



 , Nadezhda Sachivkina 2, Olga Kuznetsova 3, Ekaterina Neborak 3, Natallia Zhabo 4
, Nadezhda Sachivkina 2, Olga Kuznetsova 3, Ekaterina Neborak 3, Natallia Zhabo 41 Department of Biotechnology, Institute of Applied Sciences & Humanities, GLA University, 281406 Mathura, Uttar Pradesh, India
2 Department of Microbiology V.S. Kiktenko, Institute of Medicine, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia
3 Department of Biochemistry T.T. Berezov, Institute of Medicine, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia
4 Department of Foreign Languages, Institute of Medicine, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia
Abstract
Mycoplasmas are the smallest cell-wall-less self-replicating prokaryotes. Mycoplasma species can be found within and outside cells as “silent parasites” that live intracellularly and as membrane surface parasites. The pathogen’s impact on respiratory health seems primarily caused by its capacity to alter immune responses, cause airway inflammation, and damage epithelial barriers. Much progress has been made in understanding Mycoplasma-induced inflammation and oxidative stress. However, there are still issues in therapeutic management, such as the development of strains that are resistant to antibiotics, the shortcomings of the available diagnostic techniques, and possible long-term respiratory consequences. On the other hand, to combat oxidative stress, inflammation, and metabolic abnormalities, activation of the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is becoming a more appealing therapeutic strategy. Nrf2 activation coordinates a thorough defense through its transcriptional targets, enabling adaptability and survival under a variety of cellular stressors. Nrf2 is regarded as a therapeutic target, and pharmacological Nrf2 activators have demonstrated protective effects in multiple pathological consequences and advantages in clinical trials. In this review, we discussed the rationale for targeting Nrf2 in a series of inflammatory responses caused by Mycoplasma species.
Keywords
- Mycoplasma
- HO-1
- Nrf2
- gene expression
Mycoplasma pneumoniae plays a major role in the onset and aggravation of chronic lung pathology. Human tracheobronchitis, pharyngitis, asthma, and primary atypical pneumonia are all brought on by Mycoplasma pneumoniae [1]. The smallest self-replicating prokaryotes without a cell wall, Mycoplasmas, have a genome that is incredibly tiny, ranging from 580 to 2200 kb [2]. Only a small number of the more than 200 species of this pathogen known to affect humans, animals, arthropods, and plants. Particularly noteworthy are the pathogenic bacteria Mycoplasma pneumoniae (M. pneumoniae), M. genitalium, M. pirum, M. hominis, M. fermentans, and M. penetrans impact both human and animal systems [3]. Among these harmful strains, M. pneumoniae is the most common and well-researched species. Pneumonia due to M. pneumoniae sufferers is more common in school-age children and young adults than in newborns and elderly people [4]. Lower respiratory tract infections are thought to be a common source of morbidity and mortality in children, and this atypical pathogen is the cause of up to 40% of community-acquired pneumonia (CAP) in children older than five [5, 6]. Compared to healthy controls, respiratory samples from adults and children experiencing asthma attacks or exacerbations have greater incidences of M. pneumoniae [7]. The association between M. pneumoniae and asthma is further supported by the high rates of the bacteria in the airways of chronic stable asthmatics [8].
Chronic infections are typically caused by Mycoplasmas, and it is unclear what the majority of their pathogenic factors are and how they work [9]. Mycoplasmas have a propensity to penetrate, harm, and colonize deep tissues due to tissue necrosis, local trauma, surgery, disturbance of the mucosal surface, and compromised sterile site clearance [9]. Mycoplasmas are indeed thought to be the cause of these localized infections in a lot of cases, because it is difficult to isolate and identify them using laboratory techniques [10, 11, 12, 13]. A diagnosis based solely on serology may not be accurate since IgM antibodies might not be detectable early in the course of infection. It has been discovered that the seroprevalence of M. pneumoniae in persons with pneumoniae varies greatly, ranging from 1.9% to more than 30% [14]. Nucleic acid amplification tests (NAATs), sequencing, and proteomic investigations are examples of molecular diagnostic advancements that have helped us learn more about the pathophysiology of this organism [15, 16].
The development of protein sequence techniques has made it possible to identify potential factors that influence the pathogenicity of Mycoplasmas in both humans and animals [17]. Thus, several findings suggest that the lipoproteins on Mycoplasmas’ membrane interact with monocytes and macrophages and occasionally lead to immune system evasion [18, 19, 20]. Confocal micrographs have recently shown that, depending on the kind of cell, M. pneumonia can bind and internalize [21, 22].
