrospective cohort study | 87,677 births from 1996 through 2002 | Preeclampsia or eclampsia | Adjusted OR, 1.69, p = 0.0005 |
| Wallace et al. 2008 [36] | Retrospective study | 228 families | Preeclampsia | Significantly associated with the ADI-R communication domain (t = 2.33, p = 0.021); |
| No risk reported. |
ASD, autism spectrum disorder; TD, typical development; HR, hazard ratio; CI, confidence interval; SRS, social responsiveness scale; ADI-R, autism diagnostic interview-revised; OR, ddds ratio; RR, relative risk.
In addition, associations between preeclampsia and specific symptoms are also established both in epidemiological studies and animal models. Wallace et al. [36] observed a significant association between PE and higher communication scores using the Autism Diagnostic Interview-Revised (ADI-R) tool, which indicated a severe communication deficit in these children. However, another study found that probands with at least one prenatal factor among six, which include the PE, were associated with the severity of autistic symptoms across the assessments by the ADI-R, Social Communication Questionnaire (SCQ), and Social Responsiveness Scale (SRS), and that PE was one of the most significant factors to predict socio-communication deficits, stereotyped behaviors, and total scores of SRS [37]. The reasons for these inconsistent outcomes maybe related to the different symptoms assessing by ASD and the disparity in sample sizes. As for the animal model exploring the PE exposure and specific symptom of ASD, PE model by chronically infused with arginine vasopressin (AVP) found that PE-exposed adult males exhibited increased anxiety-like behavior and social approach while adult females exhibited impaired procedural learning and decreased excitatory synapse density in hippocampal dentate gyrus (DG), CA1, and CA3, which may indicating the microglia, which are responsible for synaptic pruning, are involved in the pathogenesis [40].
In addition to behavioral, psychiatric, and neurological risks observed in children born to mothers with preeclampsia, neuroimaging has also revealed structural changes in the brain. Offspring of PE exposure showed a significant association with cerebral palsy (adjusted OR, 1.30; 95% CI, 0.94–1.80) [11]. In a study of 7–10-year-old children from both preeclamptic and non-preeclamptic pregnancies, several brain regions, including the cerebellum, temporal lobe, brainstem, and bilateral amygdalae, were found to be enlarged relative to total intracranial volume in children exposed to preeclampsia. This was observed despite no significant differences in total intracranial volume or head circumference between the two groups [41].
These studies indicated that PE increased the risk of ASD in offspring, underscoring the importance of early ASD screening for children born to mothers with PE. Further exploration of the molecular mechanisms linking environmental factors, such as PE, to ASD is critical for future research.
Microglia, the resident immune cells of the CNS, play a dual role in coordinating neuroprotection, immune responses, and neuronal repair [42], comprising 5–12% of CNS cells [43]. Microglia are a special type of cells in the brain derived from a primitive yolk sac progenitors, which differ from other types of neural cells originating from the ectoderm [44]. Microglia arise early in the CNS and play a critical role in the development of the CNS. In the embryonic day 9.5 (E9.5), microglia appear in the neuroepithelium [45], and then rapidly proliferate to colonize the CNS, maturing into ramified microglia between E10.5 and birth [46]. Studies have found that microglia in the period of neurodevelopment, including the perinatal and early postnatal stage, have distinctly different morphologies and gene expression compared to the adult brain [47, 48, 49]. Schwarz et al. [47] found that there were more amoeboid microglia and microglia with stout processes than ramified microglia, which had longer or thinner processes in embryonic day 17, while ramified microglia were detected more in postnatal day 30. Microglia have unique gene expression profiles during different phases of development. Some genes of microglia such as Fcrls, P2ry12, are expressed very early in microglia development, whereas others including MafB, Tmem119, are only expressed in adult microglia [48, 49]. A study also found there was not substantial gene expression overlap between lipopolysaccharide (LPS)-stimulated microglia from the adult brain and microglia from a control neonatal brain [48].
Microglia are critical for normal neural development but also maintain their
role as primary CNS immune cells. Early neurodevelopment represents a
particularly sensitive window during which diverse immune-activating challenges
can induce long-term changes in brain function [47]. In neurodevelopment,
microglia play essential roles in regulating neuronal activity, synaptic
transmission, and the formation, remodeling, or elimination of synapses [50, 51].
