1. Introduction
Alpha-synuclein (-syn), a member of the synuclein family of proteins,
is a small protein with highly-conserved sequences and three well-studied domains
[1, 2]. It is ubiquitous throughout the human body, and its integral roles
include membrane fusion, endocytosis, and exocytosis, including within
non-neuronal cells, especially red blood cells (RBCs) and platelets [3, 4, 5].
Aggregates of -syn have also long been identified as a driver of a
family of progressive, degenerative central nervous system (CNS) diseases known
as synucleinopathies, most famously Parkinson’s disease (PD) but also
encompassing Lewy body dementia (LBD) and multisystem atrophy (MSA) [6].
Especially within the last two decades, multiple mechanisms of
-syn-mediated neurodegenerative diseases (NDD) have been and continue
to be both proposed and researched. However, despite its role in multiple organ
systems, research into the role of -syn in non-neuronal, peripheral
and/or hematopoietic pathophysiology has been relatively unknown until recently.
Since the hematopoietic system requires endogenous -syn for normal
function, new efforts to establish the pathways mediated by -syn in
both hematopoietic function and dysfunction have been undertaken. For example,
RBCs account for the majority of circulating -syn, while platelets
exhibit a higher protein fraction of -syn [7, 8]. Indeed, widespread
presence of -syn across cell lineages of the hematopoietic system
implicates that it is important in almost every cell line [4, 9, 10, 11]. This protein
is particularly essential to the structure and function of immune cells. Both
systemic deficiency of -syn and its aggregation in neural tissue have
been associated with significant T-cell dysregulation [10, 12], implying that
when -syn is predominantly present in aggregates, its presence in the
hematopoietic system can be affected. This suggests that
-syn-associated systemic pathophysiology within the CNS can be seen as
bimodal in nature: excess of -syn in aggregates and paucity of
-syn, also known as the synucleinopenia hypothesis [13], may both
contribute to disease.
Other systems that are affected by synucleinopathies, some of which have been
implicated in CNS disease, include the gastrointestinal (GI) tract and the
integumentary system; in this regard, the gut microbiome and its interaction with
-syn have been postulated as a potential point of origin for
neurodegenerative synucleinopathies [2, 14]. If true, these hypotheses correlate
with recent research suggesting that the gut microbiome is highly connected to
the nervous system via the “gut-brain barrier”, and with ever-increasing
evidence that multiple systems could contribute to mechanisms influencing
neurologic disease as a whole.
In summary, contrasting with the prevalent view of -syn-induced
disease as primarily neurological, potential molecular mechanisms in which
different systems cause and are affected by -syn dysregulation have
been identified. Although the structure and function of -syn are
well-studied, much work will still need to be done regarding its interaction with
non-neuronal systems in which it is necessary and those in which it is
detrimental.
2. Alpha-syn Structure and Function
Alpha-synuclein is a protein consisting of 140 amino acids with highly
evolutionarily conserved sequences, which it shares with the beta- and gamma-
isoforms in the same family, and three domains that include two regions
considered important in disease: the N-terminal and the C-terminal regions, and a
central region between them [1, 15]. The N-terminal region is primarily
alpha-helical in nature and is highly important in lipid binding (membranous
structures). Conversely, the C-terminal region has a wider range of actions that
include post-translational cellular modifications, protein chaperoning at
synapses, and docking of protein complexes; this region is also thought to
prevent aggregation in its usual conformation [16]. Multiple methods of
post-translational modification of -syn have been identified and
studied, with phosphorylation predominant in normal function as well as
preventing aggregation [1, 17]. Additional common post-translational
modifications include proteolysis/truncation, oxidation, glycation, and
ubiquitination, although phosphorylation is the best-known of these mechanisms
and appears highly necessary to retain characteristic protein flexibility [1, 17].
The configuration of -syn is largely dependent on the membrane and/or
cell type to which it binds; it is commonly found in an alpha-helix configuration
when binding to lipid membranes and in a broken alpha-helix when binding to small
vesicles [17]. While its relative structural flexibility lends itself well to
membrane trafficking, packaging, and transport, these same
characteristics—especially in the C-terminal region—cause -syn to
easily misfold and subsequently form aggregates [4, 17, 18]. Specifically,
-helical configurations have been identified as most pathologic [18].
These aggregates have long been implicated as the major drivers, if not the root
cause, of synucleinopathies such as PD and LBD [17, 18, 19].
There is also evidence that -syn contributes to mitochondrial
function, particularly in neurons, which could in turn contribute to NDD
pathology [20]. Additionally, interaction of -syn with cytoskeletal
proteins may both contribute to understanding of its full function and provide an
alternate mechanism by which it causes neurodegenerative pathophysiology [21].
The many roles that this protein plays in normal homeostasis provides ample
avenues for future research. In the following sections, tissue-specific
-syn utilization will be discussed.
