
 , Rongrong Wang 1,2,†, Qiyuan He 1,2, Yicheng Ma 3, Yanyuan Fang 1,2, Jin Yang 1,2, Hui Zhao 1,2, Lin Fu 1,2,*
, Rongrong Wang 1,2,†, Qiyuan He 1,2, Yicheng Ma 3, Yanyuan Fang 1,2, Jin Yang 1,2, Hui Zhao 1,2, Lin Fu 1,2,* , Danlei Chen 1,2,*
, Danlei Chen 1,2,*1 Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Anhui Medical University, 230032 Hefei, Anhui, China
2 Institute of Respiratory Diseases, The Second Affiliated Hospital of Anhui Medical University, 230032 Hefei, Anhui, China
3 Thoracic Surgery Department, The Second Affiliated Hospital of Anhui Medical University, 230601 Hefei, Anhui, China
†These authors contributed equally.
Abstract
Pulmonary fibrosis is a life-threatening progressive lung disease characterized by increased fibrogenesis and decreased lung function. Pulmonary fibrosis has a poor prognosis and a low patient survival rate, with no effective treatments currently available. Cellular senescence is thought to contribute to the pathogenesis of this aging-related disease. Cellular senescence and premature aging are involved in the development of pulmonary fibrosis, which affects various cellular processes such as proliferation, apoptosis, and inflammatory responses. Multiple pathways contribute to cellular senescence and participate in the pathogenesis of pulmonary fibrosis, such as tumor protein p53 (p53)/cyclin dependent kinase inhibitor 1A (p21) and cyclin dependent kinase inhibitor 2A (p16)/retinoblastoma protein (pRB). However, many unanswered questions remain concerning the relationship between cellular senescence and pulmonary fibrosis. In this review, we first summarize the common causes of lung cell senescence and pulmonary fibrosis, including aging, inflammation, chemotherapy drugs, and environmental pollutants. We also discuss the enriched signaling pathways and epigenetic factors (e.g., non-coding RNAs) that are important during cell senescence and the progression of pulmonary fibrosis. Finally, we discuss current strategies for treating pulmonary fibrosis by targeting cellular senescence, including relevant preclinical and clinical studies. This review provides new mechanistic insights for understanding the role of cellular senescence in the development of pulmonary fibrosis and its treatment by targeting cellular senescence.
Keywords
- cellular senescence
- pulmonary fibrosis
- signaling pathways
- non-coding RNAs
- senolytics
Pulmonary fibrosis is a severe respiratory disease characterized by the proliferation and fibrosis of lung interstitium. It has a complex etiology and there is currently a lack of effective treatment. Idiopathic pulmonary fibrosis (IPF) is a prevalent type of pulmonary fibrosis and is typically diagnosed in individuals aged 50 to 70 years [1, 2]. In recent years, cellular senescence has gradually attracted the attention of researchers investigating pulmonary fibrosis. Cellular senescence refers to a state of permanent growth arrest that cells enter after experiencing various internal and external stresses [3]. This is accompanied by changes in multiple signaling pathways within the cell that can potentially accelerate the progression of pulmonary fibrosis by enhancing inflammation and altering the cellular environment.
The accumulation of senescent cells in lung tissue can harm alveolar epithelial cells (AECs) and fibroblasts, thereby accelerating the development of fibrosis. The proportion of senescent cells in the lung tissue of patients with pulmonary fibrosis significantly elevated, and these cells exacerbate fibrosis by releasing various pro-inflammatory factors and components of the extracellular matrix (ECM) [4]. Furthermore, cellular senescence interacts with various pathological processes such as oxidative stress, inflammatory responses, and apoptosis to form a dense signaling network that complicates treatment [5]. Interventions that target cellular senescence have become an important research area in the treatment of pulmonary fibrosis. By targeting senescent cells or the pro-inflammatory factors they secrete, it is hoped the progression of pulmonary fibrosis can be slowed or reversed [6]. Current study found that lung epithelial cells and fibroblasts underwent senescence in pulmonary fibrosis, and removal of senescent cells can inhibit pulmonary fibrosis [6]. Recent clinical trials with senolytics (dasatinib and quercetin) have demonstrated the feasibility and tolerance in patients with IPF [7]. However, the role and mechanisms of cellular senescence in pulmonary fibrosis are not yet fully understood. Previous reviews have described different types of senescent cells in pulmonary fibrosis as well as the signaling pathways involved in senescence [8, 9]. In this review, we present a comprehensive overview of the manifestations and cell-specificity of senescence in pulmonary fibrosis. We also address in-depth the causes of cellular senescence, along with the molecular mechanisms of cellular senescence in pulmonary fibrosis. Finally, we provide a detailed account of preclinical and clinical studies targeting senescence in pulmonary fibrosis. We summarize for the first time the role of cell senescence induced by environmental pollutants (e.g., Benzo[a]pyrene, 1-nitropyrene, particulate matter 2.5) in pulmonary fibrosis, and discuss epigenetic factors (i.e., non-coding RNAs) during cell senescence and the progression of this disease. In addition to senolytic therapies that eliminate senescent cells and have been discussed in previous clinical studies and review articles, we discuss more recent preclinical drugs (tetrandrine, hydrogen sulfide, integrated stress response inhibitor, salvianolic acid B, roxithromycin) that target cellular senescence in the treatment of pulmonary fibrosis. Furthermore, we discuss the interaction between cellular senescence and pulmonary fibrosis. Altogether, this review provides an update on cellular senescence in pulmonary fibrosis, which is different from previous publications on this topic.
