







 , Shiguang Wang 2, Liya Li 1, Ruihua Li 1, Minming Fan 1, Jingjing Kong 1, Bing Gao 3, Jing Wang 3,*
, Shiguang Wang 2, Liya Li 1, Ruihua Li 1, Minming Fan 1, Jingjing Kong 1, Bing Gao 3, Jing Wang 3,* , Ran Xia 3,*
, Ran Xia 3,*1 Graduate School, Anhui University of Chinese Medicine, Special Project of Xin’an Medical and Traditional Chinese Medicine Modernization Research Institute of Great Health Research Institute, 230012 Hefei, Anhui, China
2 Department of Cardiology, The First Affiliated Hospital of Anhui University of Chinese Medicine, 230031 Hefei, Anhui, China
3 Zhongyi School, Anhui University of Chinese Medicine, Special Project of Xin’an Medical and Traditional Chinese Medicine Modernization Research Institute of Great Health Research Institute, 230012 Hefei, Anhui, China
Abstract
Heart failure (HF) continues to represent a significant global public health concern. Transient receptor potential cation channel subfamily V member 1 (TRPV1) is a calcium-permeable channel that has been linked to cardiac disease and function. However, its significance in HF and underlying processes is unknown. This study aims to determine the regulatory role of TRPV1 in mitochondrial autophagy in HF.
AC16 cardiomyocytes were exposed to angiotensin II (Ang II) to simulate pathological conditions, and changes in oxidative stress were assessed. Transverse aortic constriction (TAC) was used to create a pressure overload-induced HF mouse model, and cardiac-specific TRPV1 overexpression was achieved by Adeno-associated virus 9 (AAV9). RNA sequencing and bioinformatics analysis were performed to identify TRPV1-related mitochondrial genes. Finally, the effects of TRPV1 overexpression and sideroflexin 2 (SFXN2) knockdown on markers related to mitophagy and ferroptosis were analyzed.
In vitro, TRPV1 overexpression drastically decreased intracellular Ca2+ levels, lessened oxidative stress, and reduced Ang II-induced cell death (p < 0.05). Bioinformatics analysis identified seven mitochondrial genes associated with TRPV1, among which SFXN2 showed a strong correlation with TRPV1 (p < 0.05). Overexpressing cardiac-specific TRPV1 in the TAC model led to improved cardiac function, higher fractional shortening and ejection fraction, and reduced levels of mitophagy markers (p < 0.05). Mechanistically, TRPV1 activated SFXN2, increasing glutathione peroxidase 4 (GPX4) expression and antioxidant capacity (glutathione (GSH), superoxide dismutase (SOD)) while decreasing malondialdehyde (MDA) and ferrous iron (Fe2+) levels (p < 0.05). These protective effects were removed by SFXN2 knockdown. Furthermore, the TRPV1-SFXN2 axis suppressed mitophagy by modulating the PTEN-induced kinase 1 (PINK1)-Parkin-sequestosome 1 (SQSTM1) axis.
Our results show that TRPV1 overexpression alleviates Ang II-induced myocardial injury in HF. This protective effect is mediated through SFXN2-dependent mitophagy and ferroptosis, highlighting TRPV1 as a potential therapeutic target for HF.
Keywords
- TRPV1
- heart failure
- SFXN2
- ferroptosis
- mitophagy
Heart failure (HF) is a complicated clinical disease in which abnormal cardiac structure or function leads to impaired ventricular filling or ejection capacity [1]. Patients usually present with symptoms such as exertional dyspnea, paroxysmal nocturnal dyspnea, and orthopnea [2, 3]. Valvular heart disease, coronary artery sclerosis, hypertension, and chronic lung diseases such as acute pulmonary infarction and emphysema are potential causes of HF [4]. In addition, physiological stress, such as pregnancy, fatigue, or rapid fluid overload, can increase the burden on the heart and cause myocardial failure in susceptible individuals [5]. The current treatment strategy for HF involves a step-by-step approach. Angiotensin-converting enzyme inhibitors and diuretics are common types of medications used in basic treatment [6]. Beta-blockers, such as metoprolol, are used to treat persistent symptoms by lowering heart rate and cardiac strain [7]. In more severe situations, cardiac resynchronization treatment (CRT) can be used to promote heart contractile coordination. Implantable cardioverter-defibrillator (ICD) treatment is used to prevent sudden death in patients. In addition, the novel Sirtuin 3 (SIRT3)-targeted small-molecule agonist 2-amino-4-(3,4-dihydroxyphenyl)-6-hydroxy-pyrimidine-5-carboxamide (2-APOC) has shown potential in the treatment of HF by targeting specific ion channels [8]. This emerging evidence suggests that in-depth research on the regulation of HF-related ion channels in cardiomyocytes may provide important directions for the development of new treatment strategies.
Transient receptor potential cation channel subfamily V member 1 (TRPV1) is an ion channel protein from the transient receptor potential (TRP) family. Numerous physiological and pathological processes, including pain perception, temperature control, and the inflammatory response, are influenced by TRPV1. This molecule has a low permeability to anions but a high permeability to cations such as calcium ions. In recent years, TRPV1 has become an important target for drug research. Research has shown that aberrant expression of this gene is intimately linked to the development of numerous disorders. As an agonist of TRPV1, capsaicin can not only promote fat decomposition and improve vascular function but also activate TRPV1 in cardiomyocytes, promote energy metabolism, and reduce myocardial hypertrophy and oxidative stress caused by high salt [9]. On the basis of these important regulatory roles of TRPV1 in the cardiovascular system, researchers have investigated its therapeutic potential for cardiac remodeling and dysfunction. For example, Horton JS et al. [10] reported that TRPV1 antagonists can be used as a new treatment option for cardiac hypertrophy and HF. TRPV1, as a key regulator of the natriuretic peptide signaling pathway, may offer a novel molecular intervention strategy for cardiac hypertrophy and HF. Another rat model study revealed that moxibustion can reduce myocardial damage caused by chronic heart failure (CHF) and normalize cardiac function by regulating the expression of TRPV1, thereby reducing myocardial fibrosis [11].
Accumulating data show that ferroptosis is important in the pathophysiology of cardiovascular illnesses. Recent studies have identified key metabolic pathways that regulate ferroptosis, including iron metabolism, glutathione (GSH) metabolism, and lipid metabolism, in various cardiovascular disease models [12, 13]. Notably, autophagy, as a key regulator of ferroptosis, regulates cellular iron homeostasis by degrading iron storage proteins such as ferritin, leading to elevated intracellular iron levels and subsequent lipid peroxidation. Mitophagy, a form of selective autophagy, has emerged as a key regulator of HF progression [14]. A member of the sideroflexin (SFXN) protein family, sideroflexin 2 (SFXN2), plays a role in the metabolism of iron in the mitochondria. A recent study demonstrated a role for SFXN2 in regulating heme biosynthesis, with SFXN2-knockdown cells showing increased mitochondrial iron content. SFXN2 overexpression inhibits mitophagy in multiple myeloma cells and increases iron-mediated energy production by inhibiting PTEN-induced kinase 1 (PINK1)/Parkin-mediated mitophagy along with heme oxygenase 1 (HO1)-mediated protection against oxidative stress [15]. However, the role of SFXN2 in HF is unknown.
HF is a complicated illness with several underlying causes, such as oxidative stress, mitochondrial dysfunction, and cell death. Studies have shown that TRPV1, as an ion channel, can regulate calcium homeostasis and inflammatory responses in multiple cell types, but its specific role in HF is not fully understood. This study focused on the regulatory mechanism of TRPV1 in mitochondrial autophagy and ferroptosis to investigate its role in angiotensin II-induced cardiomyocyte injury and pressure load-induced HF in a mouse model. This analysis of the molecular mechanism of TRPV1 in HF, especially its role in the SFXN2-dependent pathway, provides a new perspective on the possibility of TRPV1 as a new target for HF therapy.
Human cardiomyocytes AC16 (CL-0790, Procell, Wuhan, Hubei, China) were cultivated in Dulbecco’s modified Eagle’s medium (DMEM, #G4512-500ML, Servicebio, Wuhan, Hubei, China) supplemented with 10% fetal bovine serum (FBS, #164210; Pricella, Shanghai, China) under standard conditions (37 °C, 5% CO2). AC16 cells were subjected to short tandem repeat (STR) analysis and Mycoplasma testing. The cells were passaged using 0.25% trypsin (#BL501B; Biosharp, Hefei, Anhui, China) once they reached 70–80% confluence. For cryopreservation, the cells were resuspended in freezing medium consisting of DMEM, FBS, and dimethyl sulfoxide (DMSO, #HY-Y0320C, MCE, Monmouth Junction, NJ, USA) at a ratio of 5:4:1. The cell suspension was transferred into cryovials and gradually cooled to –80 °C overnight before being stored in liquid nitrogen.
