








 , Hua Zhu 1, Shi Feng 1, Yonggang Zhang 1, Xiaoxing Xiong 1,*
, Hua Zhu 1, Shi Feng 1, Yonggang Zhang 1, Xiaoxing Xiong 1,*
1 Department of Neurosurgery, Renmin Hospital of Wuhan University, 430060 Wuhan, Hubei, China
Abstract
Ischemic stroke is a leading cause of mortality and disability worldwide, yet the interplay between peripheral and central immune responses is still only partially understood. Emerging evidence suggests that myeloid cells, when activated in the periphery, infiltrate the ischemic brain and contribute to the disruption of the blood-brain barrier (BBB) through both inflammatory and metabolic mechanisms.
In this study, we integrated bulk RNA-sequencing (RNA-seq), single-cell RNA-seq (scRNA-seq), spatial transcriptomics, and flow cytometry data from human and mouse models of ischemic stroke. Mouse stroke models were induced by transient middle cerebral artery occlusion (tMCAO), and brain tissues were later collected at specified time points for analysis. We examined time-dependent transcriptional changes in the peripheral blood, delineated cell-type-specific responses by single-cell profiling, and validated myeloid infiltration into the ischemic brain. We also investigated endothelial metabolic reprogramming and oxidative stress by combining scMetabolism analyses (a computational R package for inferring metabolic pathway activity at the single-cell level) with in vitro oxygen-glucose deprivation/reperfusion (OGD/R) experiments.
Cross-species bulk RNA-seq revealed a modest early immune shift at 3 h post-stroke, escalating significantly by 24 h, with robust myeloid-centric gene signatures conserved in humans and mice. Single-cell analyses confirmed a pronounced expansion of neutrophils, monocytes, and megakaryocytes in peripheral blood, coupled with a decrease in T and B lymphocytes. Spatial transcriptomics and flow cytometry demonstrated substantial infiltration of CD11b+ myeloid cells into the infarct core, which showed extensive interaction with endothelial cells. Endothelial scRNA-seq data showed reductions in the oxidative phosphorylation, glutathione, and nicotinate metabolic pathways, together with elevated pentose phosphate pathway activity, suggestive of oxidative stress and compromised antioxidant capacity. Functional scoring further indicated diminished endothelial inflammation/repair potential, while in vitro OGD/R experiments revealed morphological disruption, CD31 downregulation, and increased 4-hydroxynonenal (4-HNE), underscoring the importance of endothelial oxidative damage in BBB breakdown.
These multi-omics findings highlight the existence of a coordinated peripheral-central immune axis in ischemic stroke, wherein myeloid cell recruitment and endothelial metabolic vulnerability jointly exacerbate inflammation and oxidative stress. The targeting of endothelial oxidative injury and myeloid-endothelial crosstalk may represent a promising strategy to mitigate secondary brain injury in ischemic stroke.
Keywords
- ischemic stroke
- myeloid cells
- endothelial cells
- oxidative stress
- single-cell RNA sequencing
- blood-brain barrier
- metabolic reprogramming
- cross-species analysis
Ischemic stroke is a leading cause of mortality and long-term disability worldwide, imposing a substantial clinical and socioeconomic burden [1, 2]. Stroke is characterized by the sudden occlusion of cerebral blood flow, triggering a cascade of neuronal injury extending beyond the initial infarct core to involve systemic inflammatory processes [3, 4, 5, 6]. Peripheral immune activation and its interplay with central inflammatory networks are increasingly recognized as key contributors to secondary brain damage, yet the precise molecular drivers of this interplay remain to be fully elucidated [7, 8]. The aim of this study was therefore to investigate the interactions between peripheral and central immune cells in ischemic stroke, and to analyze the functional changes in endothelial cells.
Recent advances in single-cell transcriptomics and spatial profiling techniques have uncovered distinct cellular and molecular mechanisms that govern stroke pathophysiology [9, 10, 11, 12, 13]. Peripheral immune cells, particularly myeloid cells, play a key role in the immune response after ischemic stroke. Myeloid cells, including monocytes, macrophages and neutrophils, infiltrate the ischemic brain and disrupt the blood-brain barrier (BBB), thereby amplifying pro-inflammatory signaling and exacerbating tissue injury [14]. However, the mechanisms by which these activated immune populations rewire their metabolism and affect endothelial function remains unclear.
Metabolic reprogramming is a fundamental feature of immune cell activation and has been increasingly linked to disease pathogenesis, including stroke [15, 16]. Perturbations in glycolysis, fatty acid oxidation, and oxidative phosphorylation can shape the inflammatory behavior of leukocytes, as well as their capacity to produce reactive oxygen species (ROS) [17]. Endothelial cells also undergo metabolic shifts that subsequently influence vascular integrity and BBB permeability [18, 19]. Elucidation of these processes at the single-cell level may lead to a deeper understanding of how stroke-induced immunometabolic stress drives neurovascular damage.
In this study, we employed an integrative, multi-omics approach to dissect the cellular and molecular events underpinning the peripheral-to-central immune response in ischemic stroke. Through the integration of bulk RNA-sequencing (RNA-seq), single-cell RNA-sequencing (scRNA-seq), spatial transcriptomics, and flow cytometry data from both human and mouse models of stroke, we mapped the dynamic responses of myeloid populations and validated their infiltration into the infarcted brain. We also focused on metabolic alterations and oxidative stress responses in endothelial cells, given their central role in maintaining integrity of the BBB. Our findings shed light on conserved and cell-type-specific mechanisms that potentiate brain injury after stroke, offering potential targets to improve clinical outcomes.
Eight-week-old male C57BL/6 mice (20–25 g) were purchased from Hunan
Silaikejingda (SJA) Laboratory Animals (Changsha, Hunan, China), and individually
housed in standard plastic cages. A total of 100 mice were used in the study.
Transient middle cerebral artery occlusion (tMCAO) was performed under isoflurane
anesthesia (3–4% for induction, 1–2% for maintenance). A silicon-coated
monofilament (403756PK10, Doccol Corp. Sharon, MA, USA; diameter, 0.20
All animal experiments were conducted in strict accordance with ethical guidelines. The experimental protocol was approved by the Medical Ethics Committee of the Renmin Hospital of Wuhan University (Ethical approval: IACUC Issue No. 20241105A).
Publicly available bulk RNA-seq datasets from human ischemic stroke patients (GSE58294, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58294) and from tMCAO or sham-operated mice (GSE32529, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32529) at 3 h or 24 h post-stroke were downloaded from the GEO database. Raw or normalized count data were analyzed in R (v4.2.3) (R Foundation for Statistical Computing, Vienna, Austria) using DESeq2 (v1.38.3; Bioconductor, Boston, MA, USA) and limma (v3.54.2; Bioconductor, Boston, MA, USA) packages for differential expression [22, 23], and clusterProfiler (v4.8.1; Bioconductor, Boston, MA, USA) for Gene Ontology (GO) enrichment analysis [24].
Human (GSE285659) and mouse (GSE225948) peripheral blood scRNA-seq data were
downloaded to compare stroke and control groups. Quality control and
preprocessing were performed in Seurat (v4.3.0; Satija Lab, New York Genome
Center, New York, NY, USA) [25]. Cells with excessive mitochondrial transcript
content (
We analyzed spatial transcriptomics data from GSE233815, which was obtained as an annotated Seurat RDS file with pre-labeled cell types. Data were processed using the 10x Genomics Visium pipeline (v2; 10x Genomics, Pleasanton, CA, USA) and integrated into Seurat (v4.3.0; Satija Lab, New York Genome Center, New York, NY, USA) for spot-level clustering. Cross-referencing with single-cell data enabled the prediction of cell type. Myeloid-related genes (S100a8, S100a9, Cd14, Cd68) were mapped to delineate spatial distributions in middle cerebral artery occlusion (MCAO) versus sham brain sections.
The immortalized mouse brain endothelial cell line bEnd.3 was purchased from Procell (CL-0598; Procell Life Science & Technology Co., Ltd., Wuhan, Hubei, China) and cultured in endothelial growth medium containing 5% fetal bovine serum (FBS) at 37 °C and in a 5% CO2 atmosphere. Cells were seeded onto collagen-coated dishes at least 24 h before treatment. The bEnd.3 cell line was confirmed to be free of mycoplasma contamination and authenticated by Short Tandem Repeat (STR) profiling to ensure the correct cell line identity.