Cellular senescence is a state of cell cycle arrest, mediated
by the tumor protein p53 (p53)/cyclin dependent kinase
inhibitor 1A (p21) and cyclin dependent kinase inhibitor 2A (p16)/retinoblastoma
protein (pRB) pathways, that exhibits a senescence-associated secretory phenotype
(SASP) [10, 11]. Increased levels of senescence biomarkers are found in lung
cells and tissues taken from patients with IPF and from animal models of the
pulmonary fibrosis, including p53, p21, p16, SA-
Changes in the cells within lung tissue are an important feature of the pathological process of pulmonary fibrosis. Specifically, damage and senescence of AECs causes epithelial-mesenchymal transition (EMT), which activates and promotes the proliferation of fibroblasts [14]. Alveolar type II (AT II) epithelial cells undergo apoptosis and senescence early in pulmonary fibrosis, resulting in abnormal fibroblast growth and excessive collagen buildup [14].
IPF fibroblasts have been found to undergo senescence, leading
to reduced apoptosis and the formation of SASP [15]. Further study indicates that
IPF fibroblasts exhibit a senescent phenotype, characterized by elevated levels
of
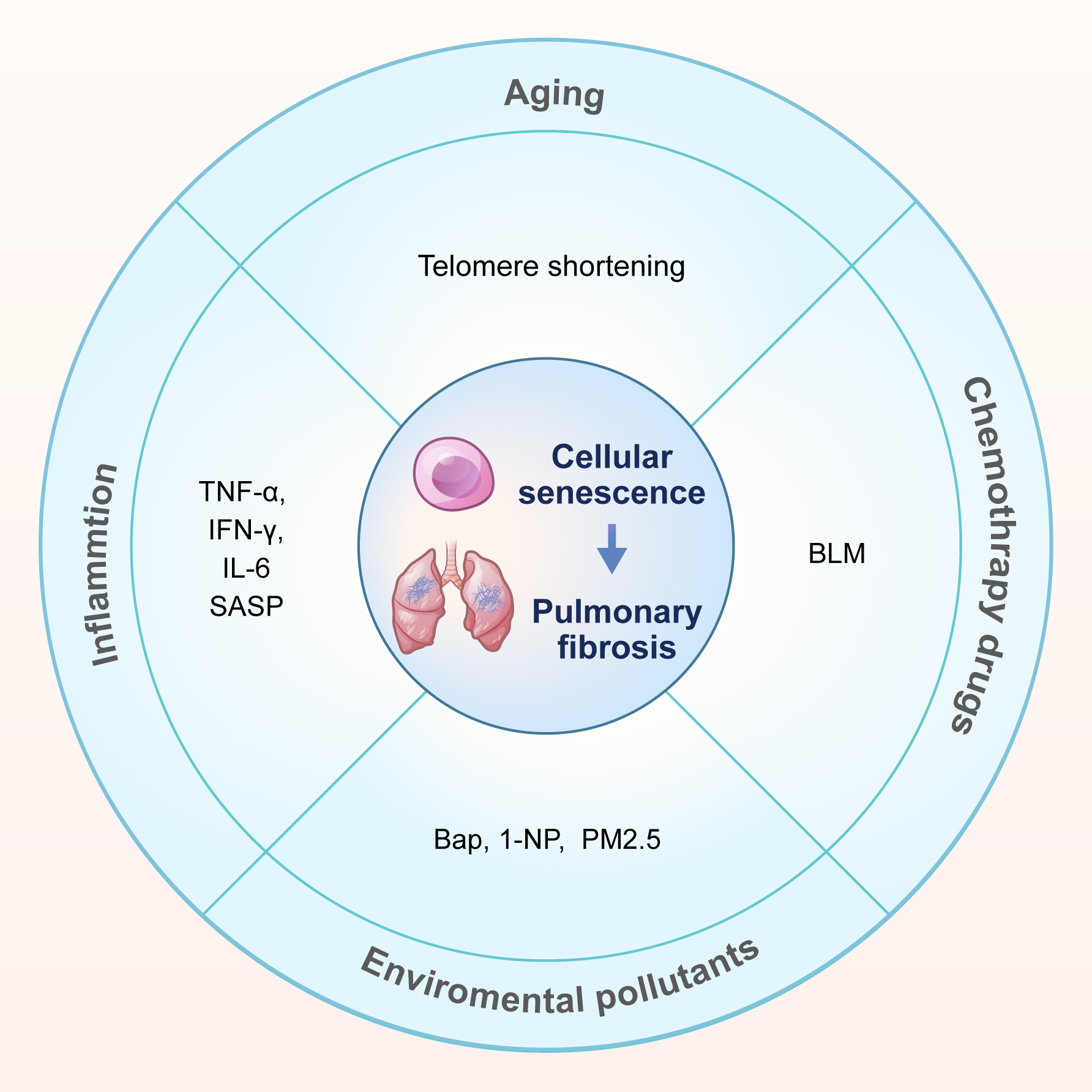
Multiple risk factors contribute to lung cell senescence during the progression of pulmonary fibrosis. This summary focuses on four key factors: aging, inflammation, chemotherapy drugs, and environmental pollutants (Fig. 1).