Angiotensin II (Ang II) is a major regulator of cardiovascular disease, causing
cardiomyocyte hypertrophy, fibrosis, and oxidative stress [16]. In this study,
AC16 human cardiomyocytes were treated with 1 µmol/L Ang II (#HY-13948,
MCE, Monmouth Junction, NJ, USA) for 24 hours to establish a cell model that
mimics the pathological conditions of HF. The small interfering RNA (siRNA)
sequences targeting SFXN2 (Supplementary Material 1), the
TRPV1 overexpression plasmid (Supplementary Material 2) and the
negative control (OE-NC, pRP[Exp]-CMV
Eight- to twelve-week-old C57BL/6J male mice (20–25 g) (total = 32) were
obtained from Liaoning Changsheng Biotechnology Co., Ltd. (Shenyang, Liaoning,
China), which maintains specific pathogen-free certification
(SCXK[Liao]2020–000). Every animal experiment was carried out in compliance with
the rules and regulations that were authorized by the Animal Ethics Committee’s
Institutional Animal Care and Use Committee of The First Affiliated Hospital of
Anhui University of Chinese Medicine (AZYFY-2024-1006). All experiments were
conducted in accordance with the 3R principles. Research was conducted at the
Anhui Provincial Key Laboratory of Meridians and Organs. The animals were
maintained at 22
The mice were randomly assigned to four groups: the sham, TAC, TAC+AAV9-cardiac
troponin T (CTNT)-control, and TAC+AAV9-CTNT-TRPV1 groups (n =
8 per group). The mice were anesthetized with ketamine (100 mg/kg) and xylazine
(10 mg/kg) (6–1712, Bingene, Beijing, China) via intraperitoneal injection. TAC
surgery was conducted as previously described [17] to create a mouse model of
cardiac hypertrophy and HF. Sham-operated control mice underwent the same surgery
but without aortic constriction. AAV9 was used to achieve cardiac-specific
overexpression of TRPV1. The AAV9-TRPV1 vector was constructed
by inserting mouse TRPV1 complementary DNA (cDNA) under the control of
the promoter (Supplementary Material 3). The AAV9-CTNT-control vector
(AAV00274Z, Creative-Biogene, Shirley, NY, USA) without any exogenous gene was
used as a control. AAV9 vectors (1
Cardiac function was evaluated using a Vevo 2100 system (VisualSonics, Toronto, ON, Canada) with a 30 MHz transducer at week 8 post-surgery. Under 2% isoflurane (#R510-22-10, RWD, Shenzhen, Guangdong, China) anesthesia and temperature monitoring, parasternal short-axis M-mode images were acquired at the papillary muscle level. Left ventricular (LV) functional parameters, including the ejection fraction (EF) and fractional shortening (FS), were analyzed via Vevo Lab software (version 3.1.1, FUJIFILM VisualSonics Inc., Toronto, ON, Canada) by an independent operator unaware of group allocation. All echocardiographic measurements were carried out by an experienced operator who was unaware of the experimental groups.
Following the manufacturer’s instructions, TRIzol reagent (#G3013-100ML; Life Technologies, Carlsbad, CA, USA) was used to separate total RNA from AC16 cells. The RNA concentration and purity were evaluated via an OD1000+ ultramicrospectrophotometer (Nanjing Wuyi Technology Co., Ltd., Nanjing, Jiangsu, China). Samples showing intact RNA bands on 1% agarose gels (ST004L, Beyotime, Shanghai, China) and RNA integrity number (RIN) values exceeding 8 qualified for library construction. The NEBNext Ultra RNA Library Prep Kit for Illumina (#E7530L; New England Biolabs, Ipswich, MA, USA) was used to produce RNA sequencing (RNA-seq) libraries in accordance with the manufacturer’s instructions. Poly-T magnetic beads were used to isolate mRNA for fragmentation. Double-stranded cDNA was generated through reverse transcription with random hexamers followed by second-strand synthesis. After that, the double-stranded cDNA underwent PCR amplification, adapter ligation, end repair, and A-tailing. A Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) was used to quantify the final cDNA libraries, and an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) was used for validation. The libraries were sequenced using 150 bp paired-end reads on an Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA). Shanghai OE Biotech. Co., Ltd. (Shanghai, China) carried out the RNA sequencing. For every group, three biological replicates were conducted.
The “limma” package (version 3.54.0) in R (version 4.0.3, R Foundation for
Statistical Computing, Vienna, Austria) was used to perform differential gene
expression analysis between the control and TRPV1 overexpression groups.
Genes with
A total of 1576 Mito-RGs were collected from the Molecular Signature Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/). The overlapping genes between 249 TRPV1-related genes and 1576 Mito-RGs were identified using the Venn online graphical tool (https://bioinformatics.psb.ugent.be/webtools/Venn/).
The sequencing datasets (containing the control group and the TRPV1 overexpression group) were used to assess the expression levels of seven overlapping genes. The raw counts were normalized with DESeq2 (version 1.30.1) in R, and the log2-transformed normalized expression values were visualized with pheatmap (version 1.0.12). The correlation matrix of the seven genes was calculated and visualized using a corrplot (version 0.84). The Spearman correlation coefficient between TRPV1 and each of the seven Mito-RGs was calculated in R using the Spearman method. Scatter plots with trend lines were generated using ggplot2 (version 3.3.3) to visualize the correlations.
Mitochondria and the cytosol were isolated from cardiac tissue via a Tissue Mitochondria Isolation Kit (#C3606, Beyotime, Shanghai, China) and a Tissue Cytoplasm and Nucleus Separation Kit (#NT-032, Invent, Wilmington, DE, USA) according to the manufacturer’s instructions. Similarly, mitochondria and cytosol from AC16 cells were extracted using the Cell Mitochondria Isolation Kit (#C3601, Beyotime, Shanghai, China) and the Cell Cytoplasm and Nucleus Separation Kit (#BB-36021, Beibokit, Shanghai, China) following the manufacturer’s protocols.
Proteins in cardiac tissue and AC16 cells (mitochondria) were extracted with
radioimmunoprecipitation assay (RIPA) buffer (#P0013, Beyotime, Shanghai,
China). After the samples were homogenized, they were incubated on ice for 30
minutes and then centrifuged at 12,000
| Antibody | Cell (Dilution; Item No.; Manufacturer) | Tissue (Cell (Dilution; Item No.; Manufacturer)) |
| Transient receptor potential cation channel subfamily V member 1 (TRPV1) | 1:500, #DF10320, Affinity Biosciences, Liyang, Jiangsu, China | 1:1000, #DF10320, Affinity Biosciences, Liyang, Jiangsu, China |
| Sideroflexin 2 (SFXN2) | 1:1000, #BS77506, Bioworld Technology, Chongqing, China | 1:1000, #BS77506, Bioworld Technology, Chongqing, China |
| Glutathione peroxidase 4 (GPX4) | 1:500, #DF6701, Affinity Biosciences, Liyang, Jiangsu, China | 1:1000, #DF6701, Affinity Biosciences, Liyang, Jiangsu, China |
| Parkin | 1:500, #AF0235, Affinity Biosciences, Liyang, Jiangsu, China | 1:500, #2132, Cell Signaling Technology, Danvers, MA, USA |
| PTEN-induced kinase 1 (PINK1) | 1:1000, #DF7742, Affinity Biosciences, Liyang, Jiangsu, China | 1:1000, #DF7742, Affinity Biosciences, Liyang, Jiangsu, China |
| Sequestosome 1 (SQSTM1) | 1:5000, #66184-1-Ig, Proteintech, Wuhan, Hubei, China | 1:5000, #66184-1-Ig, Proteintech, Wuhan, Hubei, China |
| Atrial natriuretic peptide (ANP) | / | 1:500, #DF6497, Affinity Biosciences, Liyang, Jiangsu, China |
| Beta-myosin heavy chain ( |
/ | 1:500, #ab23990, Abcam, Cambridge, Cambridgeshire, UK |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 1:5000, #380626, ZENBIO, Chengdu, Sichuan, China | 1:5000, #380626, ZENBIO, Chengdu, Sichuan, China |
| Voltage-dependent anion channel 1 (VDAC1) | 1:500, #AF5478, Affinity Biosciences, Liyang, Jiangsu, China | 1:500, #AF5478, Affinity Biosciences, Liyang, Jiangsu, China |
TRIzol reagent (#G3013-100ML; Life Technologies, Carlsbad, CA, USA) was used to extract total RNA from AC16 cardiomyocytes. An OD1000+ ultramicrospectrophotometer (Nanjing Wuyi Technology Co., Ltd., Nanjing, Jiangsu, China) was used to measure the concentration and purity of the RNA, and 1% agarose gel electrophoresis was used to confirm the integrity of the RNA. With a reverse transcription kit with dsDNAse (#BL699A; Biosharp, Hefei, Anhui, China) and a PTC-200 heat cycler (Bio-Rad, Hercules, CA, USA), first-strand cDNA was generated. The reaction proceeded for 30 min at 37 °C followed by 5 min at 85 °C. qRT‒PCR was carried out with an ABI StepOne Plus Real-Time PCR System with Taq SYBR Green qPCR Premix Universal (#EG20117M, Best-enzymes, Lianyungang, Jiangsu, China). A 20 µL PCR mixture was prepared with SYBR Green premix (10 µL), primers (0.4 µL each, 10 µM, Sangon Biotech, Shanghai, China), cDNA (3 µL), and Diethylpyrocarbonate-treated Water (DEPC-H2O) (6.2 µL, #D1007; Generay Biotech, Shanghai, China). Initial denaturation at 95 °C for 30 s was followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s as part of the amplification process. Melting curve analysis was used to establish the specificity of amplification. The expression levels of TRPV1 and mitochondria-related genes were analyzed. The internal reference gene was GAPDH. Table 2 lists the specific sequences of the primers used. The relative expression of genes was calculated using the 2-ΔΔCT method.