Mouse brain endothelial cells (bEnd.3) were washed twice with PBS (10010023, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and incubated in glucose-free Dulbecco’s Modified Eagle Medium (DMEM, PM150270; Pricella Life Science & Technology Co., Ltd., Wuhan, Hubei, China). The cells were then transferred to a hypoxic incubator set at 1% O2, 5% CO2, and 94% N2 to mimic ischemic conditions. After 6 h of oxygen-glucose deprivation (OGD), the glucose-free DMEM was replaced with complete endothelial growth medium containing 5% FBS. The cells were subsequently cultured under normoxic conditions (37 °C, 5% CO2) for an additional 12 h to simulate reperfusion (R). Control groups were washed twice with PBS and maintained in complete endothelial growth medium without oxygen-glucose deprivation.
At 72 h post-tMCAO or sham surgery, mice were deeply anesthetized and perfused
transcardially with cold PBS to clear intravascular blood. Whole brains were
dissected, mechanically dissociated, and filtered to obtain single-cell
suspensions. Red blood cells were lysed using ACK buffer if necessary. Cells were
then incubated at 4 °C for 30 min in FACS buffer (1
The euthanasia of mice was carried out in accordance with standard procedures to ensure humane treatment [26]. Mice were placed in an induction chamber and exposed to 5% isoflurane mixed with 1–2 L/min of oxygen. They were then allowed to inhale the isoflurane for 2–5 minutes until deep anesthesia was confirmed by the absence of reflex responses and evidence of stable, slow respiration. Subsequently, cervical dislocation was performed to complete the euthanasia.
All statistical analyses were conducted in R (v4.2.3) or GraphPad Prism (v9.0;
GraphPad Software, San Diego, CA, USA). Data are presented as the mean
We initially analyzed the peripheral blood transcriptomic landscape using a human ischemic stroke dataset (GSE58294) comprising four groups (control, 3 h, 5 h, and 24 h post-stroke) [27], each with 23 individuals. Principal component analysis (PCA) indicated a distinct separation of control samples from stroke samples, with the 3 h and 5 h groups clustering more closely, and the 24 h group displaying the greatest divergence (Fig. 1A). This trajectory suggests the peripheral response evolves over time, becoming more pronounced at 24 h. We analyzed differential gene expression by comparing each stroke time point (3 h, 5 h, 24 h) to controls (Supplementary Fig. 1A–C). This revealed a gradual increase in the number of differentially expressed genes (DEGs) over time. Double-volcano plots comparing 3 h vs. control, and 24 h vs. control, were used to highlight the key genes that were commonly upregulated (e.g., OLAH, ARG1, MCEMP1, CACNA1E, VSIG4) or downregulated (e.g., TIMM8A, SRCIN1, SH3GL3, FAT3, LPAR4) at both time points (Fig. 1B). Notably, the heatmap demonstrated a continued rise or fall in the expression of these genes from 3 h to 24 h (Fig. 1C).
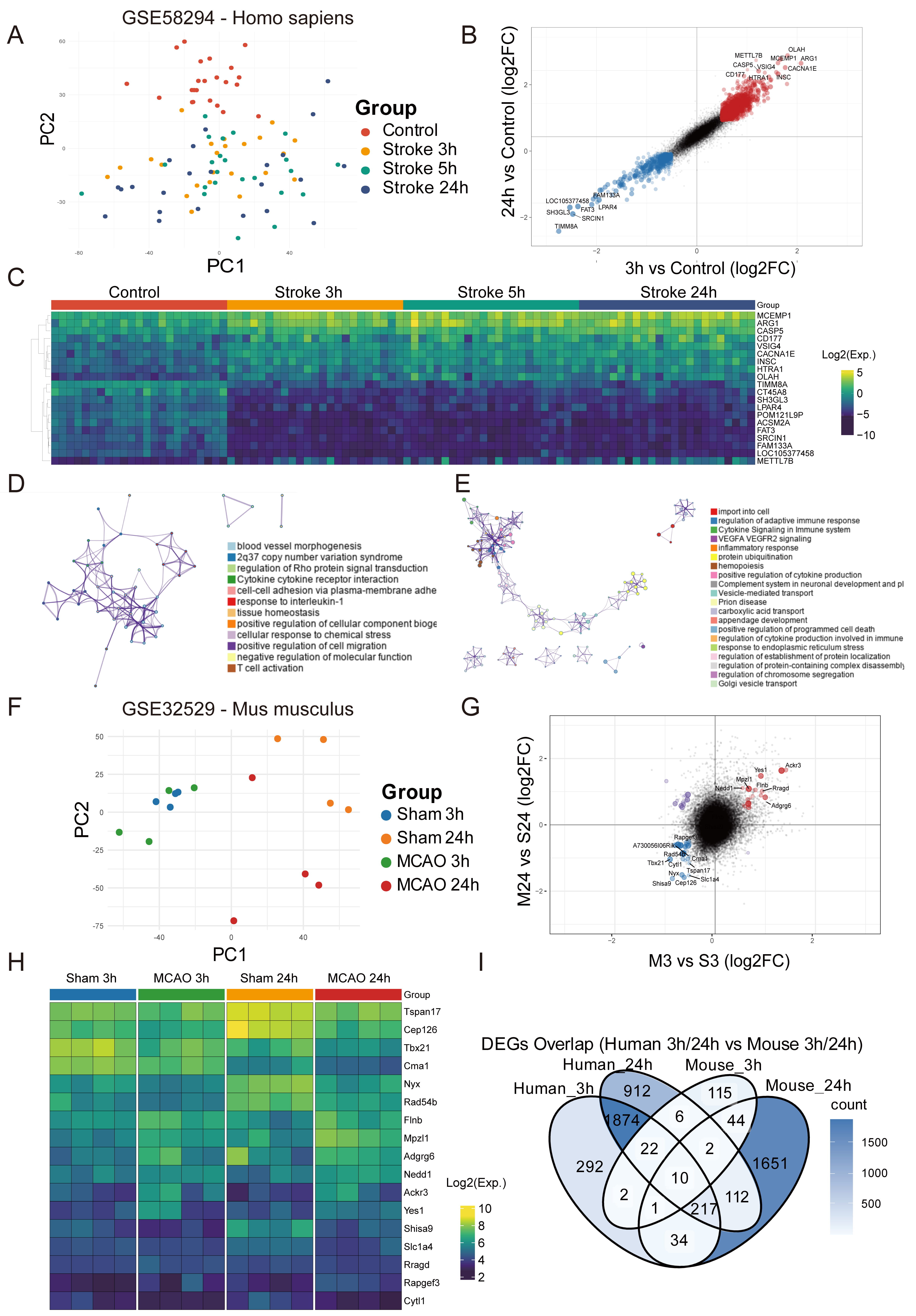 Fig. 1.
Fig. 1.
Peripheral blood transcriptomic responses in human and mouse ischemic stroke models. (A) Principal component analysis (PCA) revealed distinct clustering of control and stroke samples at 3 h, 5 h, and 24 h post-stroke in human datasets. (B) Double-volcano plots highlighting differentially expressed genes (DEGs) between 3 h vs. control and 24 h vs. control in human datasets, with annotation of the shared upregulated and downregulated genes. (C) Heatmap of key DEGs showing the temporal dynamics in expression level across 3 h, 5 h, and 24 h post-stroke in humans. (D,E) Functional enrichment analysis of DEGs at 3 h and 24 h in humans, showing pathways associated with inflammatory responses, cytokine signaling, and vascular regulation. (F) PCA plot of sham and ischemic samples at 3 h and 24 h in mouse models, revealing a time-dependent divergence. (G) Double-volcano plots showing DEGs at 3 h vs. sham and 24 h vs. sham in mouse datasets, with annotation of key genes. (H) Heatmap showing temporal changes in the expression of selected mouse DEGs. (I) Venn diagram showing cross-species overlap of DEGs at 3 h and 24 h, highlighting the conservation of transcriptional responses. MCAO, middle cerebral artery occlusion; PC1, principal component 1.