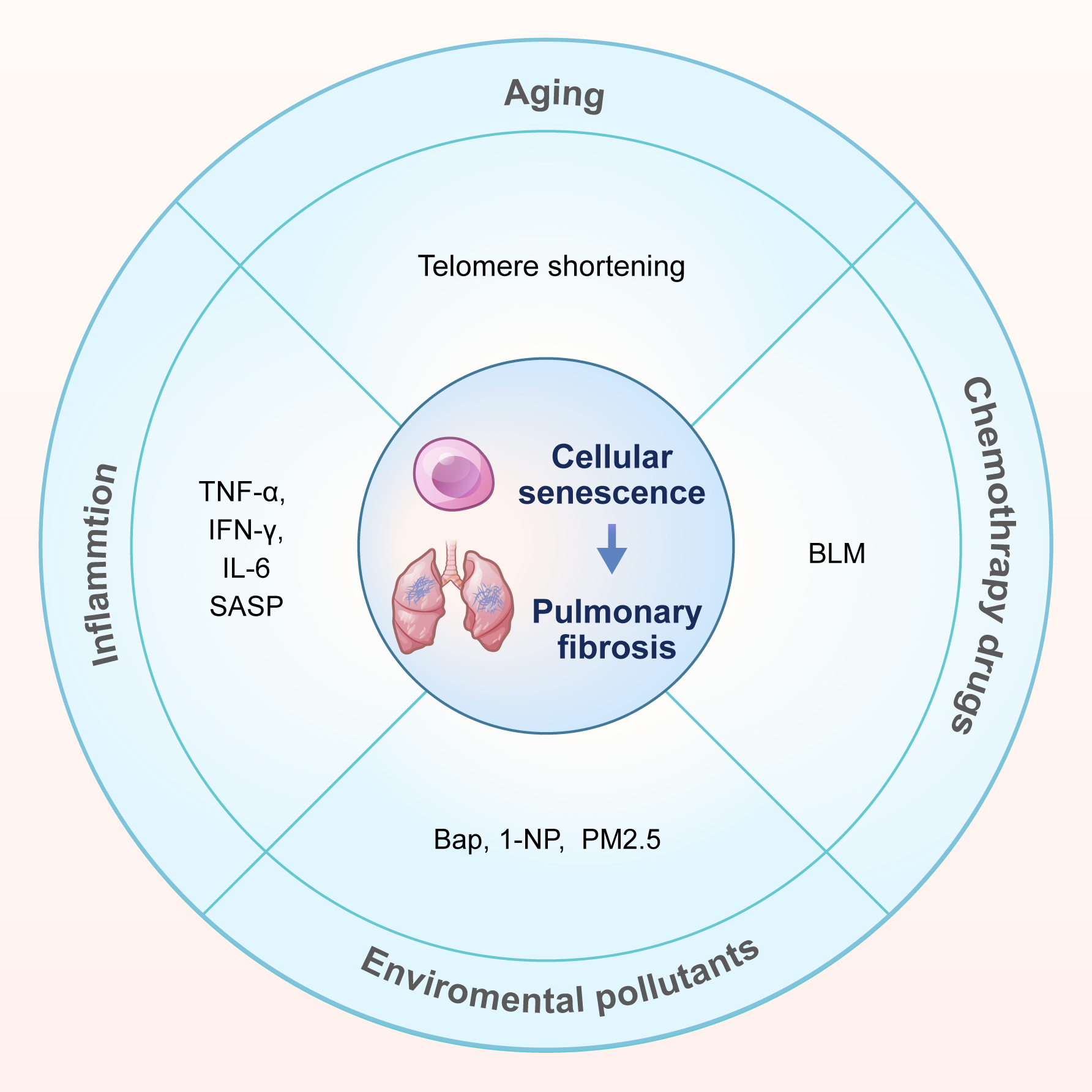 Fig. 1.
Fig. 1.
Causes of cellular senescence in pulmonary fibrosis. This review summarizes the causes of cellular senescence in pulmonary fibrosis into four categories: aging, inflammation, chemotherapy drugs (bleomycin, BLM) and environmental pollutants. Environmental pollutants include 3 types: benzo[a]pyrene (Bap), 1-nitropyrene (1-NP), and fine particulate matter (PM2.5). SASP, senescence-associated secretory phenotype.
IPF often manifests in middle-aged and elderly people, but is
rarely found in people aged
Chronic inflammation contributes to the process of cellular
senescence. The canonical inflammatory cytokines TNF-
SASP also mediates inflammatory responses that are critical for the initiation and maintenance of senescence. Abnormal accumulation of senescent cells can lead to the secretion of SASP and generate an inflammatory environment, resulting in chronic tissue damage and disease [31]. Cross-talk occurs between the canonical inflammatory response and SASP inflammation. SASP contributes to both acute and chronic inflammation, as well as playing a role in regulating immune responses [32].
Bleomycin is a DNA strand-breaking agent that induces biological effects such as DNA damage and cell senescence [33], with pulmonary toxicity being the most serious side-effect. Bleomycin induces remodeling of lung structure, epithelial cell damage with reactive hyperplasia, EMT, and fibroblast activation to promote collagen deposition [34]. Intratracheal instillation of bleomycin induces central fibrotic changes in the bronchioles, acute interstitial and interalveolar inflammation, as well as macrophage activation [35]. Consequently, Bleomycin is widely utilized to induce pulmonary fibrosis in various animal models.
Environmental pollution is a significant concern and there is increasing research to study its effects on respiratory diseases. For many years, our research group has studied the effects of exposure to environmental pollutants on respiratory diseases, including pulmonary fibrosis. Our previous study demonstrated that certain environmental pollutants can cause senescence of lung epithelial cells. In particular, benzo[a]pyrene (Bap) [36] and 1-nitropyrene (1-NP) [37, 38] have been implicated in the development of pulmonary fibrosis.