| Gene | Size (bp) | Forward primer (5′-3′) | Reverse primer (5′-3′) |
| Hu-TRPV1 | 195 | CCACTCTTCTCCCACACGAG | GGCAGGTGTCCTTTTGGAGT |
| G0/G1 switch gene 2 (G0S2) | 118 | TCCACCAAAGGAGTTTGGGA | TCCTTCCTCCCTAGTGCAAA |
| Solute Carrier Family 25 Member 24 (SLC25A24) | 121 | TATCCAGCACCTGTGGTCAG | ATTCGTCGAAAGAGGCCAAC |
| Schindler Disease (SDS) | 194 | CCCATCCTTAGCCACCTTGC | GGATGGCTGAGATGGTTCCT |
| Sideroflexin 2 (SFXN2) | 120 | AAGGGAATCTGCGTGAAGGA | GATCATCCCAGGAGCTGACA |
| Creatine Kinase Mitochondrial 2 (CKMT2) | 192 | AAAGATCACCCAAGGGCAGT | TCGGACAGCTTGTAGTAGCG |
| Tubulin Polymerization Promoting Protein (TPPP) | 113 | CCTTCTCCACTCAGCTCCAA | GGTGACCACTATGTCCTCGT |
| GAPDH | 138 | CGGATTTGGTCGTATTGG | GGTGGAATCATATTGGAACA |
A bicinchoninic acid (BCA) protein assay kit (#BL521A; Biosharp, Hefei, Anhui, China) was used to measure the protein concentration of the homogenate after AC16 cells from various treatment groups were obtained and homogenized in ice-cold phosphate-buffered saline (PBS, #G4207; Servicebio, Wuhan, Hubei, China). The malondialdehyde (MDA), superoxide dismutase (SOD), glutathione (GSH), and ferrous iron (Fe2+) levels associated with lipid peroxidation were subsequently measured using an MDA assay kit (#A003-1-2), a SOD kit (#A001-3-1), a reduced GSH assay kit (#A006-2-1), and a ferrous ion colorimetric kit (#E-BC-K773-M; Elabscience, Wuhan, Hubei, China). All unlabeled kits were provided by Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). Every test was carried out in compliance with the manufacturer’s instructions. The absorbance was measured at 532 nm (MDA), 450 nm (SOD), 420 nm (GSH), and 593 nm (Fe2+) using a microplate reader (Bio-Rad, Hercules, CA, USA). The quantitative methods were performed according to the instructions provided in the manual.
The intracellular Ca2+ concentration of AC16 cells was analyzed using a
calcium assay kit (#40776ES50, YEASEN, Shanghai, China) according to the
manufacturer’s instructions. In brief, AC16 cells were extracted via
centrifugation at 1000 rpm for 5 minutes and then rinsed with PBS. The cells were
then incubated with 5 µM Rhod-2 AM (prepared in DMSO with a 2–5 mM stock
solution) for 30 minutes at 37 °C in the dark. The cells were washed
twice with PBS, followed by centrifugation (1000 rpm, 5 min). Fluorescence images
were captured with a Leica DMI3000B inverted fluorescence microscope (Leica,
Wetzlar, Hesse, Germany) at 200
Following the manufacturer’s instructions, cell viability was evaluated using
the Calcein AM/PI Live/Dead Viability/Cytotoxicity Assay Kit (#C2015M; Beyotime,
Shanghai, China). After being washed with PBS, AC16 cells from various treatment
groups were treated with the working solution for 30 minutes at 37 °C in
the dark. PI (red fluorescence) was used to identify dead cells, whereas Calcein
AM (green fluorescence) was used to identify live cells. Fluorescence images were
taken at 200
MitoSOX Red (HY-D1055, MCE, Monmouth Junction, NJ, USA) was used to assess the
levels of ROS in the mitochondria. AC16 cells were plated, rinsed with PBS, and
then incubated with 5 µM MitoSOX Red working solution at 37 °C for
30 minutes in the dark. Nuclei were counterstained with
4′,6-diamidino-2′-phenylindole (DAPI, #C1002; Beyotime, Shanghai,
China). Fluorescence images were captured at 200
Apoptotic cells were detected via a one-step TUNEL kit (#ECK-A320; Elabscience,
Wuhan, Hubei, China). The samples were fixed in 4% paraformaldehyde (CF189021,
Solarbio, Beijing, China) (30 min) and permeabilized with 0.2% Triton X-100
(P0096, Beyotime, Shanghai, China) (5 min) in PBS. After washing, the samples
were incubated in the dark (37 °C, 1 h) with a TdT/luciferin-dutP
reaction mixture. Nuclear staining was performed with DAPI (25 µg/mL, 10
min). Images were acquired at 100
Data analysis was performed with R (version 4.0.3, R Foundation for Statistical
Computing, Vienna, Austria) and GraphPad Prism (version 8.0.0, GraphPad Software,
San Diego, CA, USA). The results represent the mean
AC16 human cardiomyocytes were treated with 1 µmol/L Ang II to simulate
pathological cardiac conditions. TRPV1 mRNA expression was significantly
downregulated according to qRT‒PCR (p
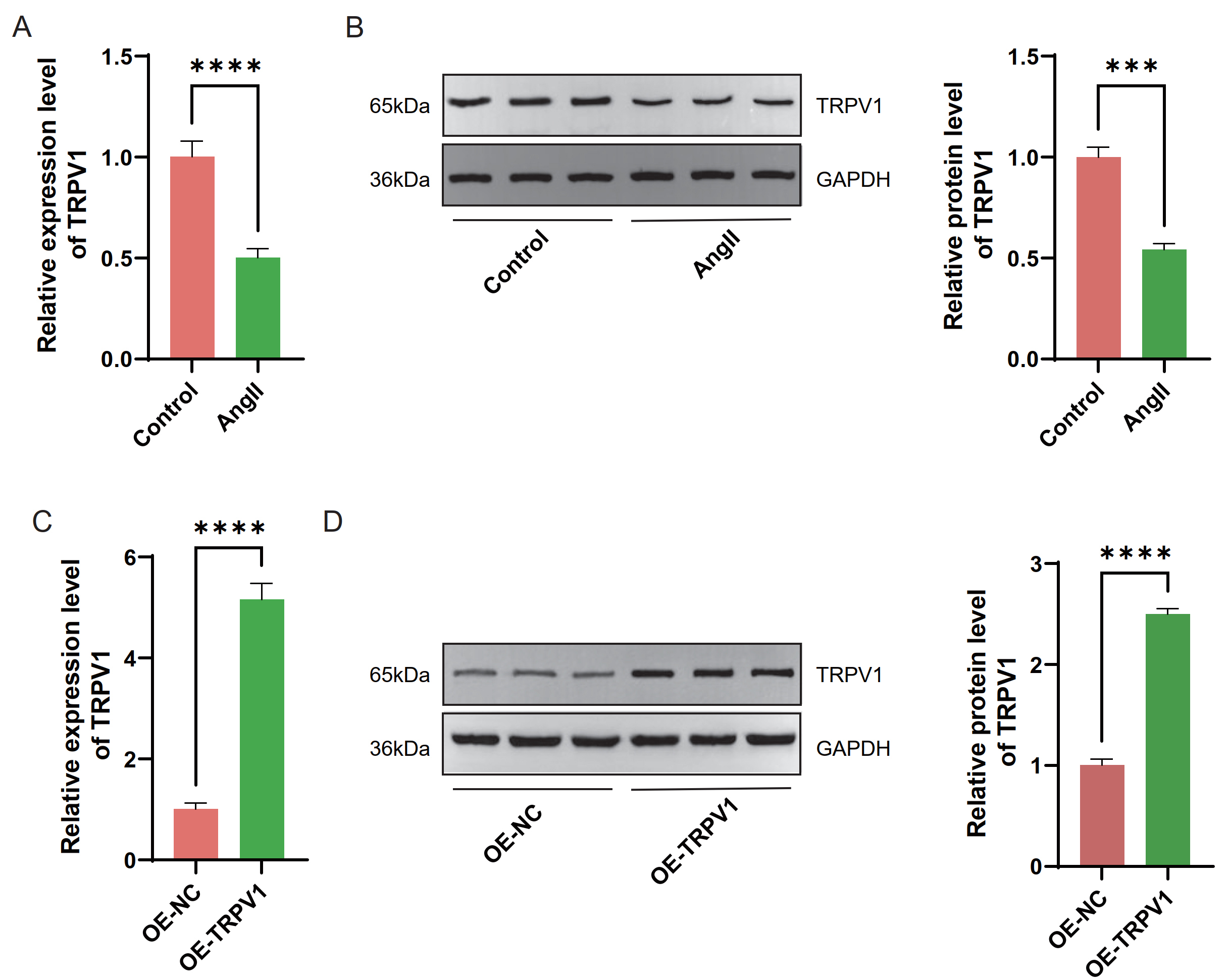 Fig. 1.