2.1 The Hematopoietic System
While research into -syn has traditionally focused on the neurologic
system, it has been found that it is important, if not essential, in the majority
of the hematopoietic system. Certain categories of cells are predominant in usage
and production of this protein. As previously mentioned, RBCs produce
-syn and require it for differentiation [4, 5, 9, 22]. Within the
erythrocyte compartment of the hematopoietic system, it has been found that
-syn is expressed as early as the erythroblast stage, as confirmed by
immunohistochemical staining of bone marrow; staining is most evident in
erythroblasts [5]. -syn is upregulated concurrently through maturation
with other maturity-marking proteins, such as hemoglobin subunits [22]. While
erythrocytes are anucleate, early erythrocyte precursors require
-syn-associated cell membrane function for effective maturation,
including the aforementioned hemoglobin expression and the later physical process
of nucleic acid expulsion [5, 23]. Changing localization of -syn on
nuclear membranes (early development) and in the cytoplasm (most prominent in
later maturation) provide evidence of its necessity; it is, unlike many proteins,
upregulated rather than downregulated with increasing maturation [22]. It is
furthermore upregulated by GATA-1, itself a prominent protein in RBC maturation
[23]. Additionally, -syn—through its widespread membrane-binding
functions including binding to phosphatidylserine—appears essential to
characteristic RBC membrane fluidity and flexibility, and likely binds to
proteins such as transferrin, which are essential to iron homeostasis and
processing [7, 22].
While not the only driver of platelet function, -syn is present in
platelets and regulates -granule release, acting not only as a
chaperone in platelet function but also as a calcium-dependent negative regulator
that prevents excessive platelet activation; also, it is present as early as the
megakaryocyte stage [3, 24, 25]. Mechanistically, these findings are likely due
to the necessary interaction of -syn with cell-surface receptors
partially responsible for platelet aggregation, such as glycoprotein Ib [26]. Platelets, although anucleate like erythrocytes, additionally express
vesicular and target soluble N-ethylmaleimide-sensitive factor attachment protein
receptors (SNARE) proteins, with which -syn likely interacts to achieve
optimal conformation and allow the release of granule contents. Such functions
have been identified in cells within multiple systems and lineages [4]. This
suggests a role in multiple phases of platelet activation, suggesting that
-syn may be necessary throughout the activation process [7]. For
instance, -syn deletion in combination with the protein multimerin-1
has been shown to contribute to bleeding diathesis [26]. Furthermore,
-syn continues to be released even after platelets have been extracted
from whole blood and stored, which provides insight into a potentially more
dynamic purpose within the megakaryocytes and platelets themselves [7]. These
roles appear to be not dissimilar to those in the erythrocyte compartment (Fig. 1).
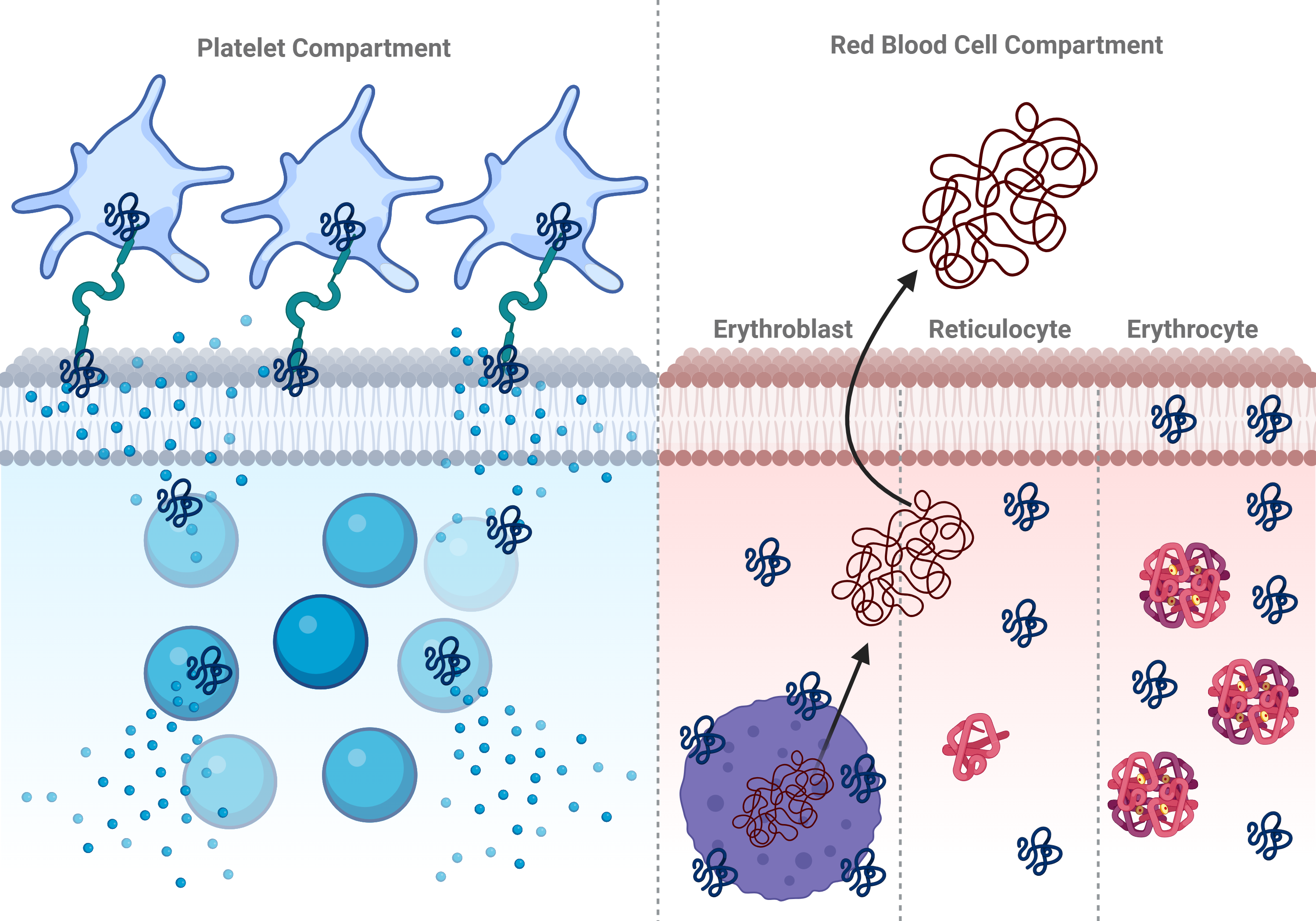 Fig. 1.