Functional enrichment via Metascape revealed that only a few pathways were enriched at 3 h, and these predominantly involved cytokine receptor signaling, cell adhesion, interleukin-1 (IL-1) responses, and tissue homeostasis (Fig. 1D, Supplementary Fig. 1E). The 24 h time point yielded more extensive enrichment categories, encompassing adaptive immune regulation, cytokine-mediated signaling, inflammatory responses, vascular regulation, and programmed cell death (Fig. 1E, Supplementary Fig. 1D,F). These terms clustered into four main functional classes, indicating the peripheral immune activity broadens and intensifies by 24 h post-stroke.
A similar approach was applied to a mouse dataset (GSE16529) obtained under multiple MCAO conditions [28]. For clarity, we focused on sham and ischemic challenge groups at 3 h and 24 h, each with four biological replicates. PCA suggested that sham and ischemic samples at 3 h remain relatively close, consistent with a less pronounced early peripheral response, whereas sham and ischemic samples at 24 h diverged markedly, reflecting a stronger systemic reaction to stroke at this later time (Fig. 1F). Double-volcano plots identified overlapping DEGs at both 3 h and 24 h post-MCAO, including both upregulated (Ackr3, Yes1, Rragd, Mpzl1, Adgrg6) and downregulated (Shisa9, Nyx, Cep126, Slc1a4, Tbx21, Cma1) genes (Fig. 1G). A heatmap of these selected genes showed distinct expression patterns across the four groups (Fig. 1H), corroborating a time-dependent transcriptional shift in the peripheral immune compartment.
Lastly, cross-species comparisons revealed relatively few overlapping DEGs at 3 h (n = 35), but many more at 24 h (n = 341), suggesting a more conserved transcriptional response at the later stage. Notably, only 10 genes (HHAT, PRKCI, RRAGD, ARSB, IL1RAP, ETF1, ACKR3, DOCK9, C3AR1, NET1) were commonly upregulated at both 3 h and 24 h in humans and mice (Fig. 1I). Collectively, these findings indicate that whereas early peripheral transcriptional changes are modest, a robust and partly conserved myeloid-centered immune response emerges at 24 h post-stroke in both species.
To further elucidate changes in peripheral blood immune cell populations following ischemic stroke, we analyzed single-cell RNA-seq (scRNA-seq) datasets from humans (GSE285659) and mice (GSE225948) [29]. The human dataset included blood samples from three stroke patients, with one set collected from the intravascular compartment of the ischemic hemisphere and the other from systemic arterial blood. Although the original dataset treated these systemic arterial samples as controls, we instead classified both sets as stroke-associated peripheral blood samples. To establish a true control group, we supplemented the dataset with healthy peripheral blood scRNA-seq data from the DISCO database (https://www.immunesinglecell.org/, disease category: “control”) [30, 31]. These human data were integrated using the Harmony algorithm to mitigate batch effects [32], as illustrated by Uniform Manifold Approximation and Projection (UMAP) projections before and after correction (Fig. 2A). Upon clustering and cell-type annotation, 6 major immune cell types were identified in human blood: myeloid cells, dendritic cells (DCs), T cells, B cells, natural killer (NK) cells, and megakaryocytes. Cell proportion analysis showed that stroke patients had significantly higher frequencies of myeloid cells and megakaryocytes compared to healthy controls, alongside notable decreases in T and B lymphocytes (Fig. 2B).
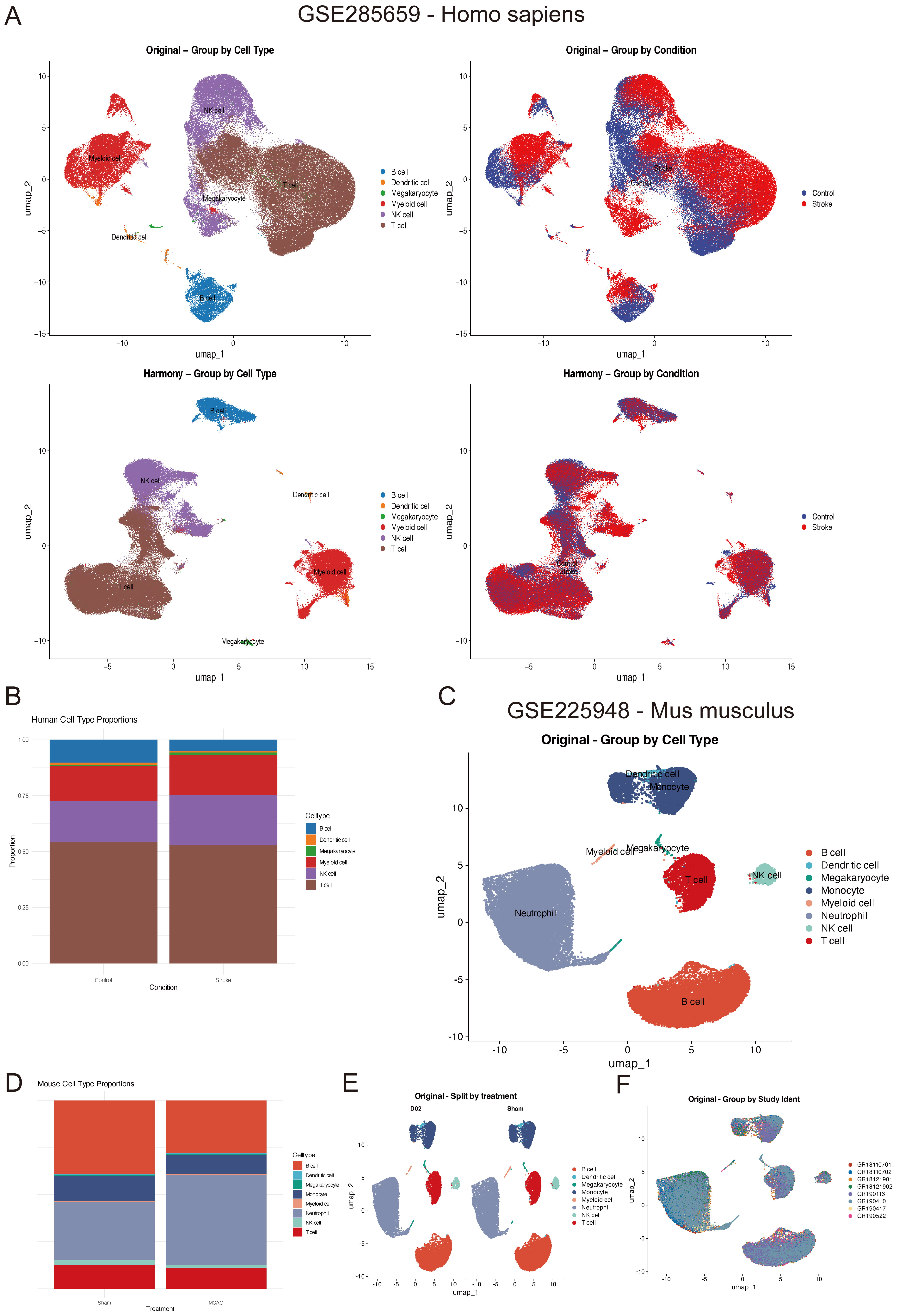 Fig. 2.
Fig. 2.
Single-cell RNA-sequencing (scRNA-seq) reveals altered immune cell composition in human and mouse peripheral blood post-stroke. (A) Uniform Manifold Approximation and Projection (UMAP) plots of human single-cell RNA-sequencing data before and after Harmony integration, illustrating improved clustering of immune cell types across healthy controls and stroke patients. (B) Proportions of immune cell types in human peripheral blood, showing increased frequencies of myeloid cells and megakaryocytes in stroke patients, and decreased T and B lymphocytes. (C) UMAP plot of mouse scRNA-seq data showing annotated immune cell clusters in peripheral blood. (D) Proportions of immune cell types in mouse blood. These mirrored the human findings, with robust neutrophil expansion and lymphocyte depletion after MCAO. (E,F) UMAP projections and cell type annotations of the mouse dataset after batch correction.