Microglia frequently interact with neurons, mediating neuronal functions
including myelination, neurogenesis, and synaptic formation and maturation
[52, 53, 54, 55] through signaling molecules such as brain-derived neurotrophic factor
(BDNF) [56], transforming growth factor-beta (TGF-
When macro-environmental challenges occurr, microglial gene expression changes in a complex manner during development [59]. Under physiological conditions, microglia self-renew through proliferation and continuously monitor the CNS microenvironment [44]. However, when exposed to threats and adverse environment, microglia can adopt an abnormal state in response to injury, ischemia, or inflammatory stimulation in the brain [42]. This homeostasis is associated with proliferation, migration to injury sites, phagocytosis of cellular debris, and secretion of neurotoxic or neurotrophic factors [64]. Signals from bacterial products such as LPS, pathological proteins, ATP, and serum factors have been identified as triggers for microglia activation [46, 65].
Brain development is particularly sensitive during early stages of life,
especially in prenatal and early postnatal periods. Disruptions during these
sensitive windows, whether due to genetic, environmental, or immune-related
factors, can have profound effects on neurodevelopment [25]. Infections or other
adverse states during pregnancy including preeclampsia can activate the maternal
immune system, leading to the release of pro-inflammatory cytokines such as IL-6
and TNF-
ASD is thought to result from complex interactions between genetic predispositions and environmental exposures [69]. Gestational factors that could affect neurodevelopment such as complications during pregnancy have been suggested to increase the risk of autism [70]. PE is one of the most severe complications of pregnancy and increases the risk of ASD.
Early neurodevelopmental deficits are considered a fundamental basis for the onset and progression of ASD [71]. These deficits disrupt normal brain development during critical periods, leading to long-lasting structural and functional changes in the brain that underlie the core symptoms of ASD [72, 73, 74]. During early brain development, neurogenesis and synaptogenesis are crucial for establishing functional neural circuits [75, 76]. Early deficits in neural circuit formation can lead to the impaired integration of sensory, motor, and cognitive information, which is characteristic of ASD [13, 76]. Early developmental vulnerabilities can be exacerbated by maternal factors including PE, which may interfere with the normal course of neurodevelopment, and therefore have a long-lasting impact on the nervous system, increasing the risk of ASD [77]. Postmortem studies of ASD brains have shown early structural brain abnormalities, such as altered connectivity in areas responsible for social cognition, sensory processing, and communication [76, 78, 79]. Neuroimaging studies in living individuals with ASD have revealed altered brain networks, including atypical connectivity patterns that reflect early developmental disruptions [25].
The pathogenesis of ASD is complex, with dysregulated neuroinflammation identified as a critical factor in its development [80]. Human cognition and social function rely on a finely tuned and balanced immune response within the CNS [81]. Emerging research using single-cell sequencing of postmortem prefrontal cortex tissues from ASD patients has revealed a significant increase in the proportions of microglia [82, 83]. Velmeshev et al. [82] used single-nucleus RNA sequencing (snRNA-seq) of cortical tissue from patients with ASD and found that the molecular state of microglia was preferentially affected in autism and microglia from ASD samples that were enriched for genes associated with microglial activation and transcriptional factors regulating developmental processes. Another study also found significant increases in reactive forms of microglia in ASD patients and an increase in microglia within gray matter laminae in ASD analyzed integrating the spatial transcriptomics data compared with control, along with an increase in reactive-state markers by highlighting the up-regulation of known pro-phagocytotic genes [83]. These findings underscore the role of neuro-inflammation in the pathogenesis of ASD [84].
Behavioral manifestations of ASD have been associated with multiple brain regions, including the amygdala, prefrontal cortex, and hippocampus [85]. Studies on structural changes of the hippocampus in the ASD have been inconsistent [86, 87, 88, 89]. Several studies observed enlarged hippocampal volume in children [86] and adolescents [87, 88] with ASD, while other studies found that the hippocampal volumes of ASD patients were smaller than those of healthy individuals [89]. In addition, studies also established a relationship between the dysfunction of the hippocampus and impairments in spatial reasoning and episodic memory, as well as social memory deficits in ASD [90, 91, 92]. The core symptoms of ASD are associated with abnormalities in neurogenesis, disrupted synaptic pruning, and dysfunction during CNS maturation [93]. Dysregulated microglia activity during critical periods of neurodevelopment can impair these processes, contributing to ASD [94, 95]. Given their integral role in coordinating neurogenesis, immune regulation, synaptogenesis, and synaptic pruning, microglia dysregulation is considered a pivotal factor in the development of ASD.
In conclusion, maternal factors, including PE, represent adverse intrauterine exposures that can impact the function of offspring microglia through various pathways, as will be explained in detail below. These effects ultimately result in early neurodevelopmental deficits, which disrupt key processes such as neurogenesis, synaptogenesis, and neural circuit formation, leading to the cognitive, social, and behavioral challenges characteristic of ASD (Fig. 1).