Fig. 1.
Hypothesized pathways and functions of alpha-synuclein in
platelet and red blood cell compartments. Depiction of the use of
alpha-synuclein in platelets (left) and red blood cells (right), including
aggregation, membrane integrity, and expulsion of genetic material and granules
into the bloodstream and surrounding tissues are depicted. Created with BioRender.com.
Lack of -syn significantly dysregulates the immune response within
almost all leukocyte categories, and as such its roles have been at least
partially described. Its deficiency prevents normal maturation and function of
both the innate and adaptive immune systems. Differentiation and effective
phenotype switching of macrophages and dendritic cells are significantly
decreased, as is their subsequent antigen presentation [27]. Within the adaptive
immune system, -syn deficiency changes and/or decreases the morphology,
regulation, differentiation, and granule release in B and T cells, which shows
not only that its release is essential to cell signaling, but that its membrane
interactions are necessary for normal cell differentiation and physiological
response [4, 10, 27, 28]. Finally, impaired immunoglobulin G (IgG) production and
B-lymphopenia in -syn knockout mice emphasize the fundamental
importance of this protein across multiple lymphoid compartments [11].
2.2 The Nervous System
The nervous system, perhaps the most commonly studied bodily system in this
context, requires properly-functioning, un-aggregated -syn to produce
synapses. Its interactions with, particularly, presynaptic elements of a synapse
is especially prominent in the dopaminergic, catecholaminergic, and glutamatergic
neurons, all of which are largely excitatory in nature; this is anatomically
readily detectable in the substantia nigra and locus coeruleus [17, 21, 29]. In
terms of the nervous system as a whole, -syn has historically been
considered largely brain-specific, although its extensive role in the enteric
nervous system will be explored later [1]. As this review describes, the
prevailing view of -syn-driven disease as brain-based with only
marginal contributions from other systems is, given the results of a growing body
of literature, likely outdated and should perhaps be set aside in favor of an
inter-systemic approach. However, -syn remains the agent of
synucleinopathies in the nervous system and its aggregation can occur and/or
perpetuate there, which means that a thorough understanding of its mechanisms in
the CNS should still be foundational in its study.
As synapses are largely driven by membrane- and vesicle-based interactions
between neural cells, native -syn in its membrane-binding and
membrane-stabilizing capacities is vital for normal brain function.
-syn binds to and stabilizes cell membranes’ lipid bilayers,
maintaining the physical membrane structures that maintain cell and vesicle
integrity as well as interacting with cytoskeletal proteins [1, 16]. Via
interactions with SNARE proteins and vesicle-associated membrane protein 2
(VAMP2), both terminal domains participate in synaptic vesicle docking and
trafficking and localize largely within presynaptic vesicles [16, 18, 30]. This
leads to well-known synapse mechanisms, in which, most commonly, vesicles
containing neurotransmitters complete their actions at the postsynaptic terminal.
However, -syn is not limited to simple synaptic membrane interactions.
It has been shown to help form fusion pores involved in the egress of cell
contents, and binds to calcium, itself involved heavily in synaptic transmission
and neuro-electrical impulses [16]. The mitochondria, which form vesicles as part
of normal maintenance, attract -syn, which has been linked to organelle
degradation and damage following normal interactions [18, 20]. In short, the
widespread necessity and localization of -syn in the CNS is consistent
with its presence and roles in other systems.
2.3 The Gastrointestinal System
The nerve plexuses that drive visceral innervation and peristalsis, known as the
enteric nervous system (ENS), require -syn to function. This protein
has been found to be distributed in the jejunum within both the neurons of the
ENS and the epithelial cells [31]. Within the colon, it is present in both the
muscular wall and the ENS, with some evidence that -syn accumulation in
these regions is a normal part of aging or reactive pathology rather than simply
a harbinger of pending NDD [32, 33]. In addition to this role, -syn can
stimulate the GI tract’s immune function, allowing recruitment of immune cells to
the gut during gastroenteritis [34, 35].