A similar analysis was conducted on the mouse dataset GSE225948, which included both peripheral blood and brain samples from MCAO and sham-operated mice. By focusing on peripheral blood data, 8 cell types were defined: neutrophils, monocytes, DCs, T cells, B cells, NK cells, megakaryocytes, and a minor “other” cluster. Consistent with the human findings, MCAO mice exhibited a robust increase in neutrophils and a significant decline in T and B lymphocytes, reflecting an acute inflammatory shift post-stroke (Fig. 2C,D). It is worth noting the original authors of the GSE225948 dataset had already addressed batch effects, resulting in comparatively uniform clustering across samples (Fig. 2E,F).
To explore transcriptional alterations within each cell type, we performed differential expression analyses in both species. In mice, marked changes were observed in DCs, megakaryocytes, myeloid cells, and NK cells, whereas T cells, B cells, and monocytes showed relatively fewer DEGs (Fig. 3A). In contrast, human stroke samples exhibited substantial gene expression changes across all 6 annotated cell types (Fig. 3B). We next mapped human and mouse DEGs to their orthologous genes and identified the cross-species overlaps for each cell type. Myeloid cells showed the largest shared set (n = 110), whereas B, T, and megakaryocyte clusters had fewer than 20 overlapping DEGs. Strikingly, DCs had no cross-species orthologous DEGs in common (Fig. 3C). GO enrichment of these shared, cell-type-specific DEGs revealed that myeloid cells were predominantly involved in chemotaxis and cell migration (Supplementary Fig. 2A,B), megakaryocytes were implicated in phagocytic processes and homeostatic regulation (Supplementary Fig. 2C,D), and B cells showed moderate enrichment in oxidative stress pathways (Supplementary Fig. 2G,H). In contrast, T cells exhibited minimal GO term clustering (Supplementary Fig. 2E,F).
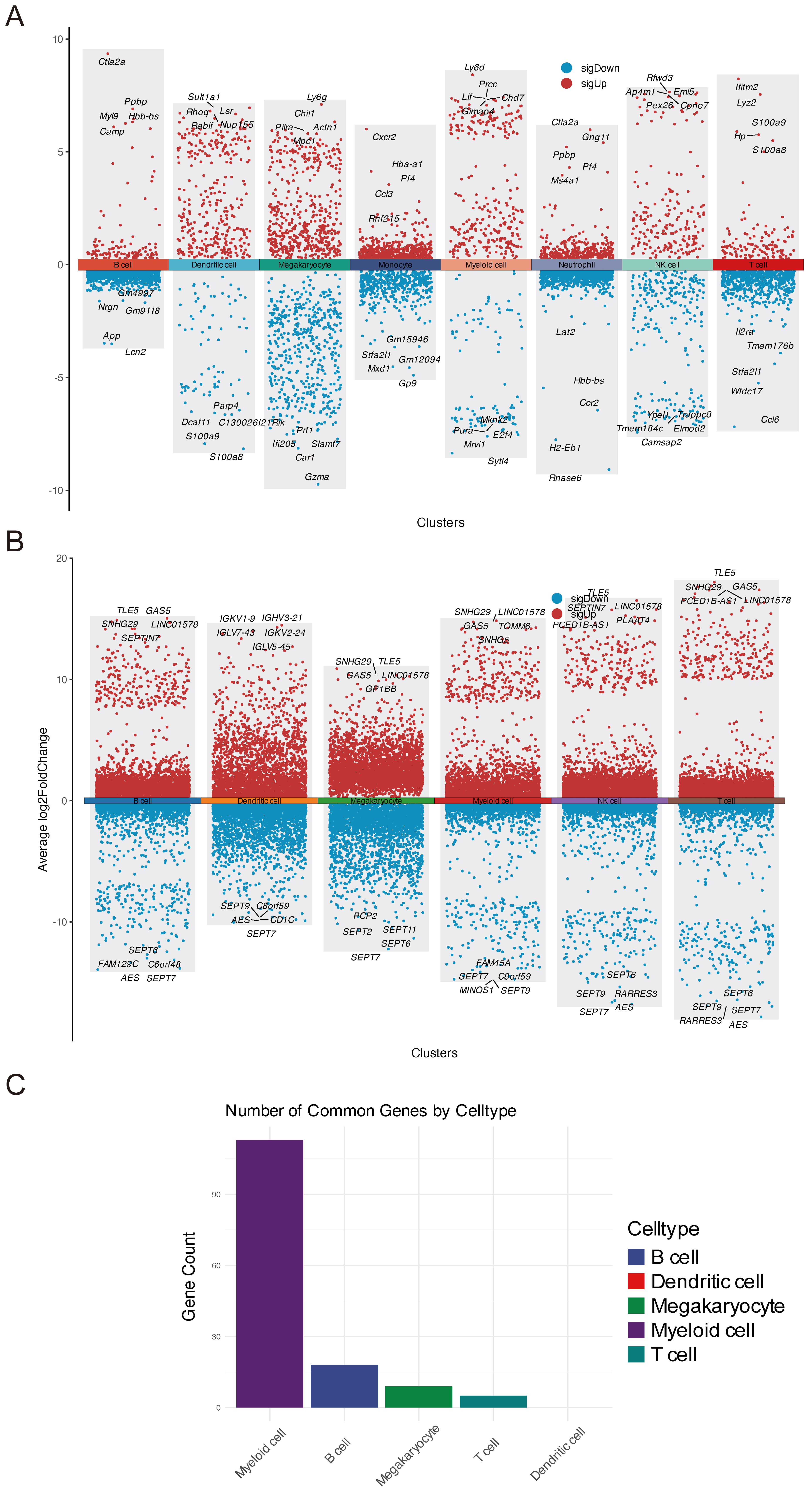 Fig. 3.
Fig. 3.
Cross-species transcriptional changes in peripheral immune cells post-stroke. (A) Heatmap of DEGs across different mouse immune cell types, showing significant transcriptional alterations in myeloid cells, dendritic cells (DCs), and megakaryocytes. (B) Heatmap of DEGs across different human immune cell types, revealing pronounced changes in myeloid cells, T cells, and B cells. (C) Venn diagrams showing cross-species overlap in DEGs for myeloid cells, T cells, B cells, and megakaryocytes.
Taken together, these scRNA-seq findings highlight a dynamic reorganization of peripheral immune cell composition following ischemic stroke, with pronounced expansions in the populations of myeloid cells and megakaryocytes, and corresponding reductions in lymphocytes. Cross-species comparisons underscore the evolutionary conservation of myeloid cell transcriptional programs, pointing to a shared innate immune mechanism driving systemic inflammatory responses to cerebral ischemia.
Building on the observed expansion of peripheral myeloid cells, we next sought to determine whether these populations actively migrate into the ischemic brain parenchyma. To this end, we examined a publicly available spatial transcriptomics dataset derived from a mouse MCAO model at 2 days post-occlusion (bregma –1.3 mm). Notably, the S100a8, S100a9, Cd14 and Cd68 transcripts were all robustly upregulated within the infarct core (Fig. 4A–D), consistent with the presence of neutrophils, monocytes, and macrophages in injured regions. These findings align well with the prominent rise in circulating myeloid cells documented earlier, suggesting a systemic-to-central shift in the inflammatory response.