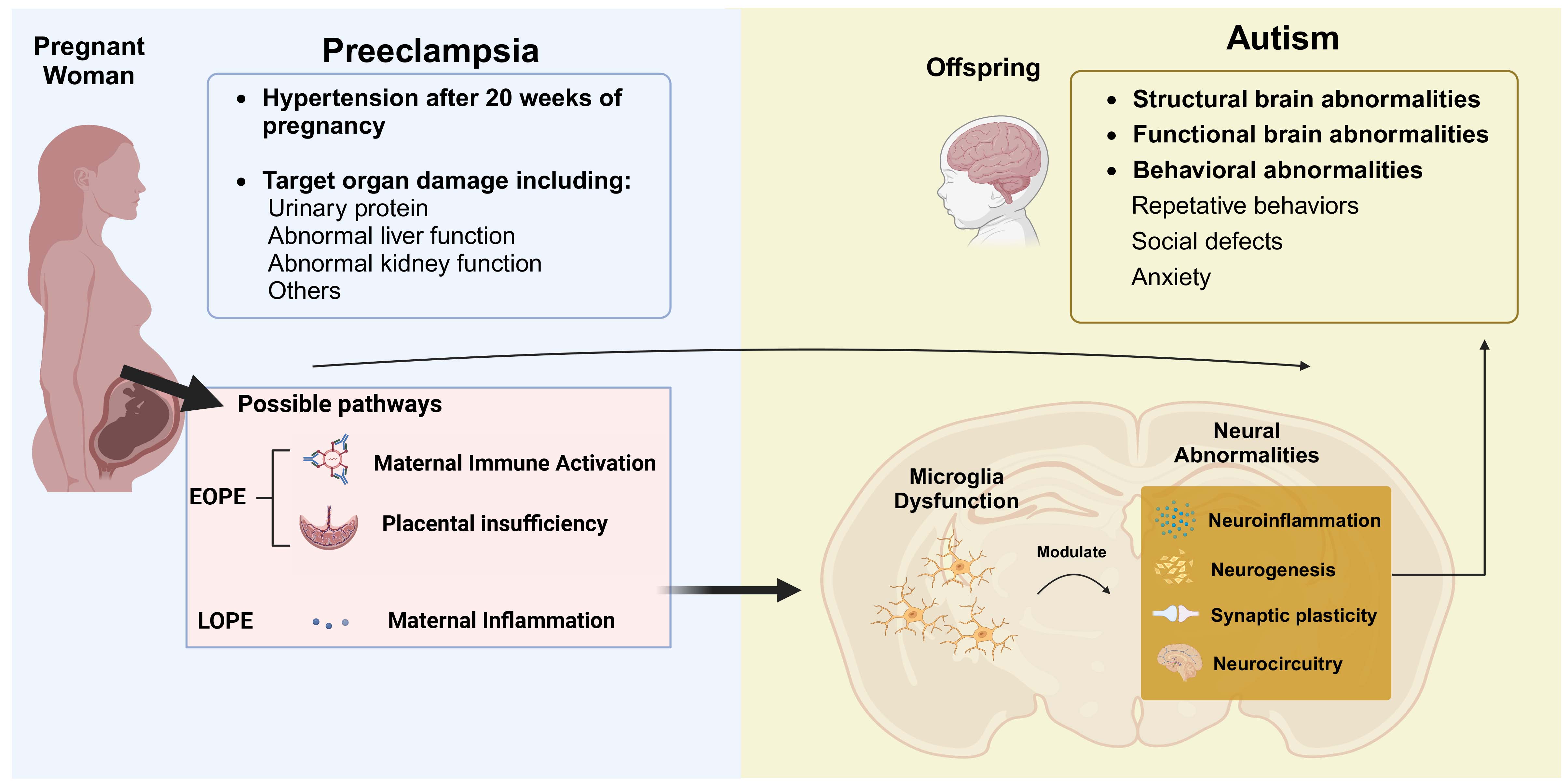 Fig. 1.
Fig. 1.
The linking mechanic connector of microglia of offspring in the maternal preeclampsia and ASD of offspring. Maternal preeclampsia is characterized by hypertension after 20 weeks of pregnancy and target organ damage, including urinary protein, abnormal liver function, and kidney dysfunction. The main manifestations of ASD include behavioral abnormalities such as repetitive behaviors, social deficits, and anxiety. The possible pathophysiological mechanisms of early-onset preeclampsia involve maternal immune activation, placental insufficiency, while late-onset preeclampsia involve maternal inflammation. These changes of PE may potentially impact the offspring’s brain, leading to microglia dysfunction. The microglia modulate neural abnormalities, including neuroinflammation, impaired neurogenesis, disrupted synaptic plasticity, and altered neurocircuitry. These disruptions contribute to structural and functional brain abnormalities in offspring, as seen in autism. ASD, autism spectrum disorder; PE, preeclampsia; EOPE, early-onset preeclampsia; LOPE, late-onset preeclampsia. Created with Biorender.com.