Bap is a human carcinogen found in large quantities in tobacco smoke. It is also a toxic environmental pollutant that is highly tumorigenic in mice [39, 40]. Benzo(a)pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE), a metabolic product of Bap, is also a toxic environmental pollutant. We recently explored the potential mechanism of BPDE exposure-induced pulmonary fibrosis. The aryl hydrocarbon receptor (AhR) was activated in the AECs of mice treated with BPDE, allowing it to bind to the promoter of the Nrf2 gene. AhR subsequently promoted BaP-induced AEC senescence and EMT, thereby contributing to pulmonary fibrosis by mediating Nrf2-p62 signaling [36].
1-NP is a significant component of fine particulate matter found in motor vehicle exhaust. It is a mutagenic and carcinogenic pollutant commonly found in the environment. 1-NP enters the body through the respiratory tract, with the lungs being a direct target [41]. In 2021, our research group reported that endoplasmic reticulum stress induced by reactive oxygen species plays a role in the EMT and pulmonary fibrosis caused by 1-NP [37]. Our 2024 study revealed that 1-NP induces mitochondrial reactive oxygen species (mtROS) overproduction, which subsequently triggers small ubiquitin-like modifier (SUMO)ylation of ALKBH5 and m6A methylation of FBXW7 mRNA, ultimately driving cellular senescence in pulmonary epithelium [38].
Particulate matter 2.5 (PM2.5) is a known risk factor for pulmonary fibrosis, causing inflammation and fibrosis in AT II epithelial cells [42]. Exposure to PM2.5 increases the expression of senescence markers (p16, p21, and p27) [43], leading to mitochondrial dysfunction in AT II epithelial cells and accelerating the progression of pulmonary fibrosis [44].
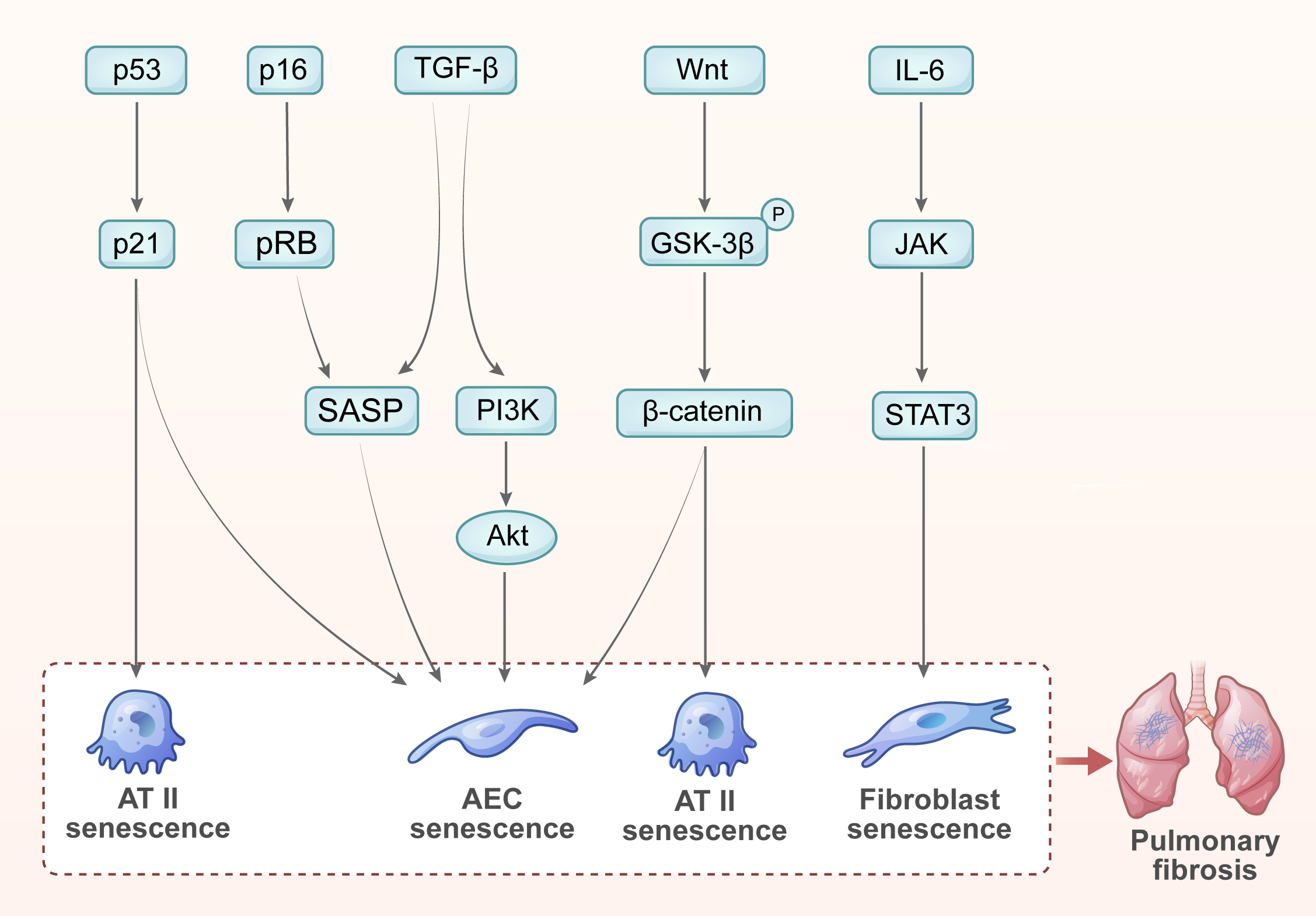
Cellular senescence significantly affects the repair processes of lung tissue and is closely associated with various signaling pathways. Here, we summarize the key signaling pathways that regulate the senescence of different cell types involved in pulmonary fibrosis (Fig. 2).