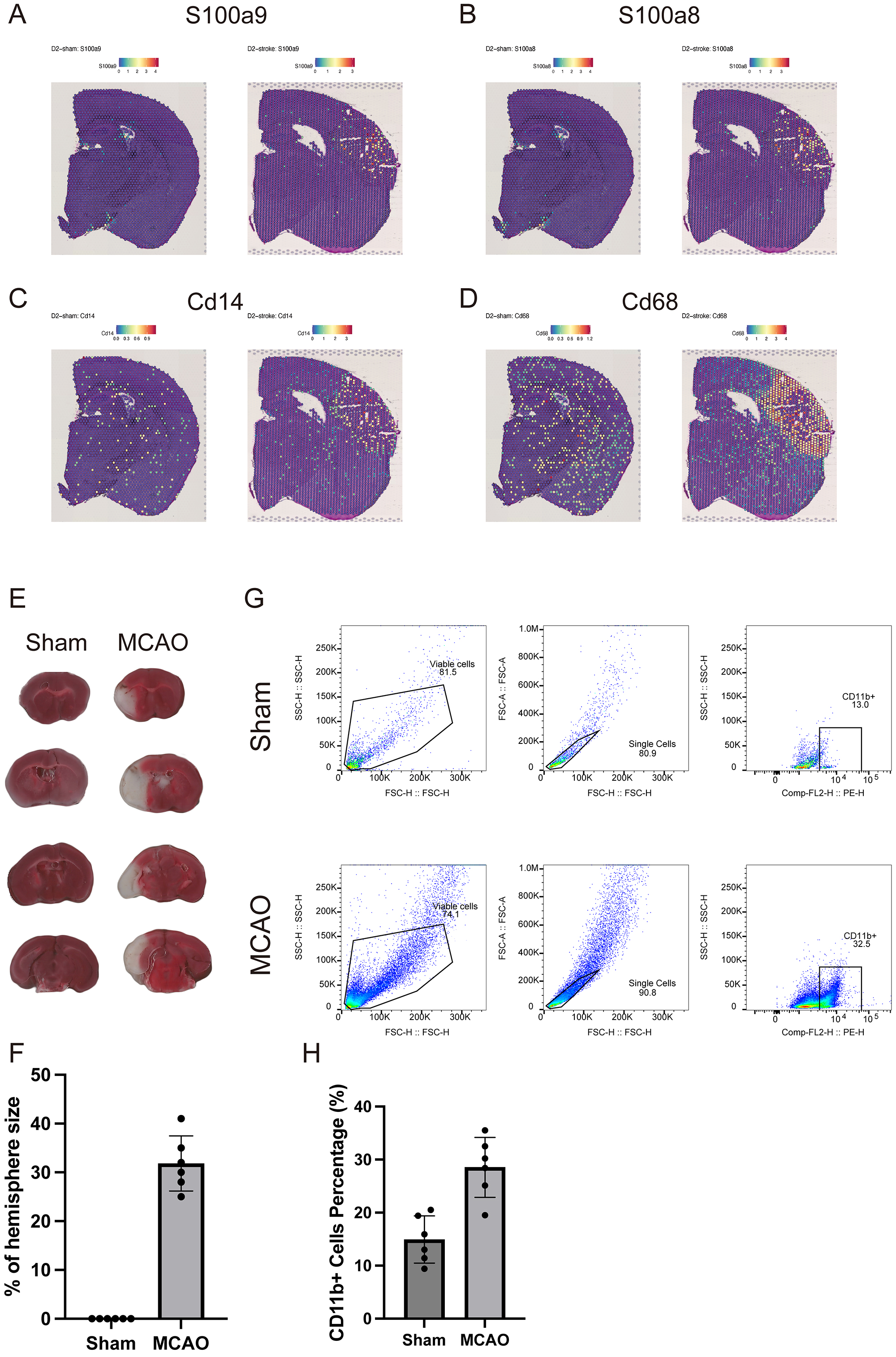 Fig. 4.
Fig. 4.
Myeloid cell infiltration into the ischemic brain confirmed by spatial transcriptomics and flow cytometry. (A–D) Spatial maps showing upregulation of myeloid-related genes (S100a8, S100a9, Cd14, and Cd68) within the infarct core in a mouse MCAO model. (E) Representative 2,3,5-triphenyltetrazolium chloride (TTC) staining of brain sections from Sham and MCAO groups. (F) Statistical analysis of infarct volume as a percentage of hemisphere size. (G) Representative flow cytometry plots gating CD11b+ myeloid cells in Sham and MCAO brain homogenates. (H) Quantification of CD11b+ cells as a percentage of total leukocytes in Sham vs. MCAO brains.
To experimentally validate this infiltration, we performed flow cytometry on whole-brain homogenates from mice subjected to MCAO, following thorough cardiac perfusion to remove intravascular cells. In sham-operated controls, CD11b+ myeloid cells constituted approximately 13% of the gated leukocyte population, whereas MCAO brains displayed a striking increase, with the CD11b+ myeloid cell content reaching almost 32% (Fig. 4E–H). This substantial enrichment strongly supports the notion that peripheral myeloid cells mobilized by ischemic stroke subsequently accumulate within the injured brain tissue, potentially exacerbating local neuroinflammation. Taken together, the spatial transcriptomic evidence and in-house flow cytometric results converge to underscore a profound myeloid infiltration process, linking peripheral immune activation to central tissue injury.
Under physiological conditions, the brain parenchyma contains relatively few immune cells, owing in large part to the restrictive BBB [33]. In the context of stroke, disruption of the BBB is a hallmark event that permits peripheral immune cells to extravasate through compromised endothelial tight junctions, thereby amplifying local inflammation [34]. To further dissect the endothelial response under these conditions, we examined a mouse ischemia-reperfusion single-cell RNA-seq dataset (GSE174574) [35]. Clustering and annotation identified endothelial cells, microglia, monocytes, epithelial cells, macrophages, oligodendrocytes, fibroblasts, neutrophils, astrocytes, and NK cells (Fig. 5A). Following stroke, the abundance of endothelial cells was notably decreased, while neutrophils, monocytes, and macrophages exhibited marked expansion, mirroring the increases observed in peripheral blood and spatial transcriptomic analyses (Fig. 5B,C). It should be noted that GSE174574 is among the earliest scRNA-seq datasets published in stroke research, with an average sequencing depth of only 1433 genes per cell. However, the uniformity of unique molecular identifier (UMI) counts among different cell types (Fig. 5D; Supplementary Fig. 3A,B) and the clear marker expression patterns (Fig. 5E; Supplementary Fig. 3C) support the reliability of cell-type annotations.
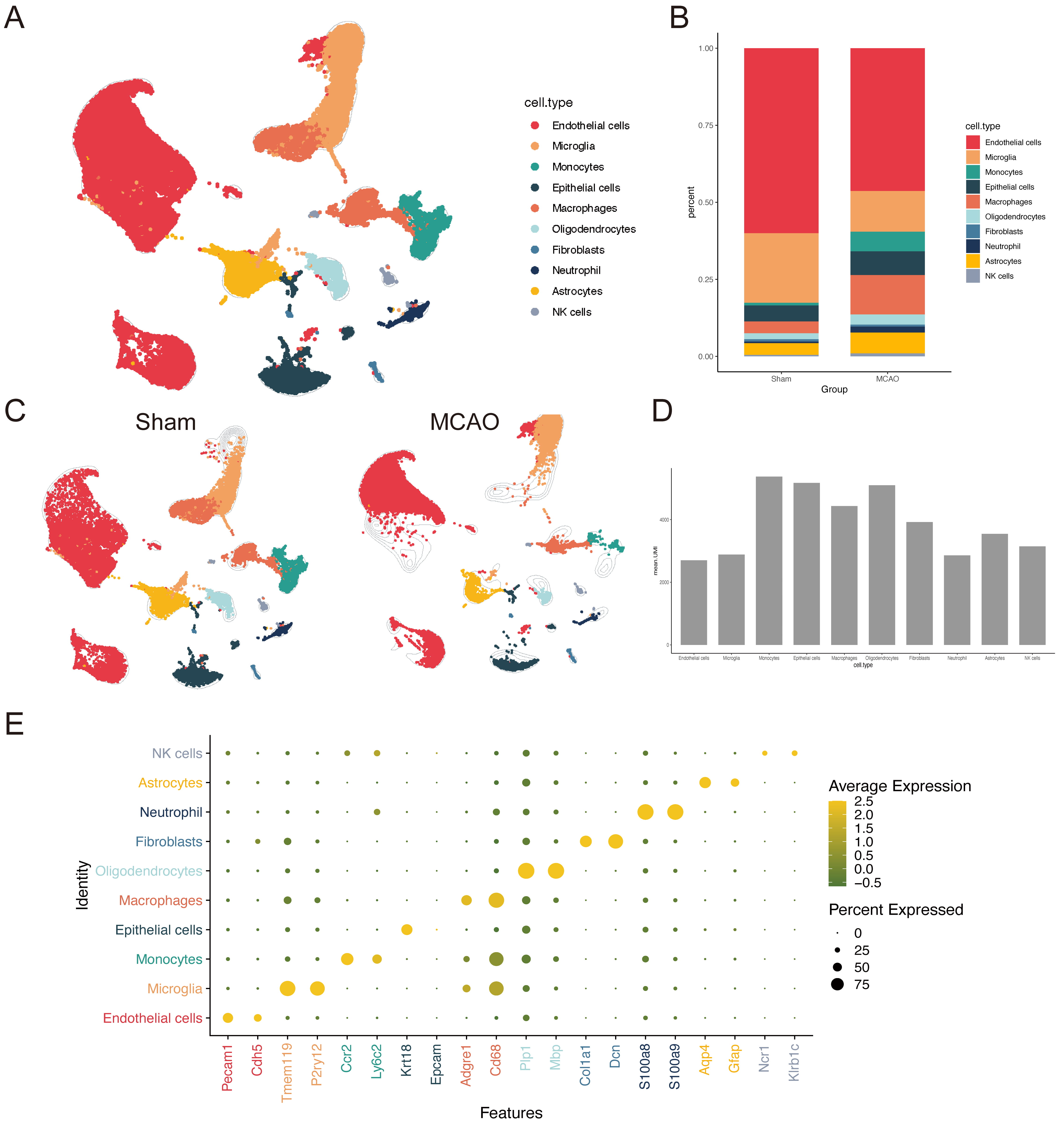 Fig. 5.