In mice, it has been shown that -syn is likely required for healthy
development of the ENS, especially in terms of cholinergic neurons. For example.
deficiency leads to evidence of poor colonic function, including constipation
[36]. Interestingly, these symptoms are similar to constipation seen in PD
patients, further indicating that disease is induced by both paucity and excess
of -syn and implying that tight regulation of its expression is
important [37, 38].
Outside of the gut, -syn may play a part in normal function of other
organs within or associated to the GI system, including the pancreas. Deletion of
the protein causes a diabetes-like phenotype in mice, while overexpression
improves glucose tolerance and insulin sensitivity, possibly through the same
transport mediators involved in membrane trafficking [39]. It has also been
described that there is a connection between -syn and insulin
resistance; since physiologic levels of -syn prevents excess insulin
secretion by modulating release of insulin granules [39, 40]. Nevertheless, the
co-occurrence of PD and diabetes, while not enough to determine causation,
suggests that the same inflammatory mechanisms may play active roles in both
etiologies, especially in the context of gut dysbiosis [40]. It should be noted
that the changes in microbiome associated with diabetes, such as a decrease in
short-chain fatty acid-producing bacteria, may themselves be independently
associated with increasing metabolic and inflammatory derangement within the gut
[41]. Given the frequency of diabetes in developed countries, potential
synergistic interaction with -syn-associated inflammatory
mechanisms—especially with age, as -syn increases systemically with
aging—presents a potential area for further study [18, 20, 32, 33].
3. Cross-System -syn-Induced Pathophysiology
Oligomers, multimers, and fibrils are known to form as the result of abnormal
-syn aggregation. However, of these oligomers are thought to be the
most toxic and to contribute most frequently to NDD, although fibrils are also
frequently found and may precede multimers formation [4, 19, 42]. It is widely
agreed that the tendency of -syn to aggregate is caused partially by
the mercurial conformation of its C-terminal component, which allows the protein
to fold abnormally with relative ease [17, 18]. While this single characteristic
is largely, although not solely, responsible for the ability to form aggregates,
likely multiple mechanisms are involved in the actual process. Phosphorylation,
for example, appears to play a part in changing the protein’s conformation [2].
Interaction with lipid membranes, one of the crucial functions of -syn,
may itself promote aggregation by changing the protein’s conformation during
normal physiologic interaction [43, 44]. Post-translational modification may also
contribute to this process [1, 44]. Additionally, other risk factors may be
environmental, such as heavy-metal toxicity, pollution, or even exposure to
pesticides [40, 45].
Once misfolded, these -syn aggregates spread throughout affected
systems in a prion-like manner, converting normal -syn to the abnormal
form potentially by hijacking normal vesicle function [19]. Initially,
-syn aggregates themselves cause intra-neuronal toxicity and neuron
degeneration [42]. Attraction of the immune system to these—in effect—foreign
bodies then create much of the early trigger and subsequent pathology
characteristic of NDD. CNS-specific macrophages, known as microglia, generate a
powerful inflammatory response locally that recruits both astrocytes and a myriad
pro-inflammatory elements from the blood, many of which are
-syn-specific, that further damages neural tissue [18, 42, 46].
Additionally, lysosomal dysfunction, leading to inability to degrade
-syn aggregates, may represent another potential contributor throughout
the entire process [47, 48].
The effects on the CNS of -syn derangement in the blood provide clues
as to how the immune system, when deprived of sufficient -syn as
aggregation disseminates, contributes to NDD patients’ physical and mental
decline. It has been reported that -syn is important to type 1
interferon signaling, an important initiator of immune signal transduction,
within the nervous system [49]. Likewise, binding of lymphocyte activation gene-3
expands -syn aggregate formation and toxicity, which suggests that
pathologic interaction between -syn and the adaptive immune system
contribute to NDD; and excess -syn may similarly activate the immune
system to attack neural tissue [12, 27, 50]. Immunosuppression has also been
correlated with NDD, potentially via loss of normal T-cell regulation preventing
-syn aggregation, although a definitive causative relationship has not
been proven [51]. Moreover, although active T cells are increased in these
diseases, overall T cell numbers are paradoxically decreased [12]. These findings
emphasize that not only is the immune system in effect weaponized to perpetuate
NDD, but its dysfunction can contribute to or possibly even initiate these
disease processes.
Different origins of -syn, and mechanisms of its transport into the
CNS, have been proposed in recent years. With the rise in our understanding of
the relationship between gut health and human health as a whole, concomitant
understanding of the relationship between the gut and -syn pathology
has similarly increased. These relationships, i.e., “the gut-brain axis”, have
led to increased focus on the gastrointestinal system as one area of research for
NDD, with the microbiome drawing increasing focus [52]. However, multiple
potential avenues must be considered (Fig. 2).