Based on the differences in the pathogenesis of EOPE and LOPE, as well as their differential impacts on neonatal perinatal outcomes, it is valuable and meaningful to further classify preeclampsia into EOPE and LOPE for a deeper exploration of their respective effects on abnormal microglia in offspring and the potential association with ASD in the offspring.
Although various epidemiological studies have found that PE increased the risk
of ASD in offspring, few studies discussed the risk of ASD by classifying the PE
as EOPE and LOPE. Wang et al. [38] conducted a population-based cohort
study consisting of 52,171 live-born pregnancies in Denmark and Sweden and found
that both EOPE and LOPE increased the risk of ASD with an adjusted HR of 1.74
(95% CI, 1.55–1.95) and 1.30 (95% CI, 1.24–1.37), respectively. EOPE was
associated with an almost 2-fold risk of ASD in offspring, and the risk was
smaller in those with LOPE. EOPE may confer a higher risk of offspring ASD
compared to LOPE, partly due to its strong association with fetal growth
restriction (FGR)—a well-established risk factor for neurodevelopmental
impairments [96], Unlike LOPE, which rarely involves FGR [18, 20], EOPE-related
placental insufficiency induces chronic hypoxia and systemic inflammation,
disrupting fetal brain development. Animal studies suggest that FGR triggers
microglial hyperactivation and excessive synaptic pruning via inflammatory
pathways, such as the microbiota-IPA-brain axis, which promotes ASD-like
behaviors [90]. Additionally, EOPE showed longer exposure time on fetal neuronal
development compared to LOPE. However, the underlying mechanism is unclear and
animal models are necessary for exploring the relationship among the two subtypes
of PE, microglial abnormalities and ASD in offspring. Studies have used several
kinds of animal models related to the pathologic mechanisms of PE, and explored
the manifestations of microglia as well as associated neural function in the
brain of offspring (Table 2, Ref. [97, 98, 99, 100, 101, 102, 103, 104]). However, these studies did not
clearly define whether the PE models established were early-onset or late-onset
and currently, there is a lack of timing to distinguish EOPE and LOPE in animal
models. Mice have a relatively short length of gestation (19
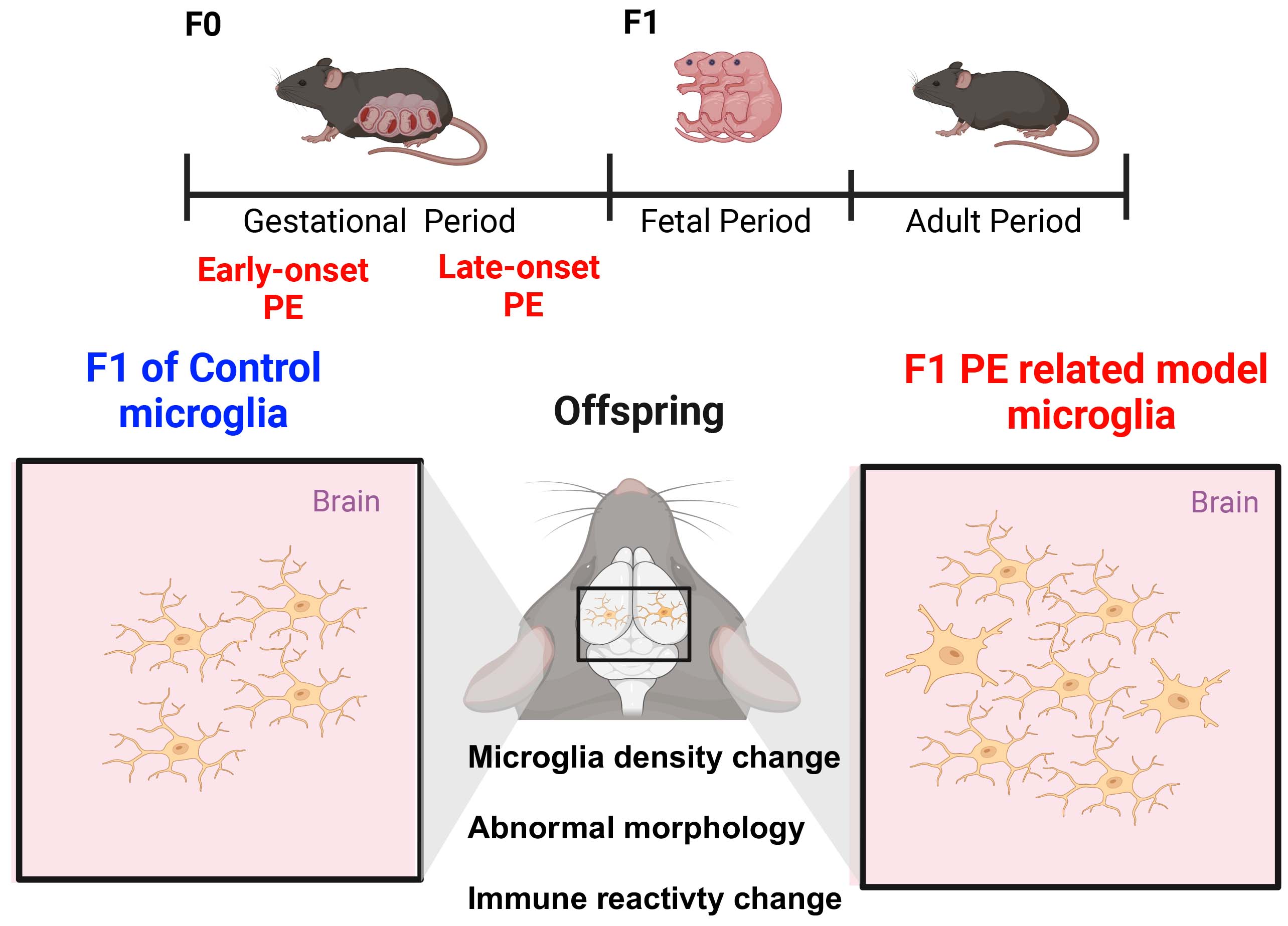 Fig. 2.
Fig. 2.
The abnormalities of microglia in the offspring of animal modeled by related pathologies of preeclampsia. The abnormalities in microglia observed in the offspring (F1) of animal models exposed to early-onset preeclampsia (EOPE) and late-onset preeclampsia (LOPE) during the gestational period (F0). F1 of Control microglia exhibit normal microglia characteristics in the brain, including typical density, morphology, and immune reactivity. In contrast, F1 PE-related model microglia show significant alterations, including microglia density change, abnormal morphology and immune reactivity. The central diagram highlights these differences, emphasizing the impact of maternal PE-related pathologies on microglia development and function in the offspring’s brain. Schematic diagrams were created using BioRender.com.