Fig. 5.
Single-cell RNA-seq reveals endothelial and immune cell interactions in ischemic brain tissue. (A) UMAP plot of scRNA-seq data from ischemic mouse brains. (B) Proportion of endothelial cells, myeloid cells, and microglia pre- and post-stroke, showing significant reductions in endothelial cells and expansions in myeloid populations. (C) UMAP plot of scRNA-seq data from ischemic mouse brains grouped by condition. (D) Bar plot showing unique molecular identifier (UMI) counts across cell types, confirming the reliability of single-cell clustering. (E) Marker expression levels also confirmed cell type identities in the ischemic brain.
We next employed CellChat to evaluate intercellular communication networks within the injured brain [36]. This analysis revealed an increased number of signaling interactions between endothelial cells and infiltrating myeloid populations, in particular macrophages and neutrophils, relative to non-injured conditions (Fig. 6A,B). Notably, chemokine pathways (e.g., C-C motif chemokine ligand [CCL]) and signaling molecules such as secreted phosphoprotein 1 (SPP1) and Protein S (PROS) were upregulated, whereas pleiotrophin (PTN) and endothelin (EDN) signals were downregulated (Fig. 6C). These findings suggest that stressed or damaged endothelial cells not only succumb to peripheral immune infiltration, but also secrete chemotactic factors that may further promote myeloid cell recruitment and migration. This heightened myeloid-endothelial interaction likely exacerbates neuroinflammation and oxidative damage, ultimately promoting endothelial dysfunction and cell death. Taken together, these data underscore the pivotal role of endothelial cells as both targets and active participants in amplifying peripheral immune infiltration and local inflammatory cascades following stroke.
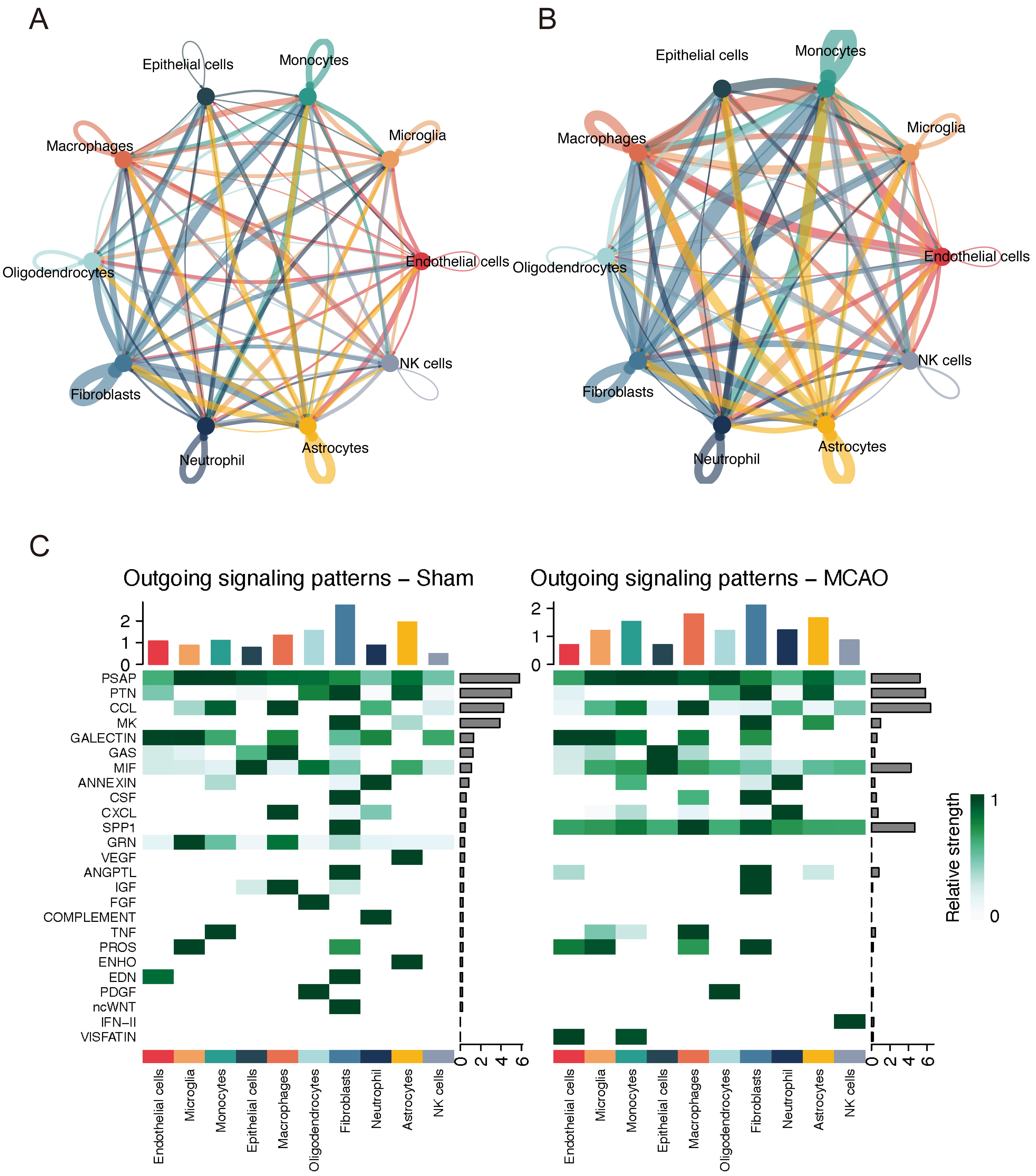 Fig. 6.
Fig. 6.
Myeloid-endothelial crosstalk intensifies inflammatory signaling after stroke. (A) CellChat network analysis showing the number of signaling interactions between endothelial and myeloid cells in Sham. (B) Signaling interactions in MCAO. (C) Heatmap displaying the strength of outgoing signaling pathways in Sham and MCAO conditions. NK cells, natural killer cells.
To determine how peripheral myeloid cells might influence endothelial function in the ischemic brain, we performed scMetabolism analysis on the GSE174574 single-cell dataset and identified numerous pathways that are directly or indirectly related to oxidative stress [37]. These include involvement in ROS generation/clearance (e.g., oxidative phosphorylation, pentose phosphate, glutathione, nicotinate/nicotinamide, selenocompound), or affect metabolism or antioxidant molecules (e.g., fatty acid, arachidonic acid, linoleic acid, purine, tryptophan, porphyrin, plus vitamin- and cofactor-related pathways) [38, 39]. Astrocytes, endothelial cells, and epithelial cells exhibited particularly high metabolic activity (Fig. 7A,B). Notably, post-stroke endothelial cells showed an increase in the pentose phosphate pathway, but decreases in oxidative phosphorylation, nicotinate, glutathione, and ascorbate/aldarate pathways (Fig. 7C). This suggests that although the cells may upregulate NADPH-generating processes to combat rising ROS levels, they are likely to be experiencing metabolic stress due to inadequate ATP production and diminished antioxidant capacity under hypoxic-ischemic conditions. In the indirect pathways, tryptophan and folate metabolism were reduced, whereas purine and porphyrin metabolism were enhanced (Fig. 7D). Collectively, these shifts imply that endothelial cells, while attempting to adapt metabolically to oxidative stress, remain in a partially compromised state that may be further exacerbated by peripheral myeloid cell infiltration.