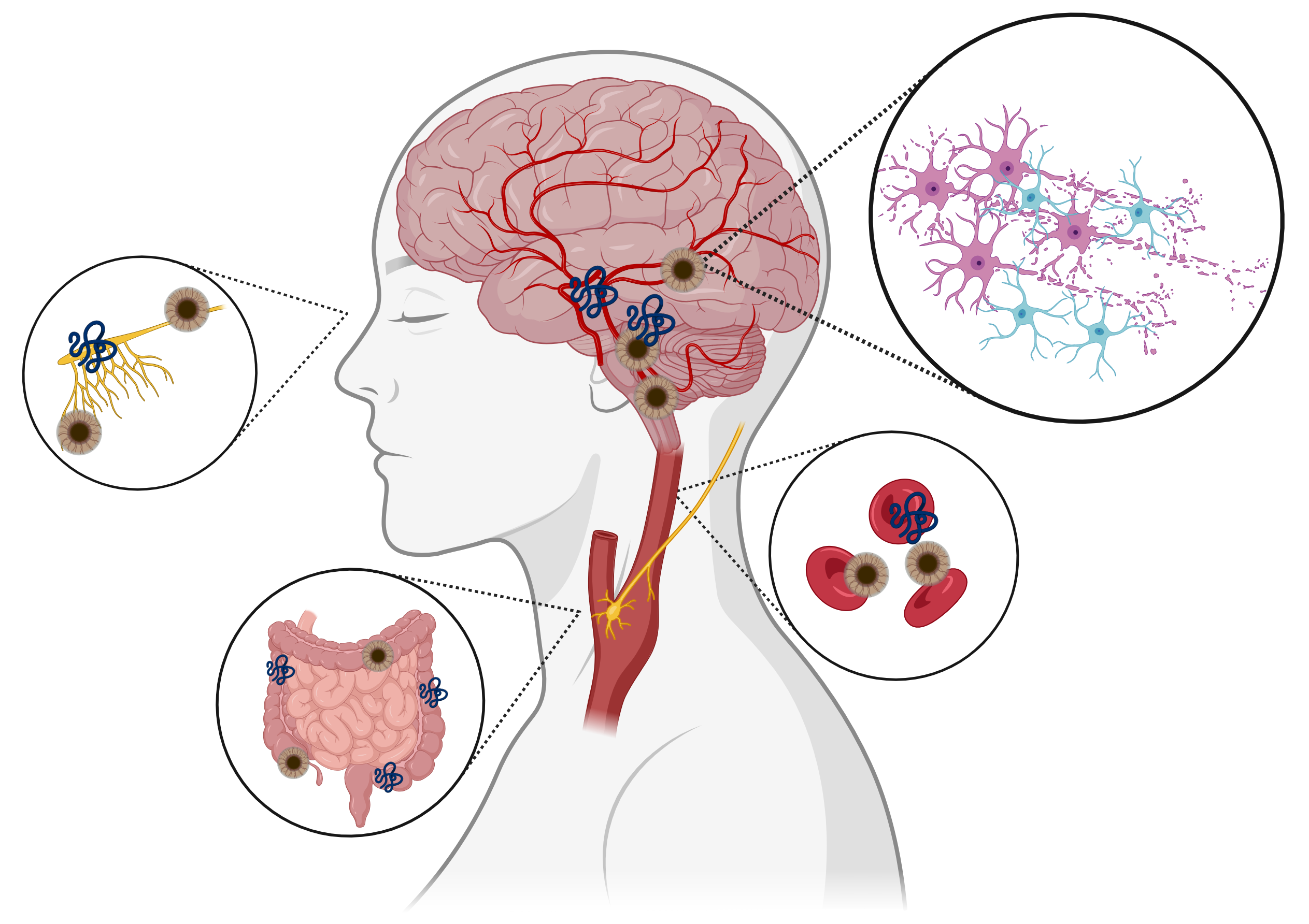 Fig. 2.
Fig. 2.
Hypothesized sources of -synuclein fibrils and
aggregates. Depiction of possible tissue sources that contribute fibrils and
aggregates of -syn to pathophysiology of Parkinson’s Disease including
the brain, the olfactory bulbs, blood cells, and peripherally via the vagus nerve
from enteric neural cells. Created with BioRender.com.
3.1 Gut-Modulated -syn Pathophysiology
As previously described, -syn is present in relatively high quantities
throughout the gut, primarily in the ENS but also within muscle and epithelium
[31, 32, 33]. It is therefore a prime candidate to interact with elements of the gut,
including the microbiome. Detrimental changes in the gut microflora typically
involve decrease of normal commensals, such as Bacteroidetes [52], in
favor of species that are typically less common, such as those in the
Bacillota phylum [39]. Microbial changes dysregulate the immune response
and disrupt normal gut homeostasis, including but not limited to development of
colitis and inducement of -syn aggregation [14, 45, 52]. In fact,
amyloid produced by certain gut bacteria, known as “curli”, can promote
-syn aggregation as well [53]. More recently, several groups have
provided more evidence for gut-first synucleinopathy. A new study demonstrated
the combined influence of -syn and tau in gut-first NDD [54], while a
separate paper provided neuropathologic evidence of Lewy body disease for gut-,
or body-first, patients in a caudo-rostral pathology [55].
The connection between the gut and the brain is not linear, and bidirectional
interactions cannot be ruled out as potential contributors to synucleinopathies
and NDD [19]. A single mechanism has not been definitively identified at this
point, but multiple potential pathways have been proposed. Of note, with an
ever-increasing amount of knowledge about the gut, the microbiome and its
proteome, and crucial interactions between the body and bacteria, research has
continued to expand on this area. However, transport of -syn in the
blood can be both beneficial to hematopoiesis yet harmful to the body. On the
other hand, -syn aggregation and genesis of disease within the CNS, at
least partially, also cannot be ruled out.