Fig. 1.
Analysis of transient receptor potential cation channel
subfamily V member 1 (TRPV1) level in AC16 cells induced by angiotensin
II (Ang II) and overexpressing TRPV1. (A) Quantitative real-time polymerase
chain reaction (qRT–PCR) detected the TRPV1 mRNA level in AC16 cells
induced by Ang II. (B) Western blot analysis of TRPV1 protein levels in AC16
cells after Ang II induction was performed and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) was used for normalization. (C) qRT–PCR detected the
TRPV1 mRNA level of AC16 cells in overexpression negative control
(OE-NC) and overexpression TRPV1 (OE-TRPV1) groups. (D) Western
blot analysis of TRPV1 protein levels of AC16 cells in OE-NC and OE-TRPV1 groups
was performed, and GAPDH was used for normalization. Each experiment was
independently repeated three times. Data are presented as mean
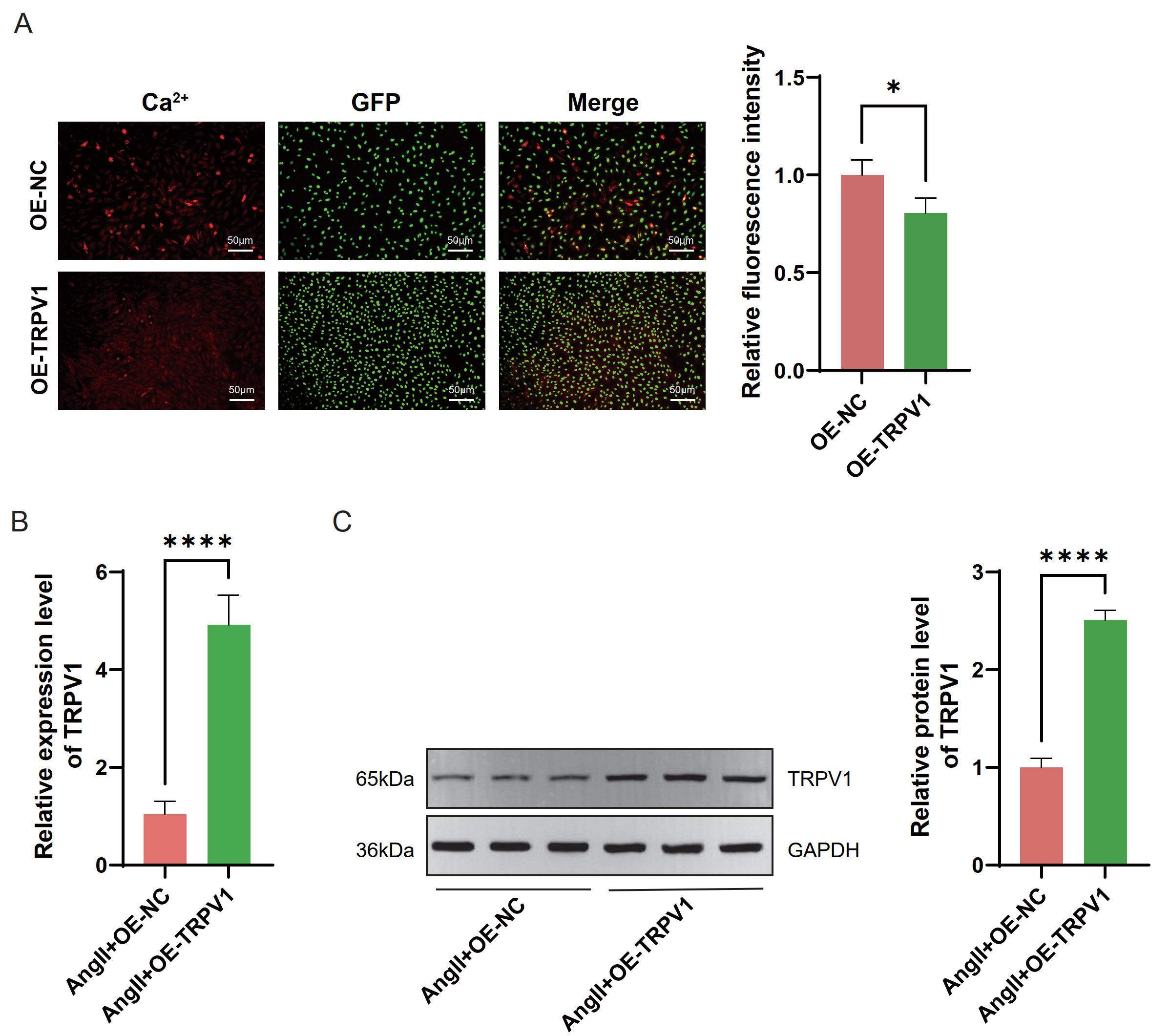 Fig. 2.
Fig. 2.
Effects of TRPV1 overexpression on intracellular
Ca2+ levels and TRPV1 expression in heart failure (HF) cell model. (A)
Representative fluorescence microscopy images showing intracellular calcium ion
(Ca2+) levels (red), green fluorescent protein (GFP) expression (green), and
merged images in OE-NC and OE-TRPV1 groups. Magnification:
200
To investigate whether TRPV1 affects cell viability under Ang II
stimulation, we performed PI/Calcein AM double staining. Compared with the
control treatment, Ang II treatment dramatically inhibited cell viability
(p
According to the screening criteria for DEGs, 195 upregulated genes and 54 downregulated genes were screened from the sequencing data (Supplementary Fig. 3). The results of the biological process (BP), cellular component (CC), and molecular function (MF) enrichment analyses revealed that the upregulated genes were associated primarily with nucleosomes (GO: 0000786), the deoxyribonucleic acid (DNA) packaging complex (GO: 0044815), protein heterodimerization activity (GO: 0046982), DNA binding (GO: 0003677), and nucleosome assembly (GO: 0006334) (Supplementary Fig. 4A). The downregulated genes were related to the extracellular region (GO: 0005576), cytokine activity (GO: 0005125), cell adhesion (GO: 0007155), and the cell surface receptor signaling pathway (GO: 0007166) (Supplementary Fig. 4B). KEGG enrichment pathway analysis revealed that the pathways enriched with the upregulated genes included the apelin signaling pathway, herpes simplex virus 1 infection, and alcoholism (Supplementary Fig. 5A). The downregulated genes were associated with KEGG pathways such as the chemokine signaling pathway, NOD-like receptor signaling pathway, influenza A pathway, and the interleukin-17 (IL-17) signaling pathway (Supplementary Fig. 5B).