 Fig. 2.
Fig. 2.
Enriched signaling pathways in cellular senescence and pulmonary
fibrosis. (1) The p53/p21 signaling pathway promotes AT II cell senescence,
leading to pulmonary fibrosis. (2) The p16/pRB signaling pathway promotes AEC
senescence, leading to pulmonary fibrosis. (3) TGF-
p53 mediates senescence by regulating the target gene p21, thus preventing cell proliferation [45]. Studies indicate that p53 activation promotes senescence markers and affects the SASP of senescent cells, thereby affecting the physiological state of nearby cells and contributing to the functional decline of tissue [46]. Activation of p53 promotes the senescence of AECs, which in turn accelerates pulmonary fibrosis. Using a model of pulmonary fibrosis, researchers found that p53 activation strongly associates with senescence in AT II cells, while p53 inhibition significantly mitigates fibrosis progression [47]. Consequently, p53 is both a regulator of cellular senescence and an important biomarker for pulmonary fibrosis.
The cell cycle inhibitor p21 is involved in cellular senescence and plays a critical role in the repair process after lung injury. Lv et al. [48] reported that p21 expression in AT II epithelial cells increased in a time-dependent manner in bleomycin-induced pulmonary fibrosis. Moreover, repeated damage led to telomere shortening and triggered p21-dependent cell senescence. Knockout of p21 can restore alveolar regeneration mediated by AT II epithelial cells in mice with chronic pulmonary fibrosis, suggesting a potential strategy for the treatment of pulmonary fibrosis [48].
p16INK4a (CDKN2A) serves as a critical cell cycle suppressor and is closely linked to cell senescence, which inhibits cell proliferation by activating the pRB, resulting in a senescent cell phenotype [49].