3.2 Proposed Pathways and Initiating Factors
The latest research regarding how -syn may move from non-neural
systems to the brain can be summarized with the terms “brain first” versus
“body first” or “gut first”, i.e., spontaneous generation within the CNS or
nearby nerves as opposed to extra-neural aggregation followed by secondary
transport to the CNS [2, 56, 57]. These categories are, of course, discrete and
generalized, but are still useful in this context. “Brain first” has been and
continues to be studied, while “body first” composes much of the current
research into synucleinopathies, but neither potential pathway can be discounted.
In fact, the connections between the central and peripheral nervous systems, and
their subsequent intertwining with non-neural bodily systems, make elaboration of
both general categories both complex yet necessary.
Within the “brain first” category, the olfactory nerve or bulb (OB) is
considered potentially crucial in genesis and/or transportation of -syn
aggregates. Reports have shown that -syn has been detected in the OB
prior to onset of NDD symptoms, particularly motor ones [40, 58]. Likewise,
non-human primate experiments have shown that seeding the OB with exogenous
-syn leads directly to an artificially-induced NDD [59]. Rapid eye
movement sleep behavior disorder, associated with excess -syn in the
area, is correlated with later development of NDD [56, 60]. Additional potential
sites for -syn propagation include the dorsal motor nucleus, nucleus
ceruleus, and amygdala, all of which are considered part of the CNS [2].
The gut-first paradigm, or gut to peripheral nerve pathway, is a strong
candidate for an NDD genesis point within the “body first” category [57]. This
hypothesis posits that following -syn aggregation within the gut,
likely as a result of gut dysbiosis and/or inflammatory disease, aggregates
travel up the vagus nerve to the CNS [18, 37, 61]. The vagus nerve is likely not
the sole pathway in this mechanism, but the autonomic nervous system (ANS)—of
which the vagus nerve is a part—and its associated organs appear extensively
involved [18, 58]. As a result, physical translocation of -syn
aggregates from the body to the CNS, where further pathology results in full NDD,
is considered the overarching mechanism.
One additional potential pathway within this category is the body fluid
circulation pathway, which is also connected to the ANS-associated organs
described in gut-first hypotheses. In this pathway, -syn has been found
in many types of circulating fluids, including but not limited to blood, lymph,
and cerebrospinal fluid [40]. Indeed, available data has described the types of
hematopoietic cells for which -syn is necessary, and its ubiquitousness
in this system is possibly both necessary for homeostasis and yet another method
of NDD perpetuation. For example, one mode of transport to the CNS may be through
-syn-laden vesicles derived from erythrocytes, or even -syn
aggregates within RBCs themselves that may appear years prior to frank disease
[2, 60, 62]. Combined with the growing knowledge of -syn’s vital role
in the hematopoietic system, such potential sources of aggregates present a rich
area for further research. These results serve to strengthen the hypothesis that
synucleinopathies may not be the result of one single mechanism or system, no
matter the strength of the system’s ability to produce aggregates, but rather
multiple systems working in tandem—especially with increasing age (Fig. 3).