To explore the possible molecular mechanisms underlying the relationship
between TRPV1 and mitochondrial function, we performed an intersection
analysis of TRPV1-related genes and Mito-RGs. Venn diagram analysis
revealed that there were 7 overlapping genes between the TRPV1-related
genes (249 genes) and Mito-RGs (1576 genes) (Fig. 3A). Among these overlapping
genes, heatmap analysis revealed different expression patterns between the groups
(Fig. 3B). Further analysis revealed differences in correlations between
different genes (Fig. 3C). For example, solute carrier family 25 member 24
(SLC25A24) and G0/G1 switch gene 2 (G0S2) were
strongly positively correlated, whereas SLC25A24 was significantly
negatively correlated with tubulin polymerization promoting protein
(TPPP) and lactate dehydrogenase A like 6B (LDHAL6B)
(p
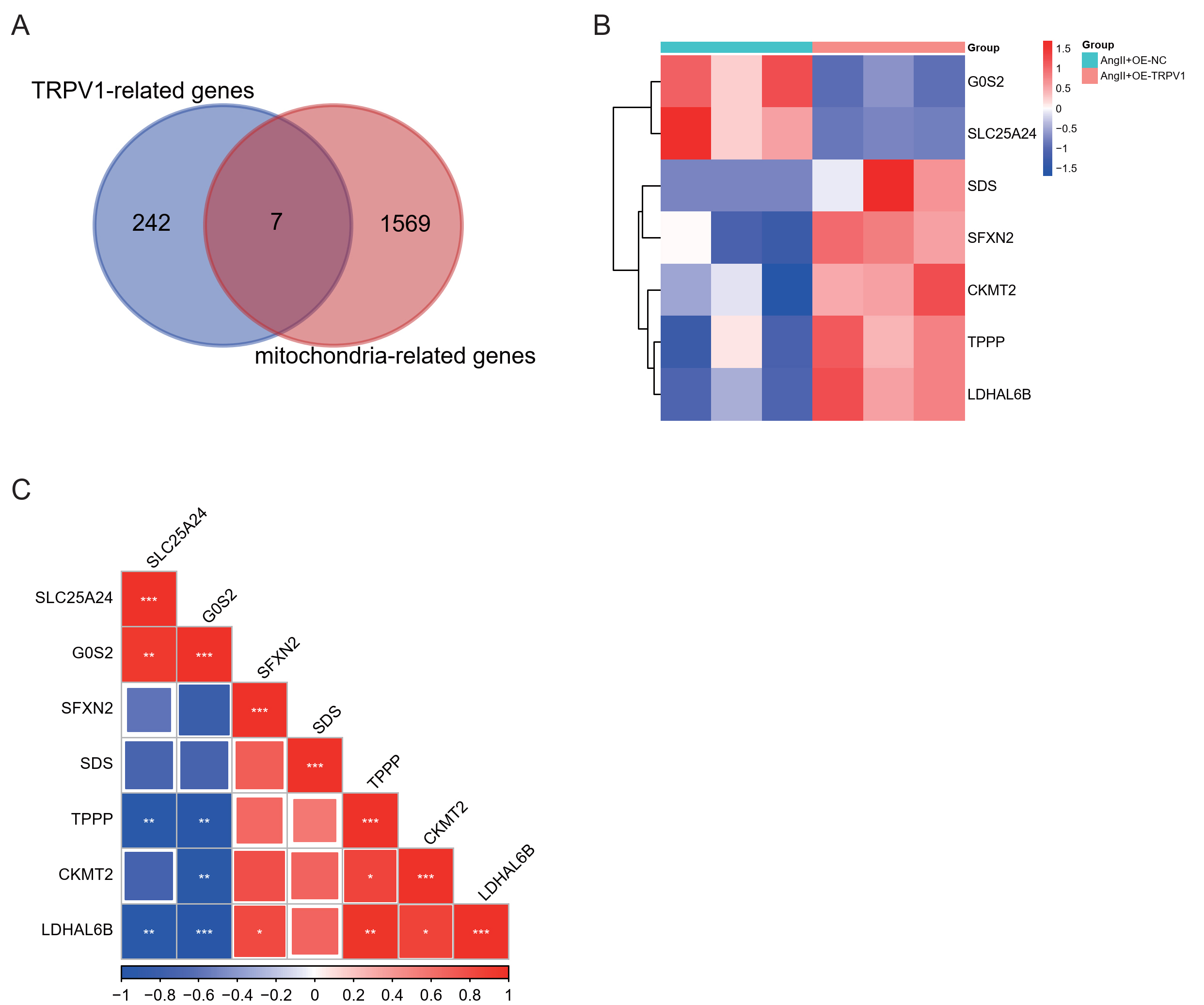 Fig. 3.
Fig. 3.
Analysis of TRPV1 and mitochondria-related gene
expression patterns. (A) Venn diagram showing the overlap between
TRPV1-related genes and mitochondria-related genes. (B) Heatmap
visualization of expression patterns for the seven overlapping genes across
different experimental groups. Color scale represents normalized expression
values, with red indicating upregulation and blue indicating downregulation. (C)
Correlation matrix showing the relationships among the seven overlapping genes.
The size and color intensity of squares represent correlation strength (–1 to
1), with blue indicating negative correlations and red indicating positive
correlations. Statistical significance is denoted by asterisks (*p
Further analysis confirmed the correlation between TRPV1 and seven
genes (Supplementary Fig. 6A–G). Strong correlations were observed
between TRPV1 and both LDHAL6B (p = 0.03, r = 0.86)
and SFXN2 (p = 0.04, r = 0.83) (Supplementary Fig.
6C,E). In vitro experiments verified the expression of these genes in
AC16 cells treated with Ang II. After TRPV1 overexpression, qRT‒PCR
analysis revealed that the expression of schindler disease (SDS),
SFXN2, creatine kinase mitochondrial 2 (CKMT2), TPPP,
and LDHAL6B was dramatically greater than that in the Ang II+OE-NC group
(p
To assess the therapeutic potential of TRPV1 in HF, we generated a
model of cardiac dysfunction caused by pressure overload using TAC. Cardiac
function was assessed using echocardiography. Compared with those in the sham
group, the cardiac function of the mice in the TAC-treated group was considerably
worse, as evidenced by a reduction in FS and EF (p
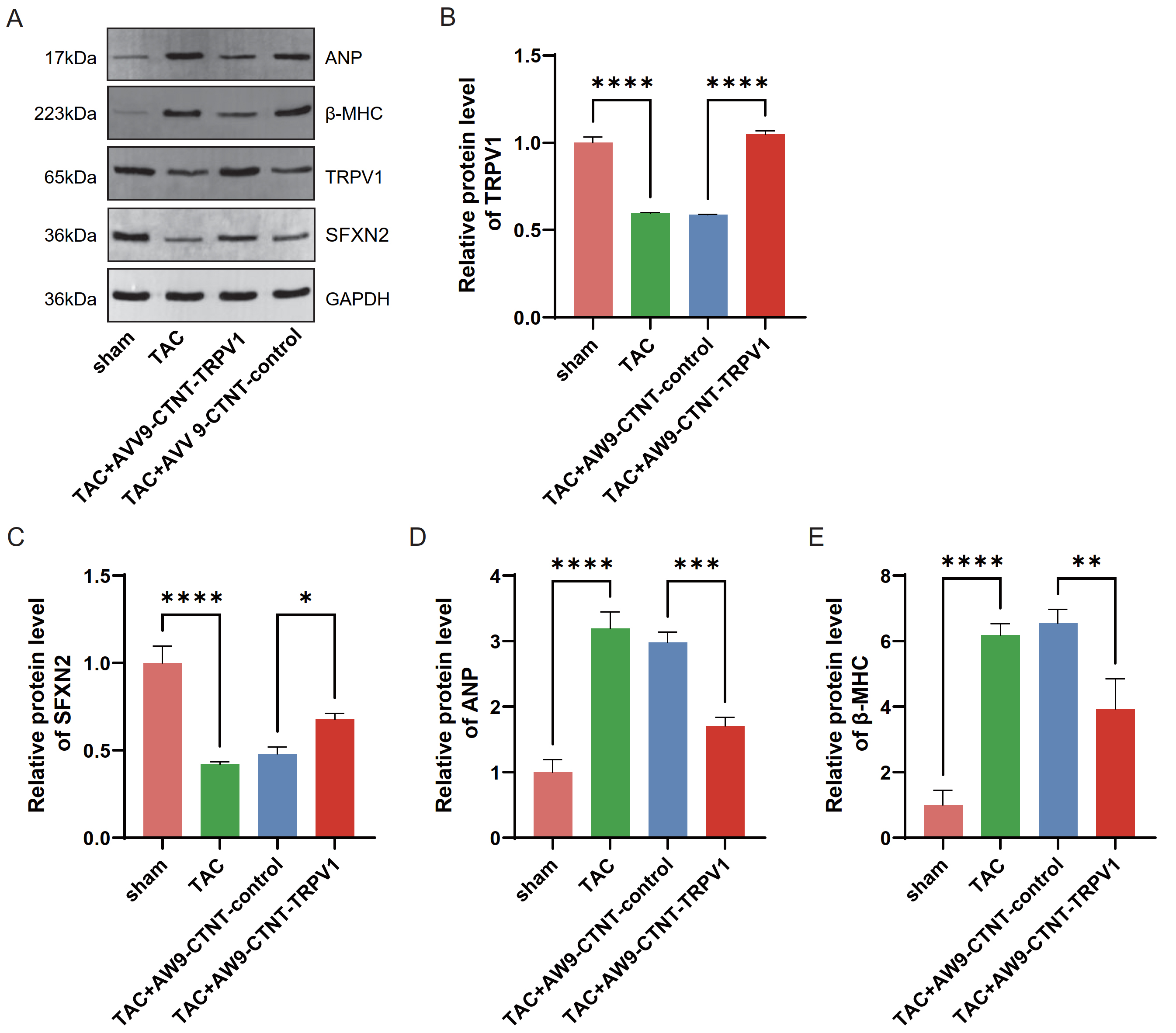 Fig. 4.
