1. Introduction
Heart fibrosis is a maladaptive remodeling process characterized by excessive
deposition of extracellular matrix (ECM) proteins and collagen fibers in the
cardiac interstitium [1]. While cardiac fibrosis initially serves as a
compensatory mechanism to uphold tissue architecture and physiological stability
following myocardial injury, this process progressively diminishes myocardial
elasticity and impairs cardiac conduction, culminating in disease-related
morbidity and mortality [2]. Although regression of fibrotic myocardium occurs
sporadically, development of precision-targeted treatments remains largely
elusive [3]. Identifying novel therapeutic targets driving cardiac fibrosis
remains an urgent priority in cardiovascular research.
Dysregulated inflammation plays a critical role in heart fibrosis [4]. For
instance, studies in small animal models have demonstrated that macrophages can
drive heart fibrotic remodeling and ventricular dysfunction, while T cells
coordinate fibrosis in non-ischemic heart failure [5, 6]. During the progression
of various forms of heart failure, cardiomyocyte death, fibroblast activation,
and inflammatory cytokine release are interconnected processes that exacerbate
tissue fibrosis [7].
Na+, K+-ATPase (NKA) serves as a classical ion pump that spans the
cell membrane and actively transports Na+ out of and K+ into the cell,
thereby maintaining membrane potential [8]. Besides its canonical ion-pumping
role, NKA also functions as a signaling transducer, modulating Src signaling
cascades, the PI3K/AKT pathway, P2X7R/K+ cascades, and cytosolic Ca2+
oscillations [9, 10]. Mature cardiomyocytes predominantly express the 1
isoform, accounting for approximately 75% of total NKA subunit
expression [11]. In the heart, NKA performs vital physiological functions. A
clinical studies have reported reduced NKA levels in left ventricular biopsies of
dilated cardiomyopathy (DCM) subjects [12]. An animal study further indicated that
administration of marinobufagenin, a natural NKA inhibitor, stimulated fibroblast
collagen production and induces cardiac fibrosis [13]. Previously, we observed
that NKA deficiency impairs mitochondrial function, accelerating isoproterenol
(ISO)-induced cardiac fibrosis and dysfunction [14]. However, whether
inflammation contributes to NKA deficiency-related cardiac fibrosis remains
unclear. Herein, we aim to elucidate the relationship between inflammation and
cardiac fibrosis under NKA-deficient conditions.
2. Materials and Methods
2.1 Chemicals and Antibodies
All compounds, including ISO (Cat No. 420355) and SLU-PP-332 (Cat No. SML3908),
were commercially sourced from Sigma-Aldrich (St. Louis, MO, USA). Mouse IgG was
sourced from Bioss Biotechnology Company (Beijing, China, Cat No. bs-0296P).
DRm217 antibody, a particular antibody targeting the DR-region
(897DVEDSYGQQWTYEQR911) of the NKA1 subunit, was prepared in
our lab [15]. The primary antibodies against -smooth muscle actin
(-SMA, Cat No. GB111364-50), CD68 (Cat No. GB113109-50), LY6G (Cat No.
GB11229-50), CD3 (Cat No. GB13014-50), and Horseradish Peroxidase (HRP)-labeled
rabbit antibodies (Cat No. GB23303) were sourced from Servicebio Technology
Company (Wuhan, Hubei, China). Primary antibody targeting estrogen-related
receptor (ERR, Cat No. CY5617) was sourced from Abways
company (Shanghai, China), and primary antibody targeting -actin (Cat
No. 66009-1-Ig) was sourced from Proteintech Biotechnology Company (Wuhan,
Hubei, China).
2.2 Animal Model Establishment
All animal procedures were approved by the Animal Care and Use Committee of
Xi’an Jiaotong University (Ethics numbers: IAUC/765/2019, #19765) and adhered to
guidelines established by the National Health and Medical Research Council of
China. Eight-week-old male wild-type (WT) and NKA1+/- mice were
subcutaneously injected with ISO (30 mg/kg) daily for 14 consecutive days.
Following euthanasia via cervical dislocation, hearts were harvested for
subsequent analyses. To evaluate the effects of DRm217, 18 male C57BL/6 mice were
randomly assigned to saline, ISO+IgG, or ISO+DRm217 groups. IgG or DRm217 was
administered intraperitoneally at a dose of 10 mg/kg every 5 days.
2.3 Histological Analysis
To evaluate histopathological changes, the hearts were rinsed with 0.9% saline
and subsequently fixed in 4% paraformaldehyde. Each heart was divided into three
segments and embedded in paraffin. Serial sections of 5 µm thickness were
prepared from the basal, mid, and apical levels of the heart. The sections were
stained with hematoxylin and eosin (H&E). A pathologist who was blinded to the
identify of experimental groups evaluated the histopathological scores based on
hyper-eosinophilic bundles, leukocyte infiltration, and cardiomyocyte necrosis.
At least 10 fields per slide were examined, with the severity of changes graded
as severe (++++), moderate (+++), mild (++), minimal (+), or nil (–). The extent
of myocardial fibrosis was assessed using Masson’s trichrome staining, while
collagen content was quantified after staining with Sirius red. Histopathological
images were captured and analyzed using a digital camera attached to a microscope
(Nikon, Tokyo, Japan). The percentage of fibrotic area in each image was measured
using ImageJ software (VERSION 1.53t, National Institutes of Health, Bethesda,
MD, USA), and calculated as the ratio of positively stained areas to the total
field area.
2.4 Immunohistochemical Staining
For antigen retrieval, paraffin-embedded sections were deparaffinized,
rehydrated, and immersed in 0.01 M citric acid buffer (pH 10.0). Sections were
treated with 3% hydrogen peroxide for 10 minutes to quench endogenous peroxidase
activity, then blocked with rabbit serum for 30 minutes at room temperature.
Subsequently, the slides were incubated overnight at 4 °C with primary
antibodies against CD68 (1:100), Ly6G (1:100), CD3 (1:100), or -SMA
(1:100). Next, slides were incubated with HRP-labeled goat anti-rabbit secondary
antibody for 1 h. Sections were then washed, stained with 3,3’-Diaminobenzidine
(DAB) (Cat No. G1212-200T, Servicebio Technology Company, Wuhan, Hubei, China), and counterstained with hematoxylin. Histopathological images were
captured and analyzed using a digital camera linked to a microscope (Nikon,
Tokyo, Japan). To quantify immunohistochemistry results, a pathologist blinded to
the experimental groups counted cells with positive immunostaining for CD68,
Ly6G, and CD3. For alpha-smooth muscle actin (-SMA) staining, the
positively stained area was analyzed using ImageJ software and expressed as the
percentage of stained cortical area relative to the total area.