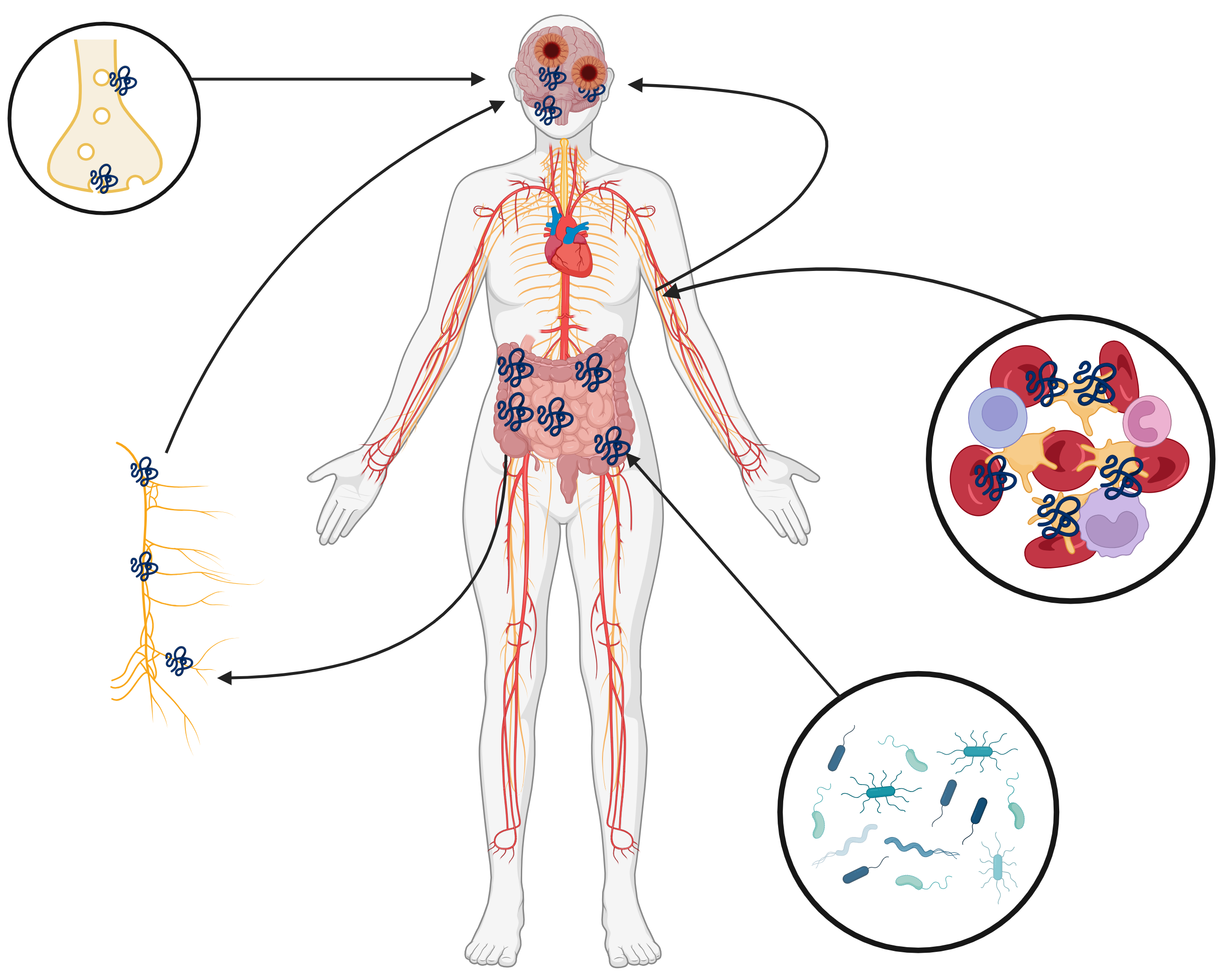 Fig. 3.
Fig. 3.
Potential systemic pathways of synucleinopathies. Stepwise
routes of alpha-synuclein aggregation and transport within the gut, hematopoietic
system, and central nervous system that may contribute to development of
pathology. Created with BioRender.com.
4. Proposed Treatments for Synucleinopathies
Given the multiple proposed pathways for synucleinopathies and resultant NDD,
much attention has been paid to mechanism-specific potential treatments. Due to
the dearth of disease attributed to synucleinopathy in extra-neural tissues, the
vast majority of effort devoted to synucleinopathy-related disease has been in
PD, and discussion of therapeutics past the preclinical stage need be seen
through the lens of this disease. An annual review of the currently active
clinical trials for PD [63], showed that most small molecules and antibodies have
been targeting the dopaminergic pathway per historical reasons. However, other
therapeutic strategies, such as targeting gut microbiota, reactive oxygen species
(ROS), and immune modulation have also been considered. Here we consider the
broadly generalizable findings of such studies with regards to non-CNS
mechanisms.
While focus on symptomatic relief through dopaminergic and other pathways
comprise a majority of these trials and will not be discussed, a substantial
subset targets -syn directly, with a multitude of completed clinical
trials (largely but not exclusively phase I) in recent years. Several general
strategies can be identified within this group. The experimental compounds
UCB0599 and anle138b have been shown to inhibit -syn misfolding and
oligomerization, respectively; the former has been tested in humans, including PD
patients, with a good safety profile. The latter study additionally shows
potential action against prion disease, as confirmed by histopathology and
decreased signs and symptoms in mice [64, 65]. Similarly, the monoclonal antibody
prasinezumab, which is specific to -syn aggregates, is the focus of an
ongoing trial [66]. Generalized translation inhibition with the compound
buntanetap shows promising results, with additional regulatory effects on
neurotoxic proteins in general [67]. Active immunization to -syn shows
sustained aggregate-specific IgG antibody responses (up to one year) against
synucleinopathy-specific -syn epitopes, although the sample size is
small [68]. Lastly, non-neurologic drugs such as the diabetes drug lixisenatide
have been tested in PD patients [69].
It should be noted that careful review of secondary endpoints and supplementary
data of some of these studies demonstrate that actual measurement of
-syn is not generally undertaken [66, 69]. The prasinezumab study
additionally excludes patients with potential genetic causes of PD, which despite
PD’s typical occurrence de novo, may skew results [66]. Thus, any
adverse events that occurred that may be due to secondary effects on extra-neural
regions are difficult to account for. Specifically, almost all patients who
received the glucagon-like peptide-1 agonist lixisenatide experienced
gastrointestinal symptoms; if a pancreatic or ENS effect were the root cause of
these symptoms, more caution would need to be exercised in further investigating
this specific treatment strategy [69]. Subanalysis of supplementary data in these
studies or revisiting biobanks for these clinical trials in the future may be
necessary to elucidate the effects of -syn-targeted therapies on
systemic concentrations of the protein.