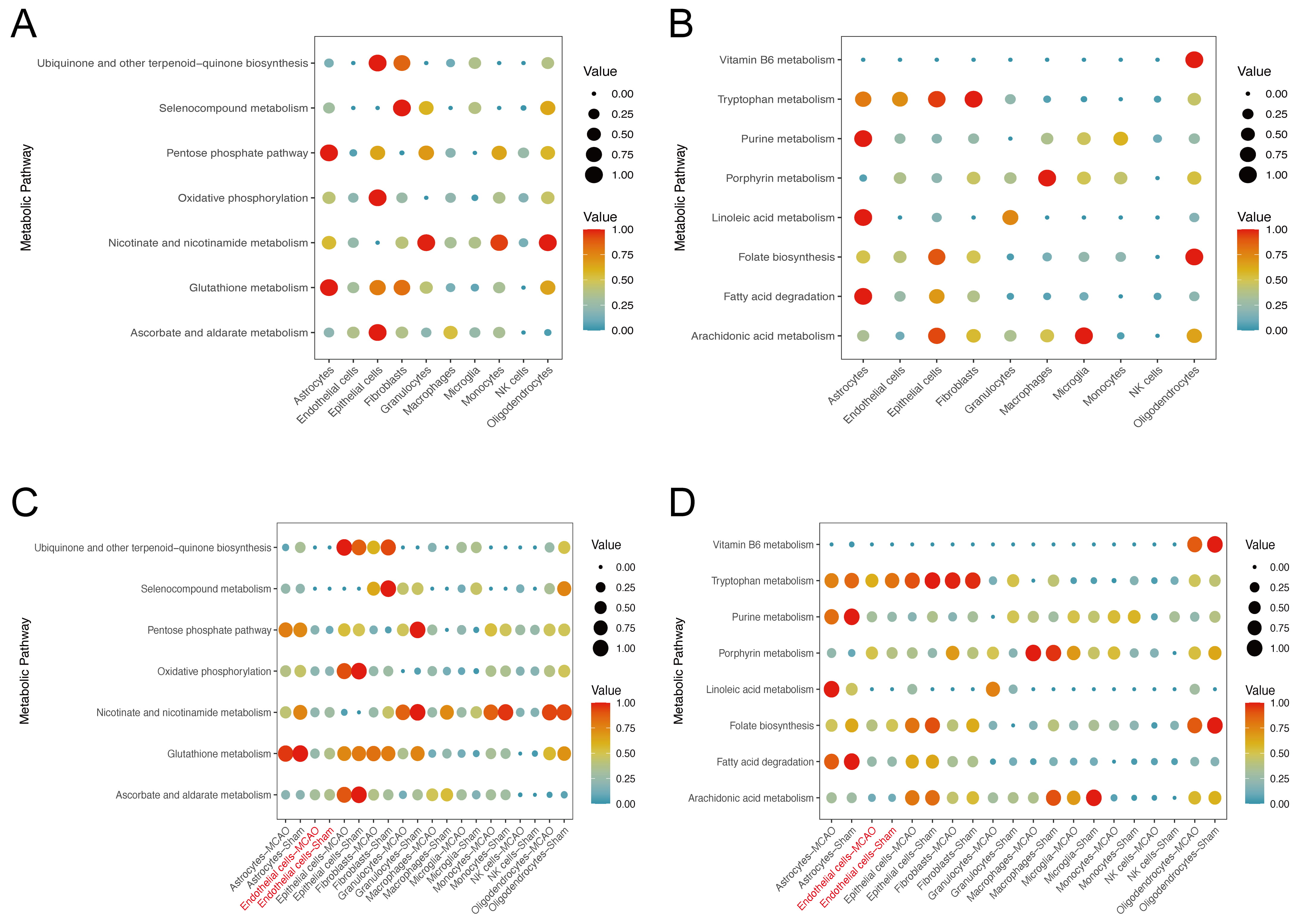 Fig. 7.
Fig. 7.
Metabolic reprogramming in endothelial cells following ischemic stroke. (A) Heatmap of scMetabolism scores for oxidative stress-related pathways in different cell types, with endothelial cells showing the highest activity. (B) Box plots comparing pathway activity in pre- and post-stroke endothelial cells. (C,D) Bar plots summarizing upregulated and downregulated pathways in post-stroke endothelial cells.
To gain further insights into oxidative stress dynamics in endothelial cells, we defined four functional modules (Antioxidant, Endothelial Function, Redox Defense, Inflammation Repair) based on a curated gene list (e.g., Hmox1, Nfe2l2, Sod1, Nos3, Edn1, Vcam1, Mmp2, Mmp9, etc.) and then applied an AddModuleScore approach (Fig. 8A) [40]. The results showed a pronounced impairment in endothelial inflammation/repair capacity, indicative of compromised redox defense and elevated inflammatory signaling (Fig. 8B). To experimentally validate these findings, we subjected cultured endothelial cells to an oxygen-glucose deprivation/reperfusion (OGD/R) protocol that models MCAO conditions in vitro. Immunofluorescence staining demonstrated marked downregulation of Claudin-5, morphological disruption, and a substantial increase in 4-HNE signal, implying heightened oxidative stress and cellular injury (Fig. 8C). Collectively, these data confirm that ischemic stroke drives endothelial oxidative damage, leading to structural abnormalities and cell death, and ultimately exacerbating BBB breakdown and neurological deficits. The findings also indicate that endothelial oxidative stress may be a promising therapeutic target in ischemic stroke.
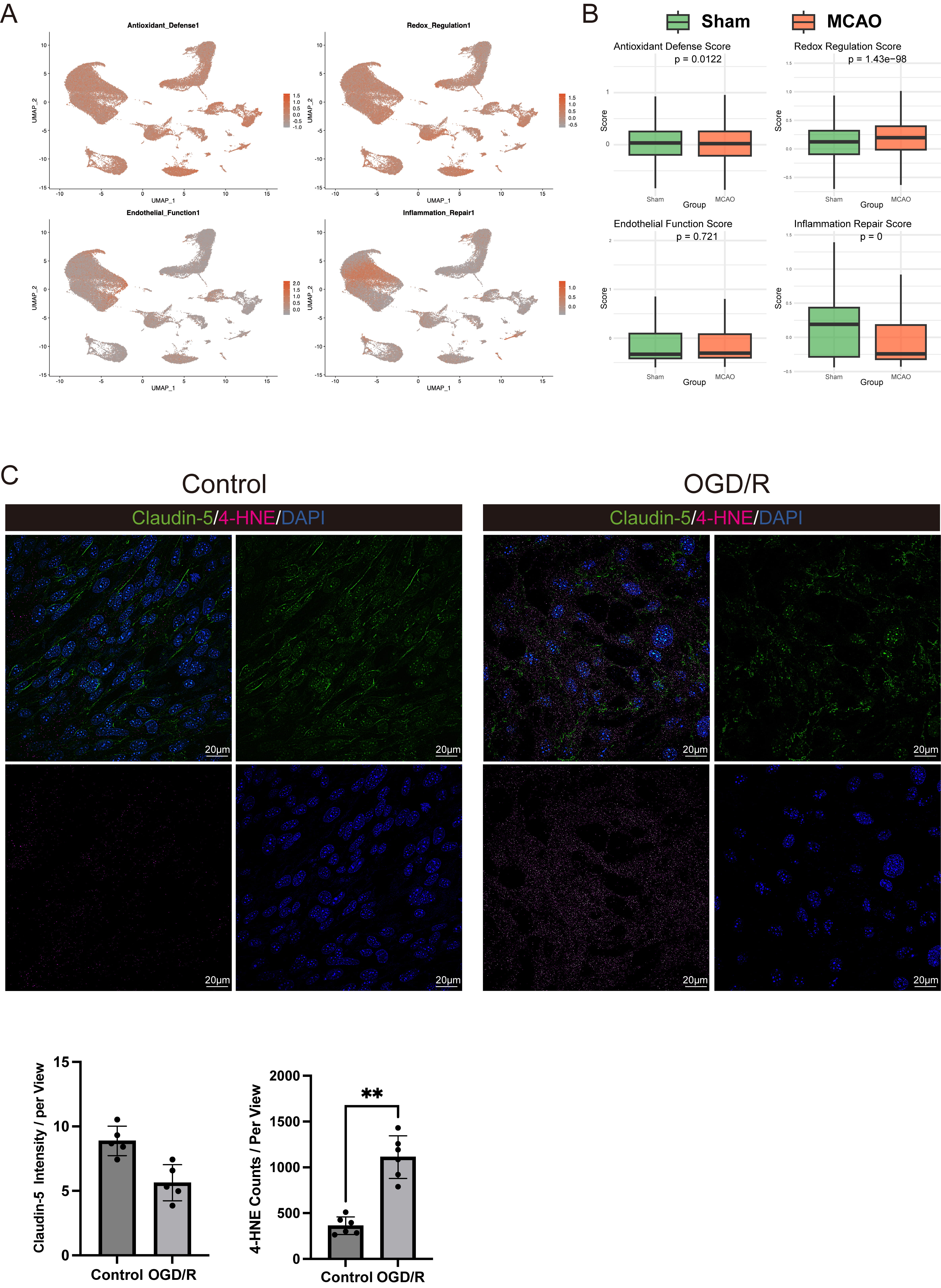 Fig. 8.