2.5 Western Blotting
Heart tissues were homogenized in RIPA lysis buffer containing a protease
inhibitor cocktail (Roche, Basel, Switzerland) and a phosphatase inhibitor cocktail
(Sigma, St. Louis, MO, USA). After incubation on ice for 30 minutes, the
supernatant was collected by centrifugation for 20 minutes at 4 °C and
12,000 rpm. Protein samples were separated by 10% Sodium Dodecyl
Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and transferred onto a
Polyvinylidene Fluoride (PVDF) membrane (Thermo Fisher Scientific, Waltham, MA,
USA). The membrane was blocked with 10% milk in TBST buffer (10 mM Tris-HCl, 120
mM NaCl, and 0.1% Tween 20, pH 7.4) for 1 h at room temperature and then
incubated with ERR primary antibody (1:1000) overnight at 4 °C. Membranes were washed three times with TBST buffer and incubated
with horseradish peroxidase-conjugated anti-rabbit IgG (1:5000) for 1 h at room
temperature. After washing, visualization was performed using an enhanced
chemiluminescence kit (GE Healthcare, Chicago, IL, USA). Protein bands were
captured using ImageQuant LAS 400 (GE Healthcare, Chicago, IL, USA) and band
intensity was quantified by densitometry analysis using ImageJ (version 1.53t,
National Institutes of Health, Bethesda, MD, USA).
2.6 Transmission Electron Microscopy
Cardiac tissues were fixed in 3% glutaraldehyde for 24 h and subsequently fixed
in 1% osmium tetroxide for 1 h. Following dehydration in an ethanol gradient and
2% uranyl acetate staining, samples were embedded in Embed 812 resin for
transmission electron microscopy (JEM-1400PLUS, JEOL, Tokyo, Japan)
ultrastructural assessment. The images were reviewed in a blinded fashion by two
radiologists.
2.7 Enzyme-Linked Immunosorbent Assay (ELISA)
The levels of interleukin-6 (IL-6, BGK08505), tumor necrosis factor-(TNF-, BGK06804), interleukin-1 (IL-1, BGK10749), and
interleukin-18 (IL-18, ab216165) were measured using corresponding ELISA kits
(Peprotech Company, Rocky Hill, NJ, USA for IL-6, TNF-, and
IL-1, and Abcam Company, Shanghai, China for IL-18) according to the
manufacturer’s instructions.
2.8 Cell Isolation and Treatment
Ventricular cardiomyocytes, macrophages, and fibroblasts were isolated from WT
or NKA1+/- mice as previously described [16]. Cardiomyocytes were
identified based on their characteristic rod-shaped morphology under light
microscopy. Fibroblasts were recognized by their characteristic spindle-shaped or
stellate morphology with cytoplasmic processes, along with relatively large
oval-to-fusiform nuclei, observed under microscopy. Macrophages were confirmed
using flow cytometry with antibodies against F4/80. To ensure uniformity across
treatments and comparisons, isolation of cardiomyocytes, fibroblasts, and
macrophages followed a strictly standardized protocol performed consistently by
the same technician for all groups. Isolated cardiomyocytes were cultured on
0.1% gelatin-precoated 6-well plates in Minimum Essential Medium (MEM, Thermo
Fisher Scientific, Waltham, MA, USA, Cat No. 11095080) supplemented with 5%
fetal bovine serum (FBS, Lonsa Science SRL, Suzhou, China, Cat No, S711-050S),
100 U/mL penicillin (Thermo Fisher Scientific, Waltham, MA, USA, Cat
No. 15070063), and 100 mg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA,
USA, Cat No. 15070063). Isolated macrophages were quantified and seeded in
RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA, Cat No. 11875093) medium
supplemented with 10% FBS, 100 mg/mL streptomycin, and 100 U/mL penicillin.
Isolated fibroblasts were quantified and seeded in DMEM (Thermo Fisher
Scientific, Waltham, MA, USA, Cat No. 11965092). The isolated ventricular
cardiomyocytes, macrophages, and fibroblast have undergone mycoplasma infection
testing. To detect the effects of ISO on cardiomyocytes, macrophages and
fibroblasts, 10 µM ISO was added to the culture medium for 48 h. For DRm217
treatment, IgG or DRm217 was suppled 15 minutes after ISO addition at a final
dose of 1 µM concentration. To detect the effects of SLU-PP-332, a specific
ERR agonist, it was suppled in cell culture medium at a final dose of 1 µM
concentration.
2.9 Cell Co-Culture
For the co-culture models of cardiomyocytes with macrophages or fibroblasts,
cardiomyocytes were pre-plated in 6-well plates at a density of 5
105 cells/mL. Macrophages or fibroblasts were seeded onto cell culture
inserts at a density of 1.0 105 cells/cm2. The inserts
containing macrophages or fibroblasts were then placed above the wells containing
cardiomyocytes. These cells were co-cultured in MEM supplemented with 10 µM
ISO or vehicle for 48 h. For the co-culture model of macrophages and fibroblasts,
fibroblasts were pre-plated in 6-well plates at a density of 5
105 cells/mL. Inserts containing previously activated macrophages were
placed above the wells containing fibroblasts. These cells were co-cultured in
DMEM medium for 48 h.
2.10 Detection of Lactate Dehydrogenase (LDH) Activity
Cellular injury was assessed by measuring LDH release. Following 48 h treatment
with ISO (10 µM), LDH activity in the culture medium was measured using a
Roche LDH assay kit (Cat No. 04744926001, Mannheim, Germany) according to the
manufacturer’s protocol and a microplate reader.
2.11 Real-Time Quantitative PCR
Total RNA was isolated from heart tissue and cells using Trizol reagent (Cat No. B610409-0100,
Sangon, Shanghai, China) and reverse-transcribed into cDNA using the
MightyScript™ RT reagent kit (Cat No. B639252, Sangon, Shanghai,
China). Quantitative polymerase chain reaction (qPCR) was performed using the
SYBR Green reagent kit (Cat No. B532955, Sangon, Shanghai, China) on a
QuantStudio™ 3 Real-Time PCR System (Bio-Rad, Hercules,
CA, USA). Primer sequences used for qRT-PCR are listed in Table 1. All
amplifications were normalized to -actin. Data were analyzed using the
comparative Ct (2-ΔΔCt) method and expressed as fold
change relative to the respective control.
Table 1.
Primer sequences used for real-time quantitative PCR.
| Gene |
Species |
Forward primer |
Reverse primer |
| Acta2 |
Mouse |
GTCCCAGACATCAGGGAGTAA |
TCGGATACTTCAGCGTCAGGA |
| Fn1 |
Mouse |
GATGCACCGATTGTCAACAG |
TGATCAGCATGGACCACTTC |
| Col3a1 |
Mouse |
TGACTGTCCCACGTAAGCAC |
GGAGGGCCATAGCTGAACTG |
| Col1a1 |
Mouse |
CGCAAAGAGTCTACATGTCTAGG |
CATTGTGTATGCAGCTGACTTC |
| -actin |
Mouse |
TGCTGTCCCTGTATGCCTCTG |
TGATGTCACGCACGATTTCC |
PCR, polymerase chain reaction; Acta2, actin alpha 2, smooth muscle,
also named alpha-smooth muscle actin; Fn1, fibronectin 1;
Col3a1, Collagen, type III, alpha 1; Col1a1, Collagen, type I,
alpha 1; -actin, beta-actin.
2.12 Statistical Analysis
Data are presented as the mean standard error. Statistical analysis was
performed using SPSS software (version 22.0; IBM Corporation, Armonk, NY, USA).
Student’s t-test was used for comparisons between two groups. Multiple
group comparisons were analyzed by one-way ANOVA followed by Tukey’s post hoc
test. A p-value 0.05 was considered statistically significant.