The expression of p16 is significantly increased in pulmonary fibrosis and correlates with the severity of fibrosis. Studies have shown that p16-positive cells accumulate in the lungs, causing cell cycle arrest and a senescence phenotype that impairs lung repair and regeneration [50]. p16 exacerbates inflammatory responses by promoting SASP. The senescent cells release pro-inflammatory factors that influence both the local microenvironment and distant lung tissues through the circulatory system, thus accelerating the progression of fibrosis [50]. Therefore, p16 is both a biomarker and a potential therapeutic target for halting its progression [51].
TGF (transforming growth factor)-
TGF-
The Wnt/
Cytokines and chemokines play important roles in the occurrence and development of pulmonary fibrosis [59]. They promote the activation of inflammatory cells and the proliferation of fibroblasts by activating inflammatory signaling pathways [60]. IL-6 was found to promote inflammation and fibrosis through the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) signaling pathway, while hesperidin reduces fibroblast senescence in vitro by inhibiting the IL-6/JAK/STAT3 pathway [61].
In summary, the pathogenesis of pulmonary fibrosis involves abnormal regulation of multiple signaling pathways that interact with and influence each other, ultimately leading to the senescence of lung cells and the development of pulmonary fibrosis (Fig. 2). Therefore, further in-depth research on the role of these signaling pathways is crucial for revealing the pathophysiological process of pulmonary fibrosis and finding new therapeutic targets.
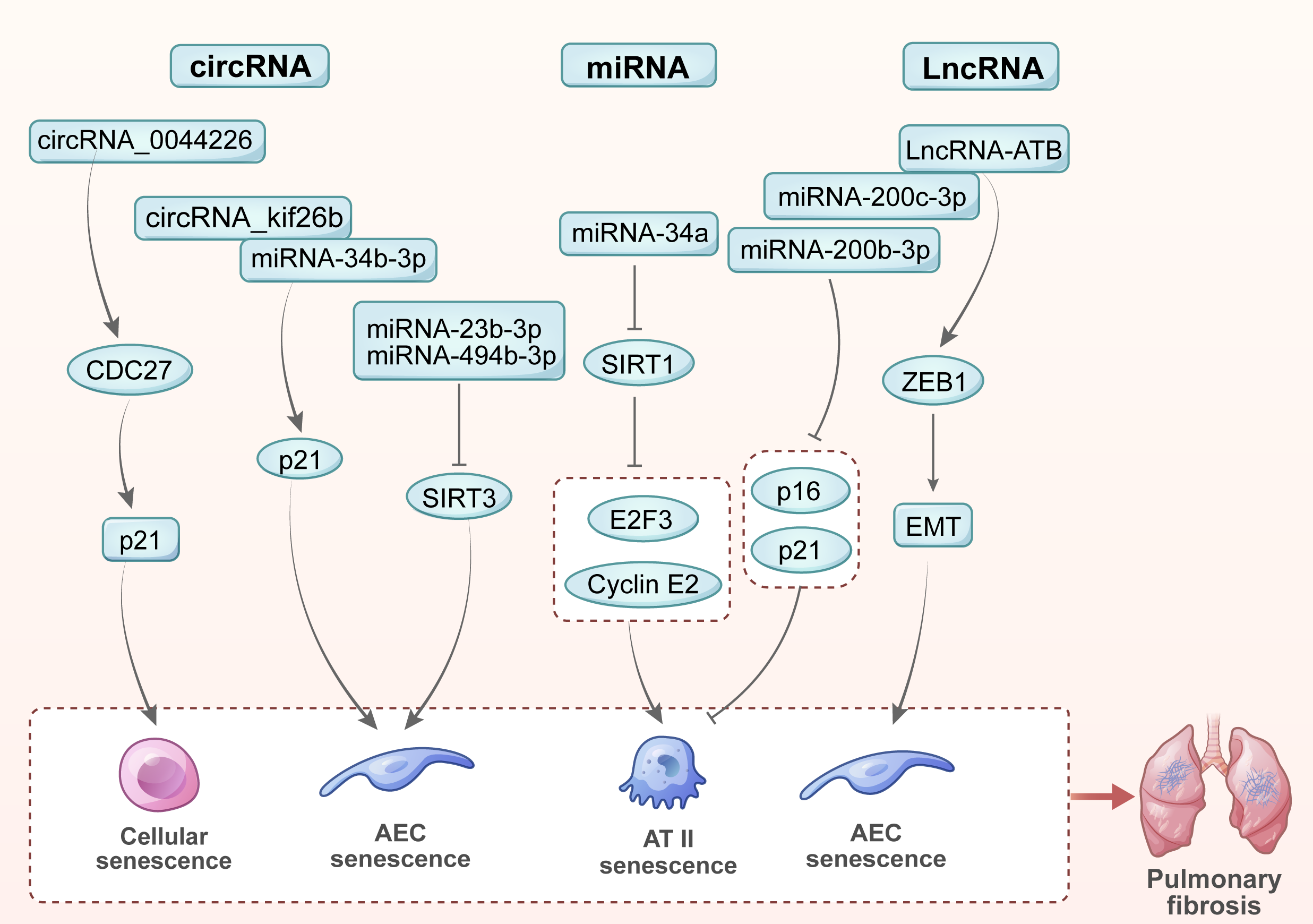
Various noncoding RNAs play important roles in cellular senescence and the development of pulmonary fibrosis including microRNAs (miRNAs), long noncoding RNAs (LncRNAs) and circular RNAs (circRNAs) [62]. In this review, we summarized the key noncoding RNAs involved in regulating cellular senescence and the progression of pulmonary fibrosis (Fig. 3).
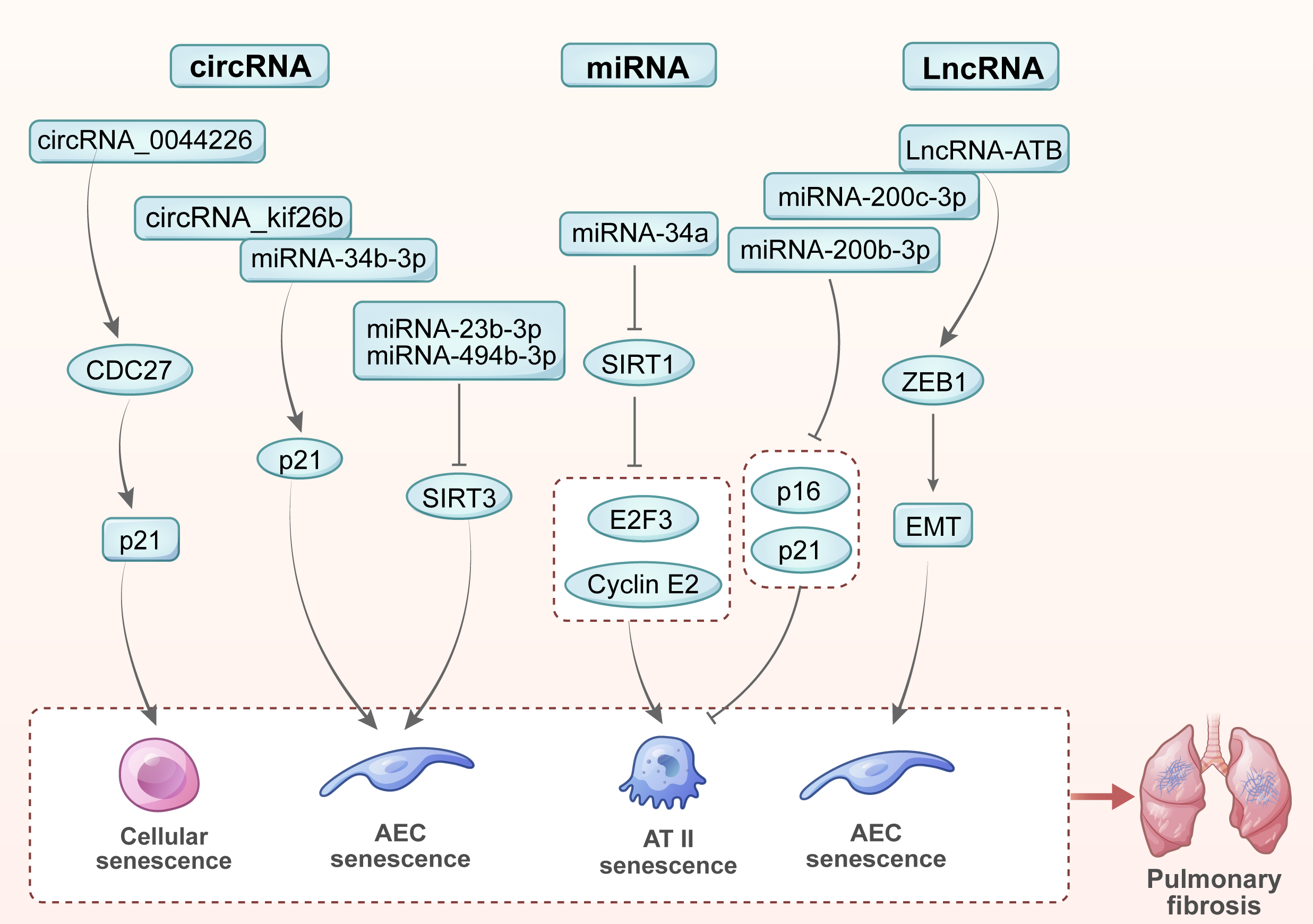 Fig. 3.