| Reference | Modeling methods | Pathology of PE | Dysfunction of microglia | Related change of neural function or behaviors |
| Katoh et al. 2022 [97] | Continuous injection of Ang II in pregnant C57BL/6 mice from gestational day 10.5. | Impaired vasoconstriction, vascular dysfunction, placental ischemia and hypoxia. | Higher solidity and circularity of microglia in the fetal brain↑, indicating a transformed amoeboid morphology and activated state. | Unexplored. |
| Hayes et al. 2022 [98] | Poly I:C (10–20 mg/kg at a volume five times the animal weight (5 mL/kg), i.p.), to pregnant C57BL/6 mice at gestational day 9.5. | MIA | Bulk RNA-seq and Single-cell RNA-seq analysis of microglia revealed that decrease of the contribution to inflammatory microglia states↓, while no distinct subpopulation change. | Dysfunctional connectivity of the ventral striatal circuit with a decrease in the glutamatergic presynaptic release probability specifically onto D2R MSNs. |
| Xia et al. 2020 [99] | Poly I:C (5 mg/kg, i.p.), to pregnant C57BL/6 mice at gestational day 12. | MIA | Total number of microglia↑, activated morphology of microglia↑ in adult hippocampus. | Fewer parvalbumin positive cells and impaired GABAergic transmission in the dentate gyrus, accompanied by schizophrenia-like behavior in the adult offspring. |
| Giambrone et al. 2019 [100] | Sprague Dawley rats by surgical reduction of utero-placental perfusion (RUPP) at gestational day 14. | Placental ischemia | Microglial density in the sub-ventricular zone ↓. | Unexplored. |
| O’Loughlin et al. 2017 [101] | LPS (50 µg/kg, i.p.), to pregnant C57BL/6 mice at gestational day 12. | MIA | Larger microglia bodies and thicker shortened processes in postnatal amygdala. | Cytoarchitecture changes of the postnatal amygdala, and may predispose offspring to amygdala-related disorders such as heightened anxiety and depression. |
| Schaafsma et al. 2017 [102] | LPS (0.05 mg/kg, i.p.) to pregnant C57BL/6 mice during gestation day 15–17 | Maternal inflammation | Inflammation of fetal microglia, reduced LPS responsiveness in total brain microglia and increased LPS responsiveness in hippocampal microglia. | Reduced basal home cage activity, impaired learning. |
| Li et al. 2014 [103] | Poly I:C (20 mg/kg, i.p.), to pregnant C57BL/6 mice at gestational day 9.5. | MIA | Microglia number in the adult hippocampus ↑. | Impaired short-term memory and altered dendritic architecture. |
| Zhu et al. 2014 [104] | Poly I:C (20 mg/kg, i.p.), to pregnant C57BL/6 mice at gestational day 9.5. | MIA | Microglia number in the adult hippocampus, thalamus, and cerebral cortex ↑. | Hyperlocomotion, deficits in social interaction and prepulse inhibition. |
Abbreviations: Poly I:C, polyinosinic-polycytidylic acid; LPS, lipopolysaccharide; MIA, maternal immune activation; Ang II, angiotensin II. Arrows indicate the direction of change: ↑ represents an increase, and ↓ represents a decrease.