3. Results
3.1 NKA1 Insufficiency Aggravats ISO-Induced Cardiac
Lesion and Fibrosis
Since animals completely lacking the 1 gene suffer from embryonic
lethality, heterozygous mice lacking only one copy of the allele
(1+/-) are a commonly used experimental model. H&E staining
revealed myocardial cell rupture, cellular vacuolization, and inflammatory cell
infiltration in WT mice treated with ISO. These pathological alterations were
more pronounced in NKA1+/- mice treated with ISO (Fig. 1A,B).
NKA1 haploinsufficiency also increased interstitial collagen deposition
under ISO treatment, as evidenced by Masson trichrome staining and Sirius red
staining (28.13 2.54% vs. 8.72 1.26% for Masson staining; 28.46
2.24% vs. 9.61 1.15% for Sirius red staining, p
0.05) (Fig. 1C,D). These results demonstrate that NKA1
haploinsufficiency exacerbates heart lesions and fibrosis under ISO challenge.
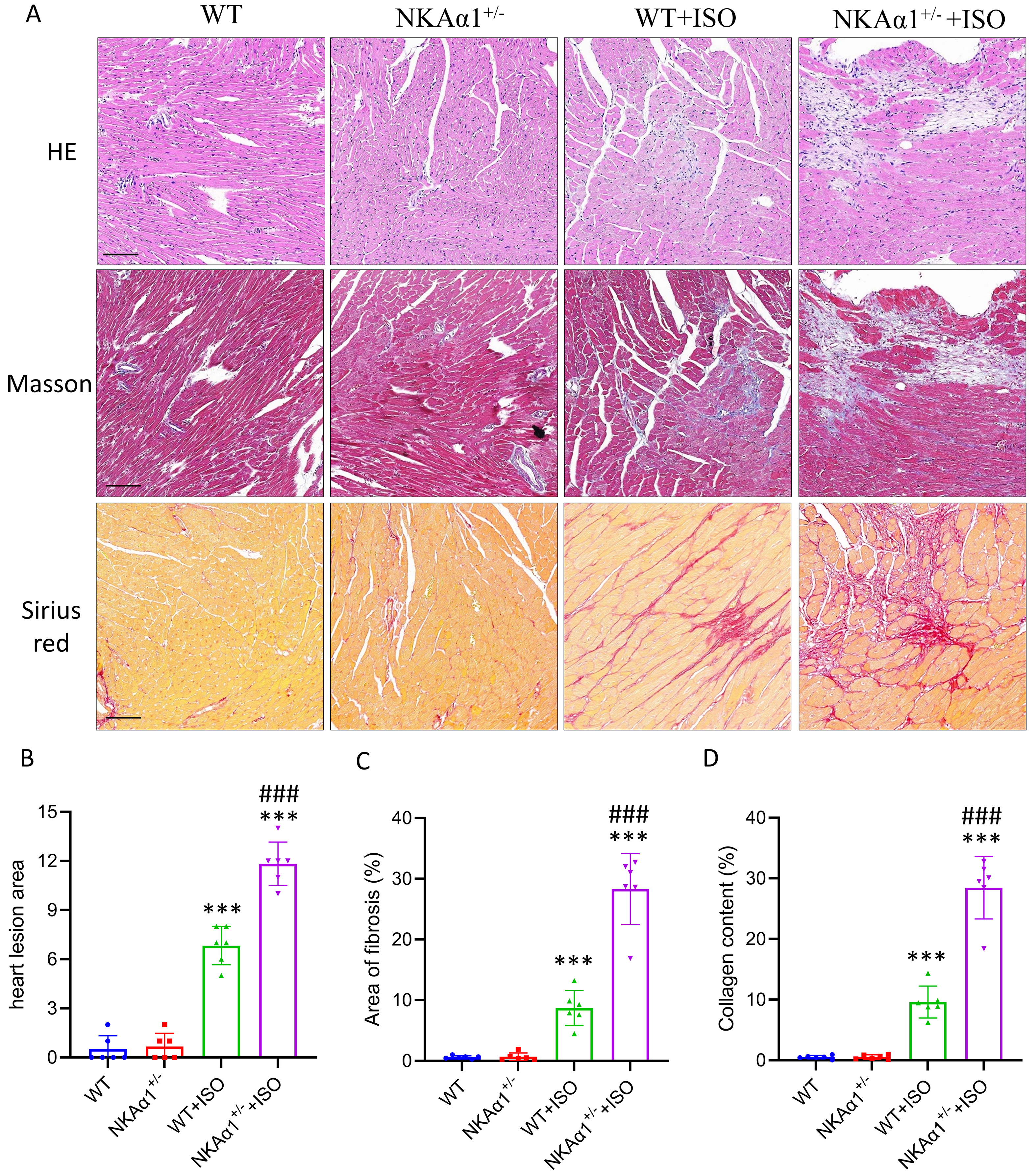 Fig. 1.
Fig. 1.
Na+, K+-ATPase (NKA) 1 haploinsufficiency
exacerbated isoproterenol (ISO)-induced heart fibrosis. (A) Representative
images of hematoxylin and eosin (H&E) staining, Masson staining, and Sirius red
staining in the different groups (scale bar = 100 µm). (B) Quantitative
assessment of the myocardial lesion area. (C) Quantitative analysis of fibrotic
(Masson blue) areas in cardiac sections. (D) Quantitative analysis of collagen
(Sirius red) areas in cardiac sections. Data are presented as means SD; n
= 6. ***p 0.001 for WT+ISO vs wild-type (WT) group and
NKA1+/- + ISO vs NKA1+/- group; ###p 0.001, vs WT+ISO group.
3.2 NKA1 Haploinsufficiency Led to Aberrant ECM Protein
Deposition and Myofibroblast Differentiation
Our previous unbiased proteomic analysis of cardiac tissues from WT and
NKA1+/- mice challenged with ISO revealed differential protein
expression patterns [14]. Re-analysis of our pre-proteomics data showed that
proteins related to organization of the ECM, including collagen types III, VIII,
XII, XIV, fibronectin 1 (Fn1), periostin (Postn), and metallopeptidase 2 (MMP2),
were significantly upregulated (Fig. 2A). qPCR results confirmed the upregulation
of collagen 1a1, collagen 3a1 (Col3a1), and Fn1 in cardiac tissue from
ISO-treated NKA1+/- mice (Fig. 2B–D). Myofibroblast
differentiation, characterized by upregulation of -SMA, represents a
pivotal event in cardiac fibrogenesis [17]. Immunostaining results showed
markedly increased expression of -SMA in hearts from
NKA1+/- mice (Fig. 2E,F). These findings suggest that
NKA1 haploinsufficiency leads to aberrant ECM deposition and
myofibroblast differentiation.