Fig. 3.
Noncoding RNAs in cellular senescence and pulmonary fibrosis. (1) miR-23b-3p and miR-494-3p promote cellular senescence and accelerate pulmonary fibrosis by inhibiting SIRT3. (2) miR-34a targets SIRT1 and two key cell cycle regulatory factors, E2F3 and cyclin E2, thereby inducing AT II epithelial cell senescence and promoting pulmonary fibrosis. (3) miR-200b-3p and miR-200c-3p hinder the process of cellular senescence and pulmonary fibrosis by inhibiting the expression of senescence markers (p16 and p21) in AT II epithelial cells. (4) LncRNA-ATB promotes EMT of lung epithelial cells by binding to miR-200c-3p and releasing ZEB1, thereby accelerating cellular senescence and the progression of pulmonary fibrosis. (5) circRNA_0044226 increases the expression of CDC27, which targets p21 to promote cellular senescence and pulmonary fibrosis. (6) By combining miR-346-3p, circRNA_kif26b jointly regulates the target gene p21 of miR-346-3p, thereby mediating the senescence of AECs and promoting the development of pulmonary fibrosis. EMT, epithelial-mesenchymal transition.
miR-23b-3p and miR-494-3p promote cellular senescence by inhibiting SIRT3. Higher levels of miR-23b-3p and miR-494-3p in extracellular vesicles derived from lung fibroblasts of IPF patients were found to correlate with increased disease severity. The vesicles deliver pathogenic substances to lung epithelial cells via paracrine signaling, inducing their senescence and speeding up the progression of pulmonary fibrosis [63].
The miR-34 family consists of three members: miR-34a is encoded on chromosome 1, while miR-34b and miR-34c are encoded on chromosome 11. The relative levels of these miRNAs were found to be significantly elevated in the AT II epithelial cells of IPF patients, suggesting they regulate senescence [64]. Further study revealed that miR-34a targets SIRT1 and two key cell cycle regulators, E2F3 and cyclin E2, in AT II epithelial cells [65].
AT II epithelial cells from IPF patients display a senescent phenotype characterized by the p16 and p21 markers [65]. Additionally, transfection with miR-200b-3p and miR-200c-3p resulted in lower expression of these senescence markers. miR-200b-3p and miR-200c-3p reduce cellular senescence by restoring the ability of AT II epithelial cells to trans-differentiate into AT I epithelial cells in vitro [66]. The effects of miR-200b-3p and miR-200c-3p on reducing cellular senescence may offer a novel approach for future anti-fibrosis treatment.
LncRNA-ATB promotes EMT in lung epithelial cells by
interacting with miR-34c-3p, thus facilitating fibrosis [67]. EMT was found to
play a critical role in a silica-induced pulmonary fibrosis model. Silica
triggers macrophages to secrete TGF-
circRNA_kif26b is homologous in humans, rats and mice. Its expression is increased in mouse lung epithelial cell line 12 (MLE12) cells treated with polystyrene microplastic (PS-MP). Moreover, circRNA_kif26b binds to miR-346-3p to jointly regulate the target gene p21. Knockdown of circRNA_kif26b attenuates PS-MP-induced AEC senescence and the secretion of SASP cytokines [68].
The level of circRNA-0044226 in the lung tissue of IPF patients was found to be significantly elevated compared to that of healthy individuals [69]. In bleomycin-induced mouse model of pulmonary fibrosis, circRNA_0044226 knockout decreased the expression of CDC27, thereby inhibiting EMT and slowing the progression of pulmonary fibrosis [70]. CDC27 primarily regulates cell cycle transitions during cell division. Also, CDC27 regulates cell senescence by targeting CDKN1A (p21: cyclin-dependent kinase inhibitor).
In summary, non-coding RNAs play crucial roles in senescence and pulmonary fibrosis by regulating the expression of specific target genes. These discoveries improve our understanding of the disease mechanisms behind pulmonary fibrosis and offer new treatment options.
Senescent AECs contribute to pulmonary fibrosis by secreting SASP. These
secretions contain pro-inflammatory cytokines and growth factors that also
activate nearby fibroblasts, leading to their transformation into myofibroblasts,
and hence the accumulation of ECM [71]. In addition, the accumulation of
senescent cells can trigger chronic local inflammation, thereby impairing lung
function and accelerating the development of fibrosis [9]. Senescent AECs promote
pulmonary fibrosis by activating the TGF-
Pulmonary fibrosis can also trigger cellular senescence. Oxidative stress and inflammation in pulmonary fibrosis can speed up the senescence of AECs [73]. For example, markers of cellular senescence are significantly increased in the lung tissue of patients with IPF patients and correlate with the severity of fibrosis. Additionally, pulmonary fibrosis causes cell cycle arrest and senescence by activating the stress response [47]. In this context, senescent cells not only lose their normal ability to regenerate, but may also exacerbate the pathological process by secreting factors that promote fibrosis.