Fig. 4.
TRPV1 overexpression regulates the expression of
HF-related markers in the TAC-induced HF model. (A) Representative
Western blot images showing protein expression of HF-related markers (atrial
natriuretic peptide (ANP), beta-myosin heavy chain (
Using the TAC mouse model, we examined how TRPV1 regulates the levels
of autophagy-related proteins. Compared with sham surgery, TAC surgery increased
the expression level of sequestosome 1 (SQSTM1) in the cytoplasm while drastically lowering
the amounts of Parkin and PTEN-induced kinase 1 (PINK1) (p
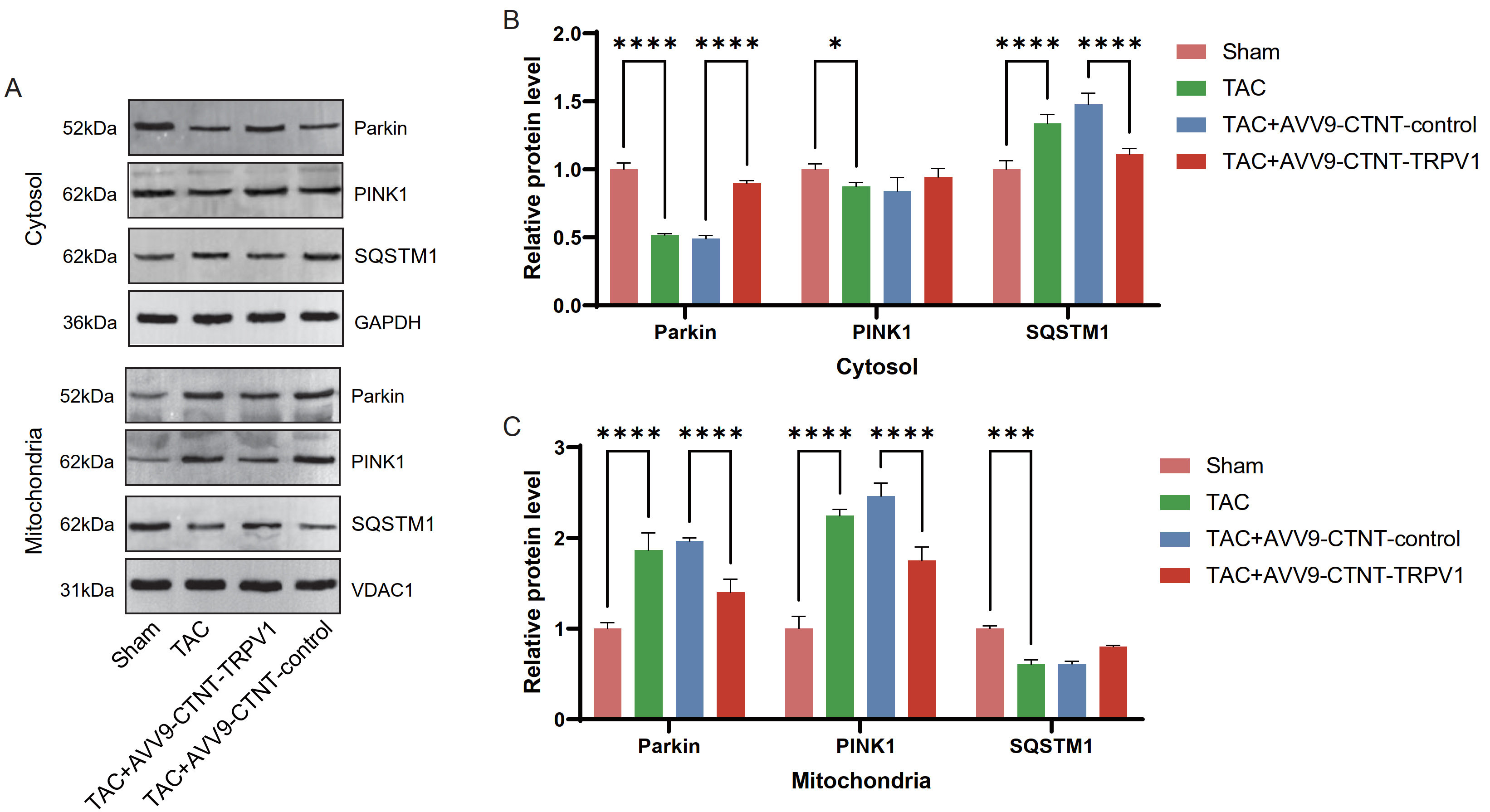 Fig. 5.
Fig. 5.
TRPV1 overexpression regulates the expression of
mitophagy-related proteins in the cytoplasm and mitochondria in the TAC-induced
HF model. (A) Representative Western blot images showing the protein expression
of mitophagy markers (Parkin, PTEN-induced kinase 1 (PINK1), and sequestosome 1
(SQSTM1)) in the cytoplasm and mitochondria fractions. GAPDH and
voltage-dependent anion channel 1 (VDAC1) were used as loading controls for the
cytoplasm and mitochondria fractions, respectively. (B) Quantification of the
cytoplasmic protein levels of Parkin, PINK1, and SQSTM1 in the experimental
groups, with GAPDH as the standard. (C) Quantification of the mitochondrial
protein levels of Parkin, PINK1, and SQSTM1 in the experimental groups, with
VDAC1 as the standard. n = 8 per group. Data are presented as mean
Previously, we analyzed and verified that TRPV1 and SFXN2
interact in HF, but the specific mechanism was unclear. Therefore, we continued
to analyze this phenomenon through in vitro experiments. To analyze the
role of SFXN2, we designed three siRNAs targeting SFXN2. The
qRT‒PCR results demonstrated that all three siRNAs effectively reduced
SFXN2 expression in cells, with Si-SFXN2#3 (referred to as
Si-SFXN2) exhibiting the highest knockdown efficacy (p
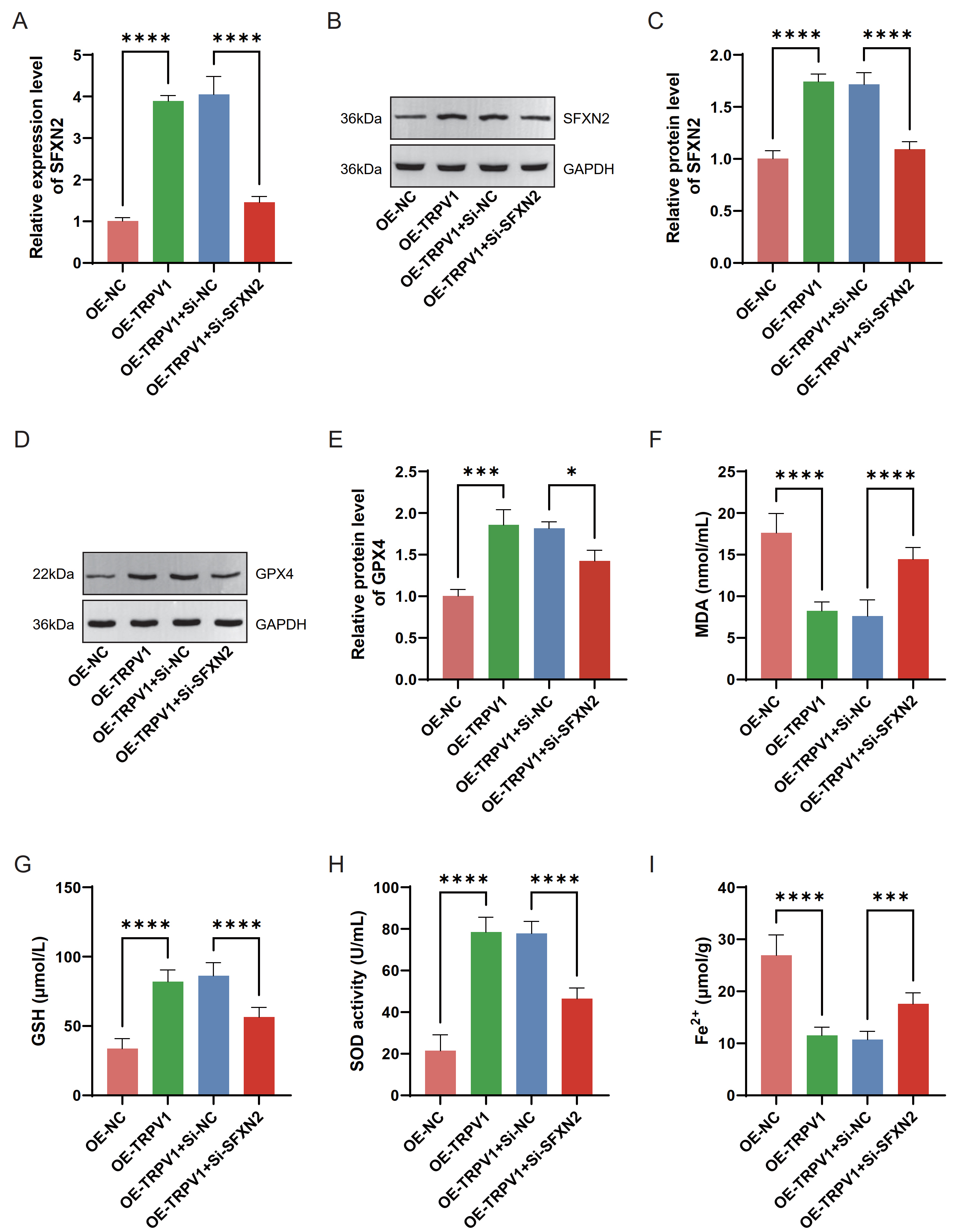 Fig. 6.