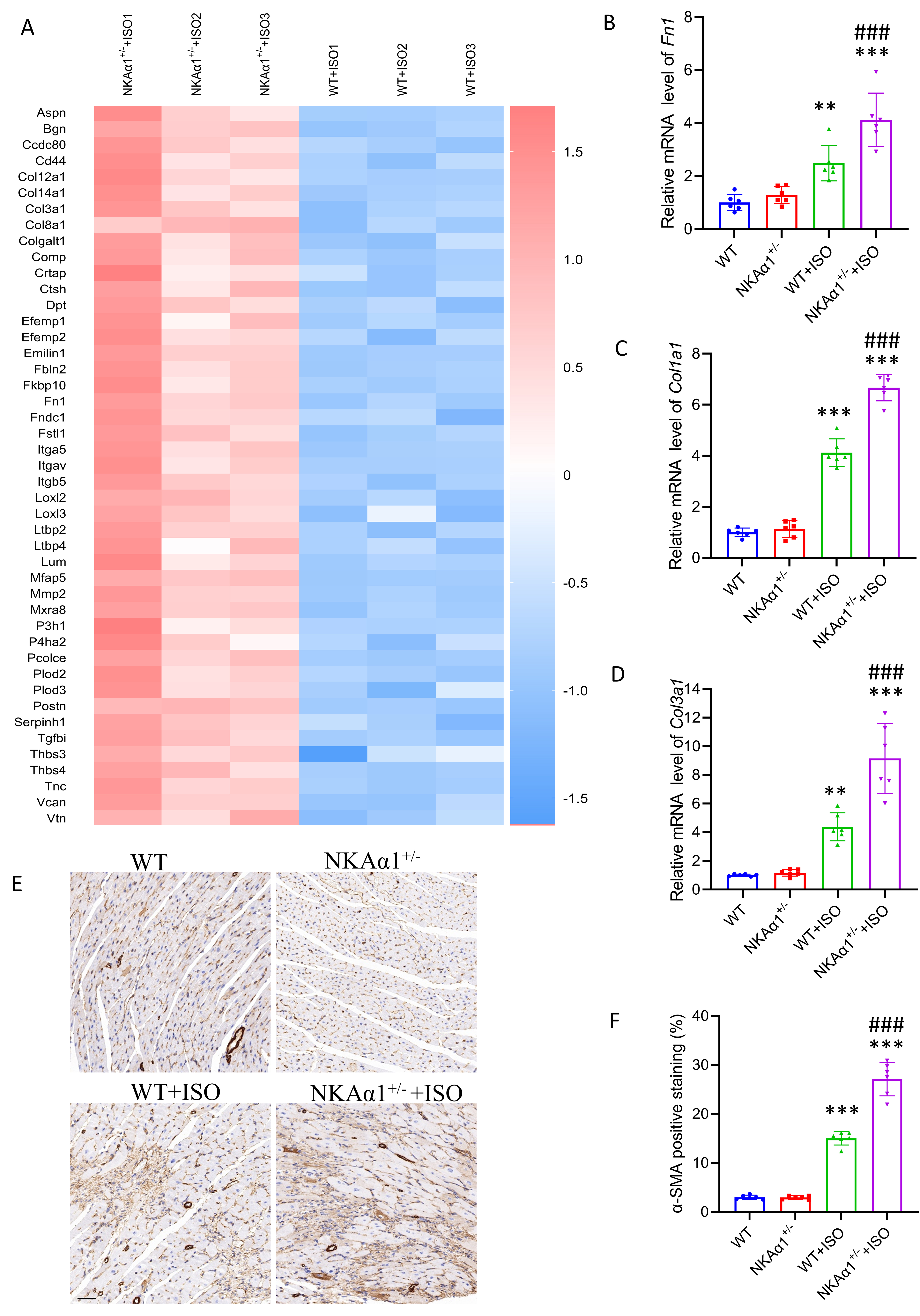 Fig. 2.
Fig. 2.
NKA1 haploinsufficiency induced the aberrant
expression of extracellular matrix (ECM) genes and myofibroblast
differentiation. (A) Proteomics analysis of cardiac tissue from ISO-treated WT
mice and NKA1+/- mice. Enrichment map of proteins associated with
ECM organization. (B–D) qRT-PCR analysis of Fn1 (B), Col1a1 (C) and Col3a1 (D)
expression in cardiac tissues from WT and NKA1+/- mice treated
with saline or ISO. (E) Representative images of -SMA immunostaining in
different groups (scale bar = 50 µm). (F) Quantitative analysis of
-SMA-positive staining areas in cardiac sections. Data are presented as
means SD; n = 6. **p 0.01, ***p 0.001, for
WT+ISO vs wild-type (WT) group and NKA1+/- + ISO vs
NKA1+/- group; ###p 0.001, vs WT+ISO group.
3.3 NKA1 Haploinsufficiency Increases Macrophage
Accumulation and the Expression of Inflammatory Cytokines in ISO-Challenged
Hearts
Analysis of our pre-proteomics data revealed increased levels of monocyte and
macrophage-related proteins, including CD14, CD68, macrophage-capping protein
(Capg), and Galectin-3 (Fig. 3A). We then further detected immune cell
infiltration in heart tissues. There was a significantly increased macrophage
infiltration in the hearts of ISO-NKA1+/- mice. The increased
macrophages were predominantly localized to fibrotic areas. No significant
changes were observed for neutrophil infiltration. Although the number of T cells
infiltrating the heart is lower than that of macrophages, it also exhibited
significant variations across groups (Fig. 3B–E). Furthermore, NKA1
haploinsufficiency increased the levels of IL-6, TNF-, and
IL-1 in the heart under ISO-induced conditions compared to controls
(Fig. 3F–H).
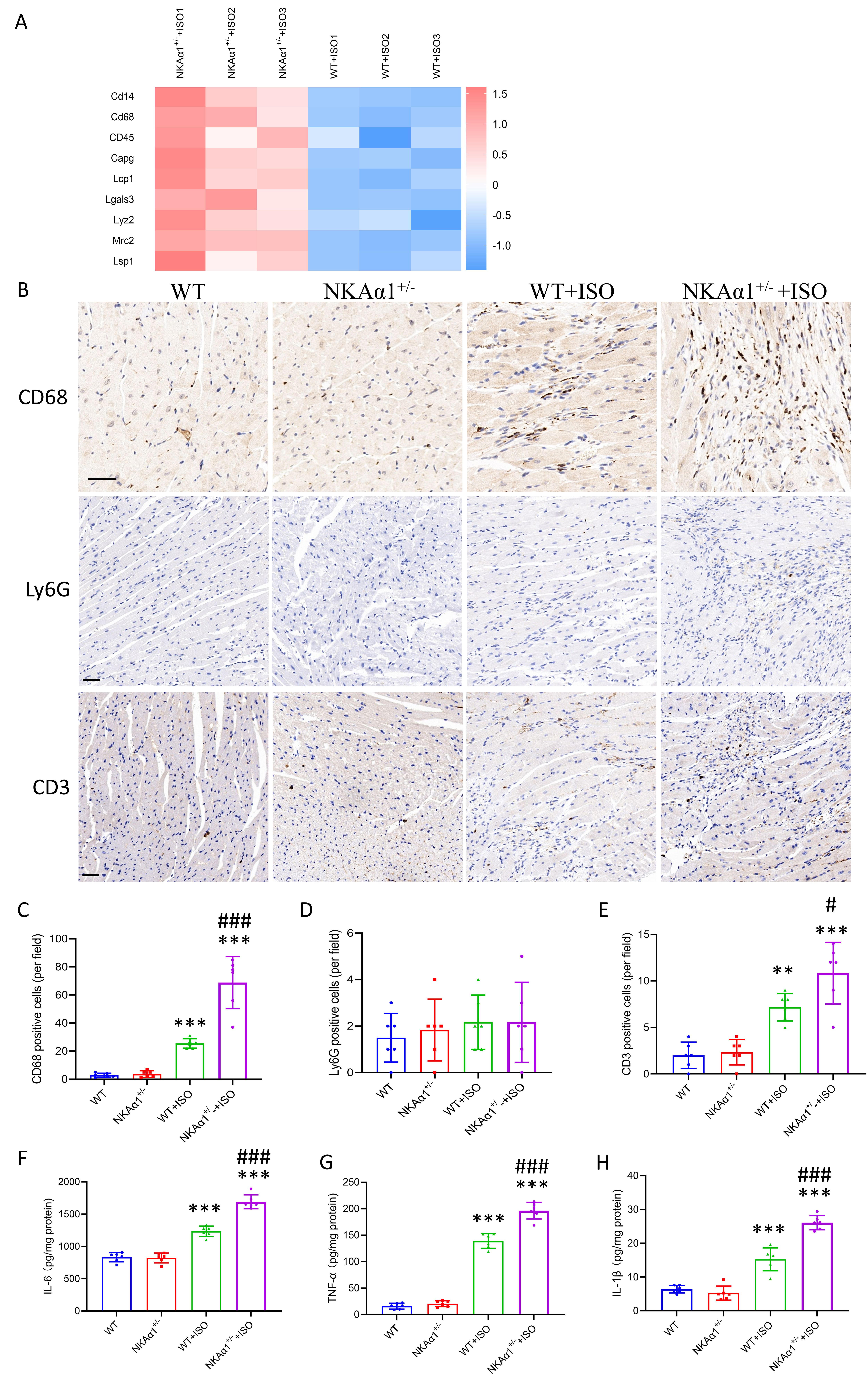 Fig. 3.