The bidirectional relationship between pulmonary fibrosis and cellular senescence highlights the need to consider the effects of cellular senescence on treatment strategies for pulmonary fibrosis. The targeting of senescent cells offers new treatment options for pulmonary fibrosis.
Numerous studies on cellular senescence and pulmonary fibrosis have shown that clearance of senescent cells is critical to the pathogenesis of pulmonary fibrosis and to the improvement of lung function. Here, we summarized the current drugs tested in preclinical studies and clinical study of pulmonary fibrosis (Table 1, Ref. [7, 12, 73, 74, 75, 76]).
| Drugs | Target of inhibition | Cell type | Reference | |
| Preclinical studies | TET | PINK1/Parkin-mediated mitophagy | AT II | [12] |
| H2S | p53/p21 pathway | AT II | [73] | |
| ISRIB | p53/p21 and p16 pathways | AT II | [74] | |
| SAB | SASP | AT II | [75] | |
| RXM | SASP | [76] | ||
| Clinical study | D+Q | cellular senescence | [7] |
TET, Tetrandrine; H2S, Hydrogen sulfide; ISRlB, Integrated stress response inhibitor; SAB, Salvianolic acid B; RXM, Roxithromycin; D, Dasatinib; Q, Quercetin; AT II, alveolar type II; PINK1, PTEN induced kinase 1.
The senescence of AT II cells is closely related to the progression of pulmonary fibrosis. Senescent AT II cells in the lung tissue of IPF patients contribute to the development of fibrosis, suggesting new treatment targets that are related to cellular senescence. Consequently, researchers are exploring treatment strategies for pulmonary fibrosis that target senescence-related pathways and eliminate senescent cells. Tetrandrine is a natural bisbenzylisoquinoline alkaloid known for its role as a calcium channel blocker and for its diverse biological activities. Using a bleomycin-induced mouse model of pulmonary fibrosis, tetrandrine was shown to have anti-fibrotic effects by delaying the senescence of AT II epithelial cells through inhibition of PTEN induced kinase 1 (PINK1)/Parkin-mediated mitophagy [12]. Hydrogen sulfide suppresses bleomycin-induced senescence of AT II epithelial cells in mice. Mechanistically, hydrogen sulfide inhibits the senescence of AT II epithelial cells and communication between senescent AT II cells and fibroblasts by blocking the p53-p21 pathway [74]. In a silicosis-induced mouse model of pulmonary fibrosis, the integrated stress response inhibitor decreased the severity of pulmonary fibrosis in mice by preventing senescence of AT II epithelial cells via inhibition of the p53-p21/p16 pathway [75].
Salvianolic acid B directly interferes with the binding of transcription factor stimulated protein 1 to the promoters of SASPs (p21 and p16). This interference blocks the senescence of AT II epithelial cells and lung macrophages, effectively reducing pulmonary fibrosis [76]. Roxithromycin was shown to reduce fibrosis in a bleomycin-induced mouse model of pulmonary fibrosis. Mechanistically, Roxithromycin inhibits cellular senescence and alleviates pulmonary fibrosis by inducing cell apoptosis and inhibiting the secretion of SASP [77].
Current treatments for IPF aim to slow the course of the disease and relieve
symptoms. Pirfenidone and nintedanib have been introduced as clinical treatments
for IPF. Pirfenidone reduces pulmonary fibrosis by inhibiting the
Wnt/
Senolytic agents are medications that target senescent cells. Clinical trials have shown these agents improved lung function in IPF patients [82]. Nambiar et al. [7] conducted an open-label, single-arm pilot study of the senolytic combination of dasatinib and quercetin in IPF patients. Their results demonstrated the feasibility of conducting further trials on the anti-fibrotic efficacy of senolytic agents. Targeting cellular senescence could therefore be a crucial focus of future IPF treatment strategies.
The mechanisms that underlie the development of pulmonary fibrosis are complex and likely to be affected by multiple factors. Although great progress has been made in understanding these mechanisms, there is still much to be learned and more in-depth exploration is required, especially in the field of environmental etiology. Cellular senescence plays an important role in the occurrence and development of pulmonary fibrosis. Further research on the molecular mechanisms behind the development, maintenance, and blocking of cell senescence in pulmonary fibrosis, together with development of new cell senescence-based treatment strategies, should open the way for novel targeted treatments of pulmonary fibrosis. In addition, combining the elimination of senescent cells with currently used clinical drugs may be an effective strategy for the treatment of pulmonary fibrosis.
Cutting-edge technologies such as scRNA-seq and spatial transcriptomics have revealed the highly heterogeneous nature of various cell types and their tissue distribution [83]. The different cell types present in the lungs are also specific in time and space during the senescence process [84, 85, 86]. The key to targeted senescence treatment of pulmonary fibrosis is the ability to accurately locate the cellular and spatio-temporal specificity of senescent cells. Future studies could employ lineage-tracing mouse models to dynamically track cell-type specific senescent dynamics during pulmonary fibrosis. The development of single-cell metabolomics technology may also provide new insights into the role played by the metabolic reprogramming of immune cells in senescence within a fibrotic microenvironment.
DC, LF, HZ, and JY participated in the review’s design. DC, LF, SW, and RW carried out the writing and revision of the manuscript. SW, and RW performed the drawing figures of the manuscript. YM, QH, and YF performed the search and sorted out related literature of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved of the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by National Natural Science Foundation of China (82100078, 82270071) and Scientific Research Project of Anhui Provincial Health Commission (AHWJ2024Aa30243).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.