Fig. 6.
TRPV1 regulates ferroptosis in HF through an
SFXN2-dependent mechanism. (A) qRT–PCR was used to detect the changes
in the mRNA level of SFXN2 after TRPV1 was overexpressed alone
or in combination with down-expressed SFXN2 in Ang II-induced AC16
cardiomyocytes. (B,C) Western blot was used to detect the protein level of SFXN2
after TRPV1 was overexpressed alone or in combination with
down-expressed SFXN2 in Ang II-induced AC16 cardiomyocytes. (D,E)
Western blot was used to detect the protein level of glutathione peroxidase 4
(GPX4) in Ang II-induced AC16 cardiomyocytes after TRPV1 was
overexpressed alone or in combination with down-expressed SFXN2. (F–I)
Commercial kits were used to detect the changes in the levels of malondialdehyde
(MDA), glutathione (GSH), superoxide dismutase (SOD), and ferrous iron
(Fe2+) after TRPV1 was overexpressed alone or in combination with
down-expressed SFXN2 in Ang II-induced AC16 cardiomyocytes. Each
experiment was independently repeated three times. Data are presented as mean
Next, we further analyzed the mechanism of action of SFXN2 and
TRPV1 in an HF model. TUNEL staining demonstrated that TRPV1
expression alone resulted in a substantial decrease in apoptosis compared with
that in the OE-NC group (p
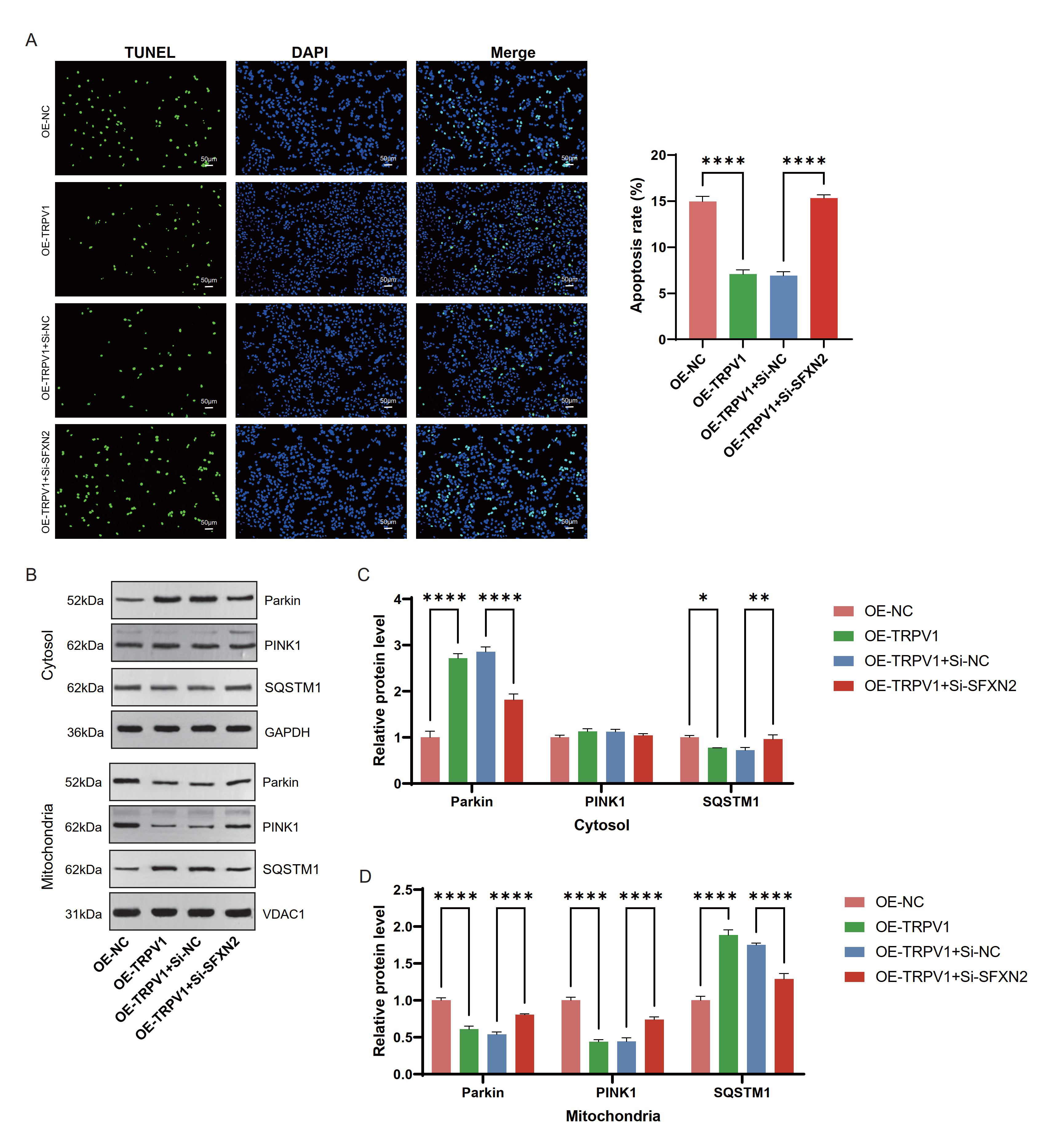 Fig. 7.
Fig. 7.
TRPV1 inhibits mitochondrial autophagy by regulating
PINK1-Parkin-SQSTM1 through upregulating SFXN2. (A) In Ang II-induced
AC16 cardiomyocytes, terminal deoxynucleotidyl transferase dUTP nick-end labeling
(TUNEL) staining was used to detect the apoptosis levels in different
experimental groups (OE-NC; OE-TRPV1; OE-TRPV1+small
interfering RNA negative control (Si-NC); OE-TRPV1+small interfering RNA
targeting SFXN2 (Si-SFXN2)). Left: Representative fluorescence
images showing TUNEL-positive cells (green) and nuclear
4′,6-Diamidino-2′-phenylindole (DAPI) staining (blue). Right:
Quantification of apoptosis rates among experimental groups. Magnification:
100
HF is a severe symptom or advanced stage of certain cardiac disorders. HF can
manifest as a decline in cardiac pumping function, decreased ejection function,
dyspnea, fatigue, edema and other symptoms. Major advancements have been made in
the treatment of HF in recent years, including sodium-dependent glucose
transporter 2 (SGLT2) inhibitors for reducing cardiac workload and increasing
cardiac metabolism, glucagon like peptide 1 (GLP-1) receptor agonists/SGLT2
inhibitors as first-line treatments for HF patients with reduced ejection
fraction (HFrEF), and
Research has shown that changes in myocardial calcium ions regulate the process of contraction and blood flow in HF. In the context of HF, the interaction between myocardial cells and the calcium ion regulatory mechanism in the body is impaired, resulting in unstable contraction and expansion of the heart [21]. This change, in turn, affects the blood transfusion capacity of the heart and thus causes HF in patients. To explore the regulatory mechanism involved in this process, we investigated the role of TRPV1 under pathological conditions induced by Ang II. Our results indicated that Ang II therapy significantly reduced TRPV1 expression in AC16 cardiomyocytes. Fluorescence analysis indicated that TRPV1 overexpression dramatically decreased the intracellular Ca2+ level in AC16 cardiomyocytes, indicating that TRPV1 is essential for preserving calcium homeostasis.