Fig. 3.
NKA1 haploinsufficiency promoted macrophage
accumulation and inflammatory factor expression in the ISO-challenged heart. (A)
Enrichment map of proteins related to immune cells. (B) Representative images of
immunostaining for CD68, LY6G, and CD3 in different groups (scale bar = 50
µm). (C) Quantitative analysis of CD68-positive cells in cardiac sections.
(D) Quantitative analysis of Ly6G-positive cells in cardiac sections. (E)
Quantitative analysis of CD3-positive cells in cardiac sections. (F–H) ELISA
results for interleukin-6 (IL-6), tumor necrosis factor-
(TNF-), and IL-1 in cardiac tissues from different groups.
Data are presented as the mean SD; n = 6. **p 0.01,
***p 0.001, for WT+ISO vs wild-type (WT) group and
NKA1+/- + ISO vs NKA1+/- group; #p
0.05, ###p 0.001, vs WT+ISO group.
3.4 Damaged NKA1+/- Cardiomyocytes Enhanced
Macrophage Activation and Fibroblast Differentiation
Since whole-body NKA1 haploid knockout mice were used in this
research, the increased deposition of ECM proteins and macrophage infiltration
might result from the direct effects of ISO on NKA1 haplo-insufficient
fibroblast cells or macrophages. Therefore, we first isolated cardiomyocytes,
fibroblasts, and macrophages from WT and NKA1-deficient heterozygous
mice and treated them with ISO. NKA1 haploinsufficiency had no
significant effect on the activation of macrophages or fibroblasts (Fig. 4A–C),
but exacerbated cardiomyocyte injury, as evidenced by the release of LDH (Fig. 4D). Subsequently, we investigated whether damaged NKA1+/-
cardiomyocytes influenced macrophage activation or fibroblast differentiation.
Macrophages or fibroblasts from WT mice were exposed to the primary cultured
cardiomyocytes isolated from WT or NKA1+/- mice and co-cultured
them in MEM medium supplemented with 10 µM ISO. Co-culture of
cardiomyocytes with macrophages significantly enhanced cytokine secretion by the
latter cells (Fig. 4E,F). Moreover, co-culture of cardiomyocytes with fibroblasts
also increased the expression of -SMA, an indicator of
fibroblast-to-myofibroblast conversion (Fig. 4G). Interestingly, when activated
macrophages were co-cultured with fibroblasts, this interaction also increased
-SMA expression (Fig. 4H).
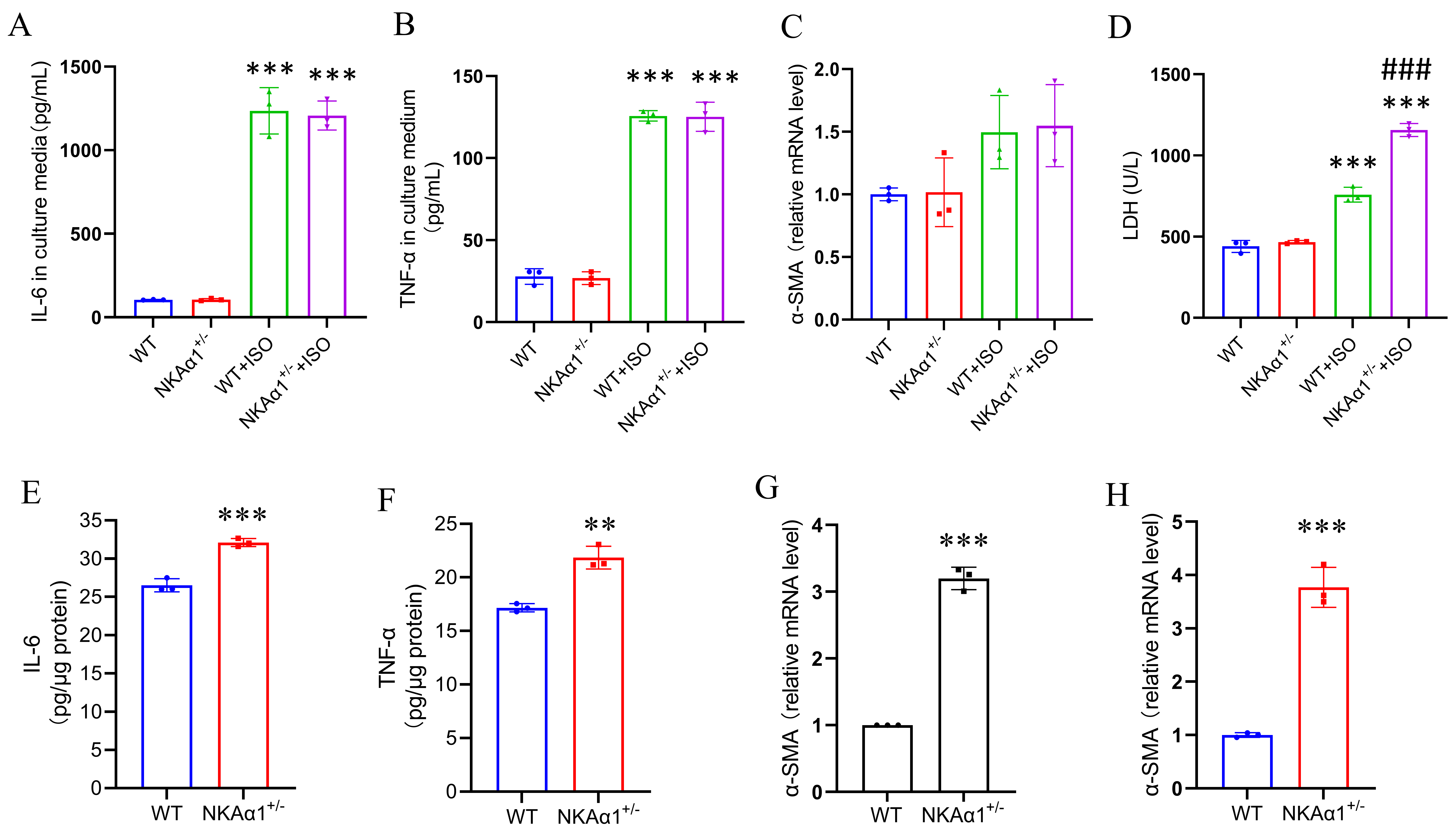 Fig. 4.