Separately from the mainstay of targeting -syn, modulation of the
immune system has had ever increasing attention in the synucleinopathy field,
particular in Parkinson’s disease. As suggested above, the interaction of the
hematopoietic compartment, and specifically of the immunity contingent, with
synucleinopathies is two-fold. First, treatment that alters -syn levels
may affect normal hematopoiesis, which is dependent partially on normal
-syn concentrations. Second, exaggerated responses in immune cells,
particularly microglia [18, 42, 46], likely contributes to pathogenesis in PD and
other NDD. With these factors in mind, the current immunomodulatory therapies
available for synucleinopathies can be divided into immune-dampening agents and
immune-stimulating factors. The former include the well-studied drugs
pentoxifylline and celocoxib as well as more experimental drugs targeting TLR2
and the NLRP3 inflammasome [70, 71, 72, 73]. Both TLR2 and the NLRP3 inflammasome complex
play important roles in inflammatory signaling and cytokine release, although
NLRP3 inhibitors have been studied largely in the context of ulcerative colitis
rather than synucleinopathies [72, 73]. Nevertheless, inflammatory bowel disease
and PD have been linked closely in recent studies, implying the utility of
connecting studies based on the former to mechanisms of the latter [61, 73].
Conversely, two extended-release GM-CSF compounds have undergone initial testing
for use in PD, which suggests a more complex role of inflammation in NDDs than
previously thought [74, 75].
Paradoxically, the fact that these seemingly opposing agents are being tested
for ostensibly the same synucleinopathy-driven pathology suggests a broadly
immunological approach may not be the most efficacious choice. On one hand,
immune dampening may delay progression or onset of disease while exacerbating
disease-related immunosuppression; on the other, attempting to reconstitute
decreased immune function may worsen or accelerate pathology. The totality of the
effect of the quantitative dysregulation of -syn, both up and down,
should ideally be considered in these broad immunomodulatory approaches, but
without further research into the secondary effects of synucleinopathies in the
hematopoietic compartment, the non-targeted effects of these approaches may in
the end cause more harm than help.
Finally, the gut-brain axis represents a third, major orthogonal approach to
studying both the pathogenesis and therapeutic milieu in synucleinopathies [52].
Gut dysfunction, such as through small intestinal bacterial overgrowth, has long
been recognized to be correlated with PD [76]. Although a somewhat indirect
mechanism, now it is thought that dysbiosis of the gut microbiome can lead to
increased neuroinflammation both by aggregation of -syn in the gut with
subsequent migration through the vagus nerve and by permeabilization of the gut
lining and subsequent escalation of systemic and downstream CNS inflammation
through cytokine action. Accordingly, several approaches to modify the root cause
of this pathway have been taken. The antibiotic rifaximin has been proposed as a
potential PD treatment and trialed in rodents [77, 78]. Alternatively,
organism-specific therapies such as fecal microbiota transplantation are gaining
traction, as are modulation of the gut microbiome and immune system [38, 79, 80, 81].
However, a still missing piece of data has been actual measurement of
-syn in non-CNS compartments. If in fact the “gut first” theory holds
true, broad analysis of intermediate steps in pathogenesis will eventually
require that some quantitative measure of -syn be undertaken.
Overall, the many different axes of therapeutic investigation in
synucleinopathies have a bright outlook, with the above trajectories as well as
others unmentioned, such as the role of reactive oxygen species in perpetuating
inflammation following -syn aggregate formation, bearing fruit in the
past decade [82]. Nevertheless, in the context of secondary effects on normal
-syn function in other compartments, especially the hematopoietic
lineages, only a small minority of effort thus far has been directed towards
understanding how the preferential shunting of -syn to oligomeric forms
causes dysregulation systemically. Further studies and therapies may benefit from
addressing the body as interconnected, synergistically functioning systems in
terms of -syn sources and pathophysiology.
5. Conclusion
It should be clear from the data presented that the crosstalk of different
systems and likely a multiplicity of pathways requiring -syn are
important to establish not only its functional role, but those physiologic axes
that require its tight regulation to function properly. Even though this protein
has shown to be of great importance in the CNS, it is readily apparent that its
function is wide-reaching and involves many systems some of which may prove to
contribute to PD and synucleinopathy pathology. It should also be clear that the
formulation of hypothesis to account for potential non-CNS sources of the disease
takes into account recent findings and thus opens up additional areas for
investigation. Thus, therapies will need to be developed that address the
multiple effects of -syn both under normal conditions and in those
instances in which its abnormal configurations drive disease symptomatology.
Abbreviations
ANS, autonomic nervous system; -syn, alpha-synuclein; CNS, central nervous system; ENS, enteric nervous system; GATA-1, GATA-binding protein 1; GM-CSF, granulocyte-macrophage colony-stimulating factor; LBD, Lewy body dementia; MSA, multisystem atrophy; NDD, neurodegenerative disease; NLRP3, NLR family pyrin domain containing 3; OB, olfactory bulb; PD, Parkinson’s disease; RBCs, red blood cells; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptors; TLR2, Toll-like receptor 2; VAMP2, vesicle-associated membrane protein 2.
Author Contributions
HHD and BZ performed the literature search and wrote the manuscript; RWM conceptualized the manuscript, wrote key sections, supervised contributions from co-authors, reviewed references, and performed critical revisions. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research received no external funding.
Conflict of Interest
The authors declare no conflict of interest.




 , Bowen Zhou 1,†
, Bowen Zhou 1,†