The microglial abnormalities in the offspring of EOPE primarily manifest as alterations in microglial number, morphological changes indicative of activation, and dysregulated inflammatory states in the brain. The specific characteristics of these abnormalities may vary depending on the modeling approach. Notably, EOPE induced by maternal immune activation is associated with an increased number of microglia, whereas EOPE resulting from placental ischemia primarily affects microglial density [97, 98, 99, 100, 104].
Most studies establishing the animal model related to the pathologic mechanisms of PE were prior to gestational day 15, which is more closely aligned with the time points defined in previous research on EOPE. Systemic activation of the immune cells using during pregnancy has been studied as a model of preeclampsia [98, 99, 101, 103, 104, 107]. Studies from animal models have shown that maternal exposure to polyinosine acid (poly I:C) from gestational day 9.5 induced increased density of microglia in diverse regions in the brain of offspring, especially in the hippocampus, and impaired short-term memory, deficits in social interaction were observed in these researches [103, 104]. In addition, poly I:C and LPS exposure separately from gestational day 12 showed changes of morphology of activation-like microglia in different regions [99, 101]. The former was observed in hippocampus and associated with impaired GABAergic transmission in the dentate gyrus, while the latter was observed in amygdala and associated with heightened anxiety and depression. These studies together indicated the activation of microglia and impaired neural function. In addition, a recent study found that poly I:C injection from gestational day 9.5 blunted the reactivity of microglia both in the development and the adult offspring, and contributed to the injury seen in striatal circuit development [98]. Disturbed neuroregulatory and immunological functions of microglia resulted from maternal immune activation harm the key processes of brain development, affecting neurogenesis, neuronal migration, myelination, neural circuit formation, and synaptic refinement, potentially resulting in long-term impairments in brain function and behavior.
The pathology of placental insufficiency of the PE may consistently influence
the microglia of offspring, which alters aggravating inflammatory functions.
Reduction of utero-placental perfusion (RUPP) is a classical placental
insufficiency animal model, which have a surgical operation at gestational day
14. A study in an animal model of RUPP in Sprague Dawley rats found increased
pro-inflammatory cytokines such as IL-1
Very few animal studies explored the impact of LOPE on microglia dysfunction and neurobehavioral abnormalities of ASD. LOPE modeled by LPS injection exhibits an abnormal microglial inflammatory state, with inconsistent changes observed between the whole brain and the hippocampus. Schaafsma et al. [102] explored the neurodevelopment outcomes by LPS administration to pregnant C57BL/6 mice during gestation day 15–17, during the period of LOPE. They found a fetal microglia pro-inflammatory response in which adult whole brain microglia displayed a persistent reduction in pro-inflammatory activation in response to a re-challenge with LPS and hippocampal microglia of these mice displayed an increased inflammatory response to an LPS re-challenge. These findings were different from the microglia changes observed in the mice using LPS earlier in the maternal period. However, more research is needed to study LOPE to help to confirm and explain these differences. Adult offspring exposed to maternal LPS administration during gestation day 15–17 showed reduced home cage activity, reduced anxiety and reduced learning performance in a T-maze. A reduced expression of brain-derived neurotrophic factor (BDNF) from microglia may play a role in these processes. Given the limited animal modeling studies on LOPE, future research should focus on exploring the underlying mechanisms and refining animal models for this condition. This could provide valuable insights into the mechanisms by which LOPE may lead to NDDs, such as autism, and microglial abnormalities in offspring.