Fig. 4.
Impaired NKA1+/- cardiomyocytes enhanced
macrophages activation and fibroblasts differentiation. (A,B) ELISA results for
IL-6 and TNF- in the culture media of macrophages challenged with ISO
or vehicle control. (C) qRT-PCR analysis of -SMA expression in
fibroblasts challenged with ISO or vehicle control. (D) Lactate dehydrogenase
(LDH) activity in the culture media of cardiomyocytes challenged with ISO or
vehicle control. (E,F) ELISA results for IL-6 and TNF- in normal
macrophages co-cultured with different cardiomyocytes under ISO-challenged
conditions. (G) qRT-PCR analysis of -SMA expression in normal
fibroblasts co-cultured with different cardiomyocytes under ISO-challenged
conditions. (H) qRT-PCR analysis of -SMA expression in normal
fibroblasts co-cultured with macrophages activated by co-culturing with different
cardiomyocytes. Data are presented as the mean SEM; n = 3. **p 0.01, ***p 0.001, for: (A–D) WT+ISO vs WT and
NKA1+/-+ISO vs NKA1+/- and (E–H)
NKA1+/- vs WT; ###p 0.001, vs WT+ISO group.
3.5 ERR Participates in NKA1
Haploinsufficiency-Induced Cardiomyocyte Death
Subcellular enrichment analysis of our pre-proteomics results revealed
significant downregulation of mitochondrial proteins (Fig. 5A). Morphological
analysis of mitochondria in heart tissues demonstrated a large number of abnormal
mitochondria in the hearts of ISO-NKA1+/- mice, characterized by
uneven size, disordered arrangement, unclear structure, and disrupted cristae
(Fig. 5B). ERR regulates a large number of genes involved in
mitochondrial function [18]. Reanalysis of our pre-proteomics data revealed that
ERR was downregulated in ISO-challenged NKA1+/- mice
(Fig. 5C). This finding was confirmed by Western blotting (Fig. 5D). SLU-PP-332
is an ERR agonist that has the highest potency for ERR. Treatment with
SLU-PP-332 partially alleviated ISO-induced cell damage in NKA1+/-
cardiomyocytes (Fig. 5E). It has been reported that ISO induces macrophage
infiltration into the heart in an IL-18-dependent manner [19]. ERR
agonists SLU-PP-332 also reduced the release of IL-18 from NKA1+/-
cardiomyocytes under ISO conditions (Fig. 5F).
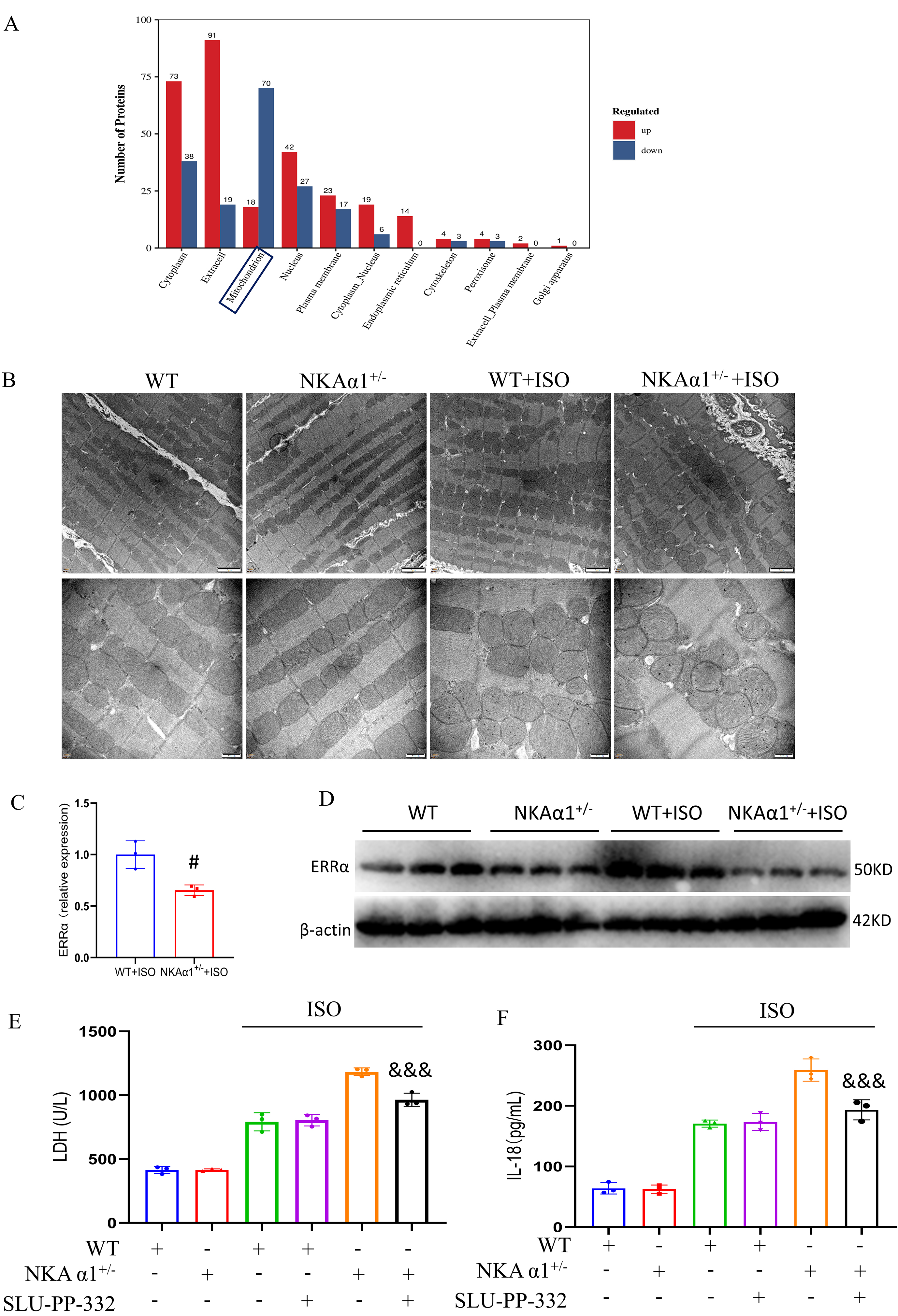 Fig. 5.
Fig. 5.
NKA1 regulated mitochondria-related myocardial cell
injury under ISO-challenged conditions. (A) Subcellular enrichment analysis of
differential proteins from proteomics results. (B) Mitochondrial morphology
observed by transmission electron microscope (upper panel scale bar, 2 µm;
lower panel scale bar, 500 nm). (C) ERR expression level determined
from proteomics results. (D) Western blotting analysis of ERR in
cardiac tissues. (E) LDH content in the culture media of cardiomyocytes treated
under different conditions. (F) IL-18 expression in cardiomyocytes treated under
different conditions. Data are presented as the mean SEM; n = 3.