Fig. 8.
In vitro validation of endothelial oxidative stress and
injury post-stroke. (A,B) Module score analysis showing impaired antioxidant,
redox defense, and inflammatory repair capacities in endothelial cells
post-stroke. (C) Immunofluorescence images of bEnd.3 cells under Control and
oxygen-glucose deprivation/reperfusion (OGD/R) conditions, with staining for
Claudin-5 (green), 4-hydroxynonenal (4-HNE) (magenta), and DAPI (blue).
Quantification of Claudin-5 intensity and 4-HNE counts per view is shown. Scale
bar = 20 µm. **p
This study integrated bulk RNA-seq, scRNA-seq, spatial transcriptomics, and flow cytometry data from human and mouse models to reveal the intricate immunometabolic changes that occur following ischemic stroke. Consistent with previous reports [14, 41, 42], we observed an early, myeloid-dominated immune response that intensified at 24 h with significant neutrophil and monocyte expansion, highlighting the role of the innate immune system in tissue injury propagation [43].
Our cross-species comparisons indicate that while early transcriptional changes are heterogeneous, the 24 h time point converges on a conserved myeloid-centered signature. This finding suggests that targeting innate immunity could be broadly applicable across species and extend to other cardiovascular and cerebrovascular conditions like heart failure, where immune dysregulation plays a key role [44]. This was also corroborated by spatial transcriptomic and flow cytometric evidence that demonstrated significant infiltration of myeloid cells into the infarct core, where they are likely to amplify local inflammatory cascades.
However, it should be borne in mind that human and mouse models exhibit significant differences in immune response and cellular interactions. Advances in multiplexed error-robust fluorescence in situ hybridization (MERFISH) and volume electron microscopy have revealed both conserved and divergent features in cortical structures [45, 46]. Notably, the cellular composition of the human brain is more type-specific, with deeper neurons showing more synaptic connections, and glial cells that interact more closely with endothelial cells than in mice. This indicates that mouse models may not fully replicate the complexities of human stroke, highlighting the challenges in translating findings from preclinical models to clinical therapies [47, 48].
Crucially, our single-cell analyses revealed that endothelial cells, which form the BBB, undergo substantial metabolic reprogramming and oxidative stress responses. Pathway disruptions, such as reduced oxidative phosphorylation and glutathione metabolism, suggest an energy-deprived and ROS-burdened endothelial phenotype [49]. The diminished antioxidant defenses in endothelial cells, as evidenced by reduced ascorbate and nicotinate pathways, may further exacerbate local ROS generation, especially in the presence of infiltrating myeloid cells. Our OGD/R experiments recapitulated this scenario in vitro, highlighting the essential interplay between hypoxic injury and immune-mediated vascular compromise.
Despite the comprehensive multi-omics approach used here, there are several limitations to our study. Rodent models may not fully capture the complexities of human ischemic stroke, particularly in the context of small vessel diseases. Furthermore, the translation of early (3 h post-stroke) findings in mice to clinical applications remains challenging due to differences in immune and cellular responses. For example, the cellular dynamics and the roles of myeloid cells in ischemic stroke may differ in humans, and the complexities of human stroke pathophysiology are not always reflected in mouse models. Additionally, the limited resolution of current technologies may obscure some important molecular interactions that are more clearly discernible in human tissue.
Taken together, our findings provide a comprehensive view of the myeloid-endothelial axis in ischemic stroke. Following peripheral expansion and activation, myeloid cells infiltrate into vulnerable brain regions, where heightened crosstalk with endothelial cells orchestrates a metabolic and oxidative storm. The resultant endothelial dysfunction fosters breakdown of the BBB, thereby perpetuating inflammation and neurological damage. In the future, therapeutic interventions that restrain myeloid cell infiltration or bolster endothelial antioxidant defenses may be effective at mitigating the severity of stroke [50].
Future research should also explore how myeloid-driven endothelial oxidative stress differs in different subtypes of ischemic stroke, particularly in lacunar strokes which have distinct pathophysiology and clinical outcomes. This could provide insights into endothelial dysfunction and immune dysregulation that is specific to small vessel disease [51]. Further investigation is needed into targeted therapies for endothelial oxidative damage and the roles of other immune cell types in ischemic stroke, such as T-cells and macrophages [52, 53, 54, 55]. Additionally, the development of more refined experimental models, including those that better simulate small vessel disease, would enhance our understanding of stroke pathophysiology.
In conclusion, our work highlights how systemic immune activation affects neurovascular homeostasis in ischemic stroke. Endothelial oxidative stress was found to be a critical bottleneck in this process. Our findings support future research on targeted therapies, such as ROS scavengers, NAD+ boosters, or anti-inflammatory agents, to mitigate both peripheral immune overactivation and endothelial vulnerability.
In summary, this integrative cross-species study analyzed data from human and mouse bulk RNA-seq, single-cell RNA-seq, supporting spatial transcriptomics, and flow cytometry. Collectively, this approach identified a robust peripheral myeloid expansion following ischemic stroke. Subsequent infiltration of these activated myeloid cells into the ischemic brain promotes heightened crosstalk with endothelial cells, triggering significant metabolic and oxidative stress responses. These endothelial perturbations are characterized by disrupted redox homeostasis and impaired inflammatory repair, thereby compromising the BBB and aggravating cerebral damage. Our findings underscore the critical role of myeloid-endothelial interactions in the pathophysiology of ischemic stroke, and suggest that targeting endothelial oxidative stress pathways may offer a promising avenue for therapeutic intervention.
All datasets used in this study are publicly accessible through the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/) (accession numbers: GSE16529, GSE58294, GSE32529, GSE285659, GSE225948, GSE233815, GSE174574) or via the original publications. Analysis scripts for quality control, normalization, and visualization are available upon request from the corresponding author.
ZC conceived and designed the study, coordinated the research efforts, and led the manuscript preparation. HZ performed the majority of the experiments and contributed substantially to data analysis. SF assisted in data collection and interpretation, ensuring the reliability of the results. YZ provided critical insights during data evaluation and helped refine the manuscript. XX contributed to the study design, supervised the project, secured funding, and finalized the manuscript for submission. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All human transcriptomic datasets were obtained from publicly available repositories (e.g., Gene Expression Omnibus, GEO) and generated in accordance with each original study’s institutional review board approvals and informed consent protocols. All animal protocols were approved by the Medical Ethics Committee of the Renmin Hospital of Wuhan University (Ethical approval: IACUC Issue No. 20241105A) and were in complete accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
The authors gratefully acknowledge the support and resources provided by the Central Laboratory of Renmin Hospital of Wuhan University. We also thank our colleagues and technical staff for their invaluable assistance throughout the research process, as well as the reviewers for their constructive feedback which greatly improved the quality of this work.
This work was supported by the National Natural Science Foundation of China (No 82371346) to Xiaoxing Xiong.
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGPT-o1 solely for spelling and grammar checks in the Methods, Results and Discussion sections. Following its use, all content was carefully reviewed, edited, and finalized by the authors, who assume full responsibility for the publication. To address potential concerns regarding AI-generated inaccuracies—particularly the risk of AI hallucinations during literature searches—please note that all references were manually verified and inserted by the authors without any AI assistance.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL37429.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.