Preeclampsia is highly heterogeneous in its clinical manifestations and pathogenesis, which may explain why early-onset (EOPE) and late-onset preeclampsia (LOPE) exert distinct effects on microglia and ASD risk. Firstly, EOPE and LOPE occur at different gestational stages, coinciding with critical phases of microglial development, neurogenesis, and brain maturation. Microglia undergo migration, differentiation, and functional maturation during pregnancy, playing pivotal roles in synaptic pruning and immune regulation [108]. Thus, the timing and duration of preeclampsia exposure may differentially impact microglial function and neurodevelopment, influencing ASD risk. Secondly, EOPE and LOPE exhibit distinct placental pathologies. EOPE, often termed “placental preeclampsia”, is linked to impaired trophoblast invasion and early placental dysfunction, leading to severe placental hypoxia. In contrast, LOPE is associated with maternal metabolic dysfunction and systemic inflammation [18, 19], which alter fetal environment, breach the fetal blood-brain barrier [67], leading to abnormal brain development. Thus, inconsistent uterine environment and placental function may contribute to different microglia response and further impact the neurodevelopment. Additionally, EOPE is strongly associated with fetal growth restriction (FGR), a known risk factor for neurodevelopmental delays [96], while LOPE is not [18, 20]. Animal studies suggest that FGR and inflammatory pathways, such as the microbiota-IPA-brain axis, may drive microglial hyperactivation and synaptic over-pruning, increasing ASD susceptibility [90]. Thus, the divergent mechanisms underlying EOPE and LOPE likely contribute to their differential impacts on microglia and ASD risk.
The pathophysiology of PE is highly heterogeneous. Current animal models are designed to replicate its diverse pathological processes by employing various approaches, including maternal immune activation, reduced utero-placental perfusion, and vascular endothelial dysfunction, often with inconsistent modeling time points. These distinct modeling strategies, which specifically mimic either EOPE or LOPE, may differentially affect microglial development and susceptibility to ASD.
Although mechanistic studies directly linking PE-specific pathways to microglial abnormalities and ASD risk remain limited, a synthesis of existing fragmented evidence highlights potential mechanisms underlying EOPE, LOPE, microglial dysfunction, and ASD pathology.
Maternal immune activation (MIA) is initially defined as those immune change triggered by infection in the pregnancy period. There is emerging evidence that EOPE can result in maternal immune activation [18, 109]. Meanwhile, the immune hypothesis is regarded as a key factor in the pathogenesis of autism, and provides an explanation for the variability in clinical phenotypes and comorbidities that affect the progression and severity of this condition [110]. It has recently been suggested as a component potentially involved in the development of ASD [111]. Pregnancy represents a critical period of immune vulnerability, where maternal immune activation may adversely affect fetal immune and nervous system development, especially in the very early period of neural development influenced by EOPE. These effects can persist into adulthood, increasing the risk of NDDs in offspring [112]. Another study has also demonstrated a more pronounced imbalance in immune cell populations in the circulatory system of patients with EOPE compared with LOPE [21].
Various MIA animal models established in a similar gestational period to EOPE, and found the changes of cell density and morphology, as well as the immune reactivity of microglia, along with neural functional and behavioral abnormality such as the social communication deficits, which is related to ASD [103, 104]. As primary immunoregulators of neural function, microglia are central to the sustained immune and/or neurodevelopmental changes associated with MIA. Microglia abnormality, driven by MIA, is thought to be a critical mechanism underlying these persistent changes in offspring [20].
EOPE is characterized with placental ischemia and hypoxia, vascular dysfunction and increased antiangiogenic factors including soluble fms-like tyrosine kinase 1 (sflt1) and soluble endoglin (sENG) [8]. The placenta is an essential organ for proper fetal development. Placental insufficiency leads to an intrauterine environment in which the fetus is at risk of inadequate oxygen and nutrient exchange [113], contributing to abnormal neuron development [4]. Villamor et al. [10] conducted population-based cohort and sibling-controlled studies and found that placental abruption was associated with increased HR of ASD in a general cohort analysis, and that syndromes linked to defective placentation are associated with an increased incidence of ASD in the offspring.
Although there is still a lack of specific and in-depth basic studies on the maternal placental dysfunction, the microglia abnormality and development of ASD, data from human studies have found that placental insufficiency of severe PE appears to be responsible for the increase in neurodevelopmental disorders and the risk of developmental delay (DD) [34]. The abnormity of microglia may serve as a bridge to the development of PE and ASD because of the similar changes of microglia both on the offspring exposed to PE and individuals with ASD, including evidence from animal models of placental insufficiency showing abnormal microglia and neuronal function related to ASD [97, 100].
LOPE is often associated with maternal conditions such as obesity, diabetes mellitus, and other metabolic diseases, which are considered to be characterized by chronic, low-grade systemic inflammation [21]. Systemic chronic inflammation is characterized by persistent, sterile, non-resolving inflammation that increases with age, and is implicated in many common disease states including PE [112]. Valencia-Ortega et al. [22] conducted an age-matched study and reported that maternal serum concentrations of IL-6 were significantly higher in late-onset pre-eclampsia, compared to early-onset preeclampsia or uncomplicated pregnancy. Moreover, IL-6 was only significantly associated with term pre-eclampsia, while the first and second trimester levels of IL-6 were not associated with pre-term preeclampsia, suggesting that elevations in IL-6 may be a late-stage feature of pre-eclampsia [114, 115].