#p 0.05, vs WT+ISO group; &&&, p 0.001, vs
NKA1+/-+ISO group.
3.6 Treatment With NKA1 DR-Region Antibody Alleviates
Macrophage Infiltration and Heart Fibrosis
The DR region (897DVEDSYGQQWTYEQR911) of NKA1 has been
identified as an activation site for NKA [20]. Our group and others reported that
DR-region-specific antibodies enhance the activity and level of membrane
expression of NKA1 [15, 21]. We subsequently evaluated the effects of
DRm217, a specific DR-region monoclonal antibody, on ISO-treated mice.
Histopathological analyses revealed that DRm217 attenuated heart lesions,
fibrosis, and macrophage accumulation under ISO-challenged conditions (Fig. 6A–D). The levels of inflammatory cytokines, including IL-6, TNF-, and
IL-1, were also downregulated by DRm217 treatment under ISO-insulted
conditions (Fig. 6E–G).
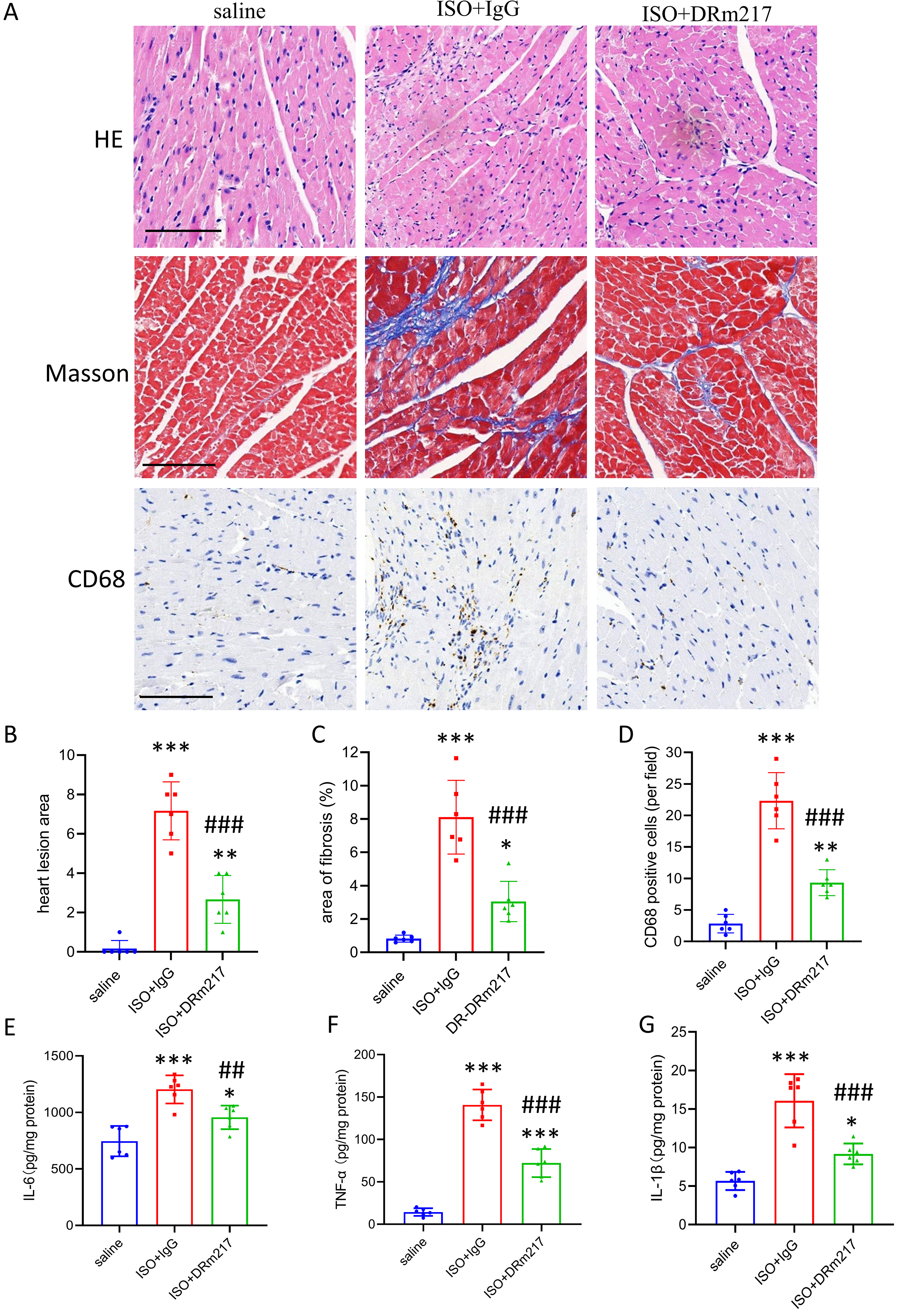 Fig. 6.
Fig. 6.
DRm217 treatment mitigates ISO-induced fibrosis and macrophage
activation under ISO-challenged condition. (A) Representative images of H&E,
Masson staining, and CD68 immunostaining in different groups (scale bar, 100
µm). (B) Quantitative analysis of the myocardial lesion area. (C)
Quantitative analysis of fibrotic (Masson blue) areas in cardiac sections. (D)
Quantitative analysis of CD68-positive cells in cardiac sections. (E–G) ELISA
results for IL-6, TNF-, and IL-1 in cardiac tissues from
different groups. n = 6. Data are presented as the mean SD; *p 0.05, **p 0.01, ***p 0.001, vs saline group;
##p 0.01, ###p 0.001, vs ISO+IgG group.
4. Discussion
NKA is a multi-subunit protein complex contains three essential
components: -catalytic subunit, -regulatory subunit, and
-modulatory subunit. Cardiac tissue expresses abundant NKA,
which plays a critical role in maintaining Na+ and Ca2+ ion gradients,
generating action potentials, regulating cell volume, and promoting cell survival
[22]. Dysregulation or deficiency of NKA has been observed in various heart
diseases, such as acute myocardial infarction, hypertension, and heart failure
[12, 23, 24], indicating its critical contribution to the advancement of
cardiovascular pathologies. NKA is closely associated with fibrosis. Cardiotonic
steroids (CTS), a class of NKA ligands and inhibitors, have been reported to
stimulate fibroblast collagen production and induce heart or kidney fibrosis [13, 25]. Activation of the NKA/Src signaling pathway can initiate miR-29b-3p
dysregulation and cardiac fibrogenesis [26]. Furthermore, knockdown of the
NKA1 subunit in A549 cells resulted in elevated expression of
profibrotic factors [27]. However, the relationship between inflammation and
NKA-related tissue fibrosis is still unclear. In the present study, we
demonstrated that NKA1 haploinsufficiency exacerbates ISO-induced
cardiac lesions, fibrosis, and inflammation. Ventricular tissue from heart
failure and dilated cardiomyopathy patients exhibits reduced expression of
NKA1 subunits [12, 28]. There have also been reports of elevated plasma
catecholamine levels in patients with heart failure [29]. Perhaps the interaction
between these two factors may promote heart fibrosis and dysfunction. The current
study also found that DRm217, a proven activator of NKA, prevents ISO-induced
cardiac lesions, fibrosis, and inflammation under ISO-challenged conditions.