Oxidative stress contributes to the progression of HF through many pathways,
which can cause myocardial cell damage and mitochondrial dysfunction and induce
inflammatory responses by activating the nuclear factor
kappa-light-chain-enhancer of activated B cells (NF-
Through sequencing data, we screened 195 upregulated and 54 downregulated
TRPV1-related DEGs from AC16 cardiomyocytes with TRPV1
overexpression. According to the enrichment analysis, these genes were connected
to pathways such as the apelin signaling pathway, herpes simplex virus 1
infection, alcoholism, the IL-17 signaling pathway, influenza A, the chemokine
signaling pathway, and the NOD-like receptor signaling pathway. Through a variety
of signaling pathways, TRPV1 may be crucial to the onset and progression
of HF, according to the roles of these enriched pathways. The apelin signaling
pathway is one such pathway and has been extensively recognized to be linked to
HF [24]. Apelin binds to the apelin receptor (APJ), regulates cardiac function
and vasodilation, and has a crucial protective effect [25]. The pathological
process of HF is often accompanied by an inflammatory response, and chemokines
such as C-C motif chemokine ligand 2 (CCL2) and C-X-C motif chemokine ligand 12
(CXCL12) play key roles in myocardial injury, fibrosis and remodeling [26, 27].
Influenza virus infection can also induce viral myocarditis, which can lead to HF
in severe cases [28]. NOD-like receptor family pyrin domain containing 3 (NLRP3)
inflammasomes in NOD-like receptors (NLRs) promote myocardial fibrosis and
ventricular remodeling by releasing interleukin-18 (IL-18) and interleukin-1 beta
(IL-1
Studies have revealed that mitochondrial dysfunction is directly associated with the incidence and progression of HF, with dysfunctional mitochondrial energy metabolism, gene mutations, oxidative stress damage, and calcium homeostasis imbalance being major variables that cause HF [31, 32]. In-depth studies into the association between mitochondrial malfunction and HF may lead to the discovery of novel therapeutic targets. In this study, seven genes related to TRPV1 (G0S2, SLC25A24, SDS, SFXN2, CKMT2, TPPP, and LDHAL6B) were identified from Mito-RGs. Research has revealed substantial positive associations between the TRPV1, SFXN2, and LDHAL6B genes. qRT‒PCR was used to validate the changes in the expression of these genes in Ang II-treated AC16 cells. Among these genes, SDS, SFXN2, CKMT2, TPPP, and LDHAL6B were significantly overexpressed after TRPV1 overexpression. These results suggest that TRPV1 has a potential connection with mitochondrial regulation. Given that SFXN2 plays a crucial role in controlling iron metabolism and mitochondrial activity and that the relationship between LDHAL6B and mitochondria has not been explored in detail, SFXN2 was selected as a key downstream target of TRPV1 for further study. SFXN2, an evolutionarily conserved protein found in mitochondria, plays a role in the metabolism of iron within the mitochondria. Our study revealed that TRPV1 regulates SFXN2 expression in cardiomyocytes, suggesting a potential link between calcium signaling and mitochondrial iron metabolism in HF.
The TAC model is a common experimental model of left ventricular hypertrophy
(LVH) and HF and has been widely used to study the processes of compensatory or
maladaptive cardiac remodeling, cardiac hypertrophy, fibrosis, and dysfunction
[33, 34]. To evaluate the therapeutic potential of TRPV1 in HF induced by
pressure overload, we used this well-established model in our study. The
increased ejection fraction and fractional shortening in the TAC-induced HF model
demonstrated normalized cardiac function in this study due to TRPV1
overexpression. TRPV1 overexpression reduced the TAC-induced increase in
cardiac stress indicators (ANP and
Excessive ROS generation during HF development is caused by mitochondrial dysfunction and is a major factor in the promotion of oxidative damage and disruption of cellular homeostasis [37]. When mitochondrial function is impaired, cardiomyocytes are unable to effectively clear damaged mitochondria, resulting in ROS accumulation and subsequent cell death. Recent studies have shown that intracellular stress caused by lipid peroxidation, oxidative stress, and inflammatory responses triggers autophagy in endothelial cells and cardiomyocytes, particularly by affecting cardiac iron metabolism [38, 39]. This dysregulation of iron homeostasis may lead to ferroptosis, a newly recognized form of regulated cell death. In HF with preserved ejection fraction (HFpEF), the inhibition of ferroptosis has been shown to alleviate pathological cardiac remodeling, including cardiac fibrosis and decreased capillary density [40]. Notably, targeting the ACSL4-ferroptosis-ferroptosis signaling axis may represent a promising therapeutic strategy for HF [41]. For example, phosphoglycerate mutase family member 5 (PGAM5) has a protective effect against ferroptosis through a reduction in oxidative stress mediated by the kelch-like ECH-associated protein 1 (KEAP1)/nuclear factor erythroid 2-related factor 2 (NRF2) pathway [42]. Moreover, GPX4, a key antiferroptosis protein, can be regulated through the autophagy‒lysosomal pathway, in which excessive autophagy may lead to GPX4 degradation and subsequent ferroptosis [43]. Therefore, understanding the role of ferroptosis in HF may help develop new targeted therapies.
Recent studies have proposed a complex relationship between mitophagy and
ferroptosis in HF. The PINK1-Parkin-SQSTM1 pathway has been shown to regulate
mitochondrial quality control through selective mitophagy [44]. PINK1
phosphorylation recruits Parkin to depolarized mitochondria, thereby activating
SQSTM1-mediated clearance. Notably, this mechanism has been implicated in
ferroptosis, as autophagy can promote ferroptosis by disrupting iron storage
proteins, leading to increased intracellular iron accumulation and lipid
peroxidation [45]. This connection is further supported by another study showing
that under conditions of oxidative stress, such as hypoxia/reoxygenation injury,
disturbances in iron metabolism and GSH homeostasis promote ferroptosis through
alterations in GPX4 and iron transport-related proteins [46]. Similar regulatory
mechanisms have been observed for other molecules, such as nucleolar protein
family member 4 (NPLOC4), which promotes mitochondrial function through
endoplasmic reticulum oxidoreductase 1 alpha (ERO1
Our findings revealed that TRPV1 expression was downregulated in Ang II-treated cardiomyocytes and the TAC-induced HF model, whereas the upregulation of TRPV1 expression significantly normalized cardiac function and alleviated pathological remodeling. Mechanistically, TRPV1upregulated SFXN2 expression, increased GPX4 levels and antioxidant capacity, and regulated PINK1-Parkin-SQSTM1 distribution in the cytoplasm and mitochondria. Taken together, these findings identify TRPV1 as a novel cardioprotective factor through SFXN2-dependent ferroptosis regulation and mitophagy in HF.
All data are available upon the reasonable request to the corresponding authors.
PL: methodology, writing, review and editing, and approval for final version. LYL and RHL: investigation, data curation, formal analysis, and approval for final version. MMF, JJK and BG: investigation, data curation, software, and approval for final version. SGW: methodology, writing, review and editing. JW: conceptualization, review and editing. RX: methodology, review and approval for final version. All authors contributed to the article and approved the submitted version. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was authorized by the Anhui University of Chinese Medicine First Affiliated Hospital Animal Ethics Committee’s Institutional Animal Care and Use Committee (AZYFY-2024-1006). All experiments were conducted in accordance with the 3R principles.
We would like to thank the platform support provided by Anhui Provincial Key Laboratory of Meridians and Organs.
The present research was funded by the National Key R&D Program of China (2022YFC3500502) and Anhui Provincial Scientific Research Planning Project (2023AH050796).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL37052.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.