Substantial evidence indicates that chronic maternal inflammation, triggered by
pregnancy complications such as PE, is a major environmental risk factor for ASD
in offspring [5]. Population-based studies have linked elevated maternal
interleukin (IL)-6 and C-reactive protein (CRP) levels during pregnancy to
alterations in neonatal brain structure and functional connectivity [116].
Furthermore, children exposed to high intrauterine tumor necrosis
factor-
According to the animal study, injection of 0.05 mg/kg LPS on gestational days
15, 16, and 17 significantly elevated TNF-
Early intervention is important for improving behavior expression of children with ASD. There were inconsistent findings supporting that very early interventions on infants and toddlers with ASD have limited impact on neurodevelopmental outcomes by age 3 years [118]. As the neurodevelopmental and environment mechanisms underlying autism are better understood, a broader array of targets within the developmental cascade to autism is expected to emerge, including those linked to parental risk factors [93].
Given that PE is recognized as a risk factor for ASD of offspring, early prevention or management of PE starting from the perinatal period may represent a potential strategy for reducing the risk of ASD development. Currently, the management of preeclampsia involves preconception counseling, perinatal blood pressure monitoring and control, pharmacological therapy for high-risk women and close postpartum blood pressure monitoring [8]. Due to differences in the timing of diagnosis and underlying mechanisms between EOPE and LOPE, the approaches for intervention and treatment may also vary. In patients with EOPE and severe features, close monitoring of maternal and fetal well-being is crucial, and delivery should be considered at any point if there is deterioration in either maternal or fetal condition, as EOPE is strongly associated with neonatal mortality [8].
As the pathological processes of maternal PE and neural development often begin earlier than the clinical manifestations, identifying high-risk cases and applying preventive measures becomes even more critical. Moreover, the subtypes of PE must be considered in order to select and combine appropriate preventive and therapeutic strategies. For example, low-dose aspirin is now recommended for the prevention of PE in high-risk women [119]. Sildenafil, which is believed to reduce placental ischemia associated with the pathology of EOPE, has shown promise [120]. Metformin, an insulin sensitizer, has been associated with a reduced incidence of hypertensive disorders in pregnancy and can also be used as a preventive measure for PE, especially in women with diabetes mellitus, obesity, or metabolic abnormalities, who are at higher risk for developing LOPE [121].
In summary, the prevention of both EOPE and LOPE requires identifying high-risk factors for PE. However, the different subtypes of PE may indicate distinct pathologies, helping clinicians choose drugs targeting the associated mechanisms. Furthermore, addressing metabolic and vascular abnormalities may reduce the occurrence of LOPE. Since the role of microglia as a molecular link between maternal preeclampsia and offspring autism remains unclear, future research should focus on exploring the underlying mechanisms to identify molecular targets in microglia. This could provide a foundation for developing early preventive strategies targeting microglia.
Several therapeutic strategies have been explored to mitigate the impact of PE
on fetal microglial function and neurodevelopmental outcomes. Anti-inflammatory
agents, such as low-molecular-weight heparin (LMWH) and aspirin, have been shown
to attenuate placental inflammation by suppressing the TNF-
PE creates a pro-inflammatory intrauterine environment that can profoundly affect fetal brain development. Chronic maternal immune activation and inflammation associated with PE leads to dysregulated microglia dysfunction, which is strongly implicated in the pathogenesis of ASD. Microglia dysfunction impacts NSC proliferation, synaptic pruning, and neuroimmune modulation, contributing to structural and functional brain abnormalities. Postmortem and animal studies consistently reveal microglia abnormality and neuroinflammation in ASD, highlighting their potential role as therapeutic targets. Given that different pathologies of EOPE and LOPE, it is possible that the effects of EOPE and LOPE on offspring neurodevelopmental outcomes may vary depending on the severity and clinical course of the disease.
Future research should focus on elucidating the molecular pathways connecting two subtypes of PE and microglia activation, as well as identifying biomarkers for early detection of ASD in high-risk populations. Early interventions targeting neuroinflammation and microglia dysregulation have promise for reducing the burden of ASD in children born to PE-affected pregnancies.
Contribution to the conception and design of the manuscript—YZ, SYW, XTF, XC, XHL; writing—original draft preparation—YZ; writing—review and editing—YZ, SYW, XTF, XC; supervision—XHL; project administration—XHL. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We acknowledge BioRender.com for providing the tools used to create the figures in this manuscript.
This work was supported by the Hangzhou medical health technology project (Z20250245, XHL).
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGpt in order to improve language, which was mainly used in the “Abstract”, “Introduction” Sections, and check spelling and grammar in the main body of the manuscript. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.