These results suggest that regulation of NKA can influence heart fibrosis.
Inflammation serves as a critical driver of maladaptive cardiac remodeling
following heart injury [30]. The molecular and cellular mechanisms that underlies
inflammatory responses vary across different cardiac diseases [31]. In various
models of cardiac disease, heterogeneous inflammatory cells infiltrate to the
heart. For example, significant infiltration of neutrophils and inflammatory
monocytes occurs in infarcted myocardium [32]. The present study revealed that
macrophages are the most abundant infiltrating cell type in the myocardium upon
ISO stimulation, consistent with a prior report [20]. NKA1 exacerbated
macrophage infiltration and the expression of inflammatory factors in mouse
hearts. Notably, infiltrated macrophages and activated myofibroblasts accumulated
mostly in the cardiac lesion area, as evidenced by the results of CD68 and
-SMA immunohistochemical staining. This prompted us to investigate
whether the activation of macrophages and fibroblasts was a direct consequence of
NKA1 deficiency under ISO-treated conditions. We treated macrophages
and fibroblasts isolated from WT and NKA1+/- mice with ISO. Our
findings indicated that NKA1 deficiency does not directly activate
macrophages or fibroblasts under ISO conditions. However, NKA1
deficiency accelerated cardiomyocyte death in response to ISO insult.
Cell-to-cell communication represents a fundamental characteristic of adult
complex organs. Interactions between different cardiac cell types play a crucial
role in cardiac fibrotic pathogenesis [33]. Co-culture of NKA1+/- cardiomyocytes with macrophages increased the secretion of cytokines by the
latter cells. Furthermore, the activated macrophages promoted differentiation of
fibroblasts. Cells exchange information using chemical, electrical, and
mechanical signals, which travel either via direct contact or through the
secretion of local mediators like cytokines and growth factors [34]. Through
intercellular cross-talk, it appears that damaged NKA1+/-
cardiomyocytes can induce harmful inflammatory responses and thus exacerbate
cardiac fibrosis.
Previous studies have demonstrated that suppression of NKA1 increases
the susceptibility of cells to stress-induced death [35, 36]. This observation
was further substantiated by the experimental results of the present study. Our
proteomics analysis revealed that NKA1 deficiency under ISO-challenged
conditions significantly reduced the expression of mitochondrial proteins.
Ultrastructural analysis of mitochondrial morphology further confirmed that
NKA1 deficiency markedly accelerated mitochondrial injury in myocardial
cells under stress conditions. ERR is a well-established transcription
factor that plays a pivotal role in regulating cellular metabolism and
mitochondrial function [37]. Our proteomics and Western blot results showed that
ERR was downregulated in ISO-challenged NKA1+/- mice.
Furthermore, cellular experiments showed that ERR agonists partially
alleviated ISO-induced cell damage in NKA1+/- cardiomyocytes. It
was previously reported that ISO-induced macrophage activation in the heart
occurs in an IL-18-dependent manner [20]. ERR agonists also reduced
IL-18 expression in ISO-insulted NKA1+/- cardiomyocytes. These
results suggest that low ERR expression contributes to the increased
susceptibility of NKA1+/- cardiomyocytes to ISO-induced damage, as
well as to the increased release of inflammatory factors. Pan Z et al.
[38] previously showed that the EGFR/Src pathway regulates ERR
expression. Other reports also indicate that microRNAs, such as miR-135a and
miR-137, can modulate ERR expression [39, 40]. Whether NKA signaling
pathways are linked to ERR expression warrants further investigation.
Immune cell infiltration is a significant driver of cardiac fibrosis. Diverse
immune populations, including macrophages, T lymphocytes, monocytes, and
neutrophils, infiltrate cardiac tissue and participate in injury repair and
structural remodeling processes [41]. Concurrently, mitochondrial dysfunction,
manifested through impaired metabolism, reduced energy production, elevated
mitochondrial reactive oxygen species (mtROS), and disrupted calcium homeostasis,
also plays a pivotal role in cardiac pathogenesis [42, 43]. Emerging evidence
strongly supports an interconnection between immune dysregulation and
mitochondrial impairment [44]. On one hand, leaked mitochondrial components, such
as mtDNA or mtRNA, can activate inflammatory pathways like cGAS-STING and TLR2/4
[45]. Furthermore, abnormal metabolites and an altered myocardial
microenvironment (e.g., lactate accumulation and decreased pH) resulting from
mitochondrial dysfunction can drive metabolic reprogramming in immune cells such
as macrophages and T lymphocytes [46]. This reprogramming promotes the secretion
of pro-inflammatory cytokines, such as IL-6, IL-1, and TNF-,
thereby triggering inflammatory cascades. On the other hand, inflammatory
cytokines like TNF- or IL-1 facilitate mitochondrial
permeability transition pore (mPTP) opening, induce mtDNA damage, and disrupt
energy metabolism [47]. Although therapeutic strategies targeting inflammatory
mediators show promise for cardiac remodeling, clinical trials of specific
chemokines/cytokines have achieved limited success [48]. Interventions directly
targeting mitochondria are also increasingly recognized as compelling therapeutic
strategies for heart disease [49]. In our study, ISO-challenged mice exhibited
concurrent mitochondrial impairment and heightened inflammation, effects
exacerbated by NKA1 deficiency. Given the interplay between
mitochondrial damage and inflammatory responses, dual-targeted therapies may
synergistically prevent myocardial fibrosis.
Postmortem analyses of human failing hearts show ~40%
reductions in NKA expression/activity [12]. We previously demonstrated that
NKA1 deficiency under ISO stimulation enhances glycolysis while
suppressing TCA cycle activity and oxidative phosphorylation (OXPHOS) [14]. Here,
we observed mitochondrial swelling, ultrastructural abnormalities, and increased
macrophage infiltration in ISO-treated NKA1+/- hearts. We highly
suspect that injured NKA1+/- cardiomyocytes release mitochondrial
components and metabolites that activate infiltrating immune cells. Li et
al. [44] linked mitochondrial genes (FBXO7, PGS1) to immune infiltration in
septic cardiomyopathy. Our work identifies ERR as a critical regulator
of NKA1+/- cardiomyocyte injury and inflammatory factor release,
suggesting its potential role in immune cell recruitment during ISO-induced
cardiomyopathy in NKA deficiency condition. DRm217-mediated NKA activation
protected against ISO-induced cardiac lesions, fibrosis, and inflammation. Some
studies suggest that enhanced protein degradation serves as the primary mechanism
underlying NKA1 reduction [50, 51]. Therefore, inhibiting NKA
degradation represents another promising therapeutic approach for preventing
cardiac fibrosis.
However, our study has certain limitations. First, we used whole-body
NKA1 haploid knockout mice in vivo; these findings should be
further validated using cardiomyocyte-specific NKA1+/- haploid
knockout mice to eliminate interference from other factors. Second, although
reduced ERR expression correlated with ISO-induced
NKA1+/- cardiomyocytes injury, the mechanism linking NKA
deficiency to ERR downregulation and the precise role of in
ERR mitochondrial damage require further investigation. Third, it
remains to be determined whether macrophage cytokine secretion is triggered by
mitochondrial debris or metabolic byproducts from injured NKA1+/-
cardiomyocytes.







 , Tao Liu 1,2
, Tao Liu 1,2