The pathophysiology of respiratory infections caused by M. pneumoniae is complex and includes both direct cytotoxic effects on host cells and indirect processes that cause the host’s immune responses to become dysregulated [23]. M. pneumoniae effectively invades and adheres to pulmonary epithelial cells, resulting in impaired mucociliary clearance, significant cell damage, and disruption of the epithelial barrier [24]. M. pneumoniae can use its apical structure to attach to the surface of host cells during infection, after which it can absorb nutrients from the cells [25]. Apical structural attachment and gliding have the potential to physically harm host cells [25]. Furthermore, upon attachment to host cells, modifications in M. pneumoniae’s pathogenicity have been documented [26]. Increased production of superoxide and hydrogen peroxide radicals, the development of the toxin associated with community-acquired respiratory distress syndrome (CARDS), and modifications to M. pneumoniae’s lipid proteins are among the virulence-related regulators [26]. When exposed to noxious stimuli, neutrophils are usually the first cells to extravasate into tissues. They are extremely sensitive to a wide range of stimuli, especially infections and molecular patterns linked to injury [27]. By promoting the recruitment of more granulocytes and phagocytic monocytes, these prodigious leukocytes serve as the first line of defense against microbial threats. They also effectively control infection and preserve mucosal integrity through processes like degranulation reactions, oxidative bursts, and the release of neutrophil extracellular traps (NETs) [27]. Significant neutrophil recruitment and infiltration in the lungs 24 hours after M. pneumoniae infection, a notable rise in the neutrophil ratio in bronchoalveolar lavage fluid (BALF), and a sizable neutrophil infiltrate visible in lung histopathology are further indications from animal studies that Mycoplasma pneumoniae (MP) infection causes a strong early neutrophil response. Numerous signaling pathways are involved in neutrophil recruitment and migration following M. pneumoniae infection [28, 29]. Pro-inflammatory cytokine production is linked to the symptoms of pneumonia brought on by M. pneumoniae, which imply that M. pneumoniae-induced exaggerated immune responses are a significant contributor to pneumonia development [30]. Although M. pneumoniae infections usually resolve on their own in healthy people, they can have more serious effects in other groups, such as those with weakened immune systems or underlying respiratory disorders [31].
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor
made up of 605 amino acids and features conserved domains ranging from Neh1 to
Neh7, which play a crucial role in its function. The N-terminal domain interacts
with (Kelch-like ECH-associated protein 1) Keap1 through binding motifs like ETGE and DLG to stabilize and ubiquitinate
Nrf2 [32]. DNA binding through the basic-region leucine zipper (bZIP) domain is
carried out by the Neh1 domain [33]. The interaction of Nrf2 with other
coactivators is then mediated by the Neh3 to Neh5 domains [34]. Ubiquitination
through
The Nrf2 and nuclear factor kappa B (NF-
According to reports [45], several Mycoplasma species stimulate different immune cells and produce pro-inflammatory cytokines. It has long been unclear what causes inflammatory reactions in Mycoplasma species, as they lack immune cell stimulators such as peptidoglycan and lipopolysaccharide (LPS). Regardless of whether they are affixed to the surface of eukaryotic cells or invade them, certain Mycoplasmas disrupt and modify the host cell’s biological pathways at the functional and/or regulatory level [45]. After infection, the host organism initiates a series of reactions involving multiple signaling pathways to protect itself from such harmful outcomes. These reactions ultimately lead to the activation of the immune response, which triggers processes that promote inflammation. In response, Mycoplasmas evolved defense mechanisms that allow them to evade immune regulation and infiltrate many bodily regions, including mucosal surfaces. However, Mycoplasmas are frequently able to adapt because of the lag between initial triggering and the emergence of a full-scale reaction [45, 46].
As previously stated, Mycoplasma adheres to the outer cellular membrane, which causes specific bacterial proteins (lipoproteins (LPs) or lipopeptides) to interact with particular cellular receptors on the target cells’ surface. In this context, various investigations have discovered a variety of Mycoplasmas LPs that can communicate with the host organism’s leukocytes and epithelial cells [47, 48]. The first lipopeptide expressed in Mycoplasmas that was shown to bind toll-like receptors (TLRs) was M. fermentans’ macrophage-activating lipopeptide-2 (MALP-2) [49]. Heterodimers of TLR 1/2 or TLR 2/6 were then demonstrated to bind to triacylated or diacylated lipopeptides, respectively [50, 51]. There are no Mycoplasma genomes that have the Lnt gene, which codes for the enzyme responsible for N-acylation [52]. However, according to a study on the proportion of N-amide to O-ester linkages in M. gallisepticum and M. mycoides, triacylated lipoproteins may be present [53]. Additionally, M. mycoides proteins’ resistance to Edoman degradation suggested the presence of N-acylation [54]. The diacylated lipoprotein subunit b of the F0F1 ATP synthase (MPN602) in M. pneumoniae is known to trigger inflammatory reactions via TLR2 [55]. Interestingly, leukocyte infiltration in the respiratory tract can be promoted by specific lipopeptides that have been identified and purified from Mycoplasmas, suggesting that these factors may potentially have an effect when the entire Mycoplasma organism is absent [56].
The presence of lipoproteins in non-pathogenic Mycoplasma species, however, points to the possibility of an additional mechanism by which M. pneumoniae triggers inflammatory reactions. It was already been reported that M. pneumoniae causes significant inflammatory reactions, even in macrophages generated from mice with TLR2 deletion [57].
Certain Mycoplasma species, notably M. pneumoniae, generate
the cytotoxic CARDs toxin. Another report demonstrated that NLRP3 inflammasome
activity is regulated by CARDS toxin (MPN372) (Fig. 1) [58]. This work showed
that CARD toxin increases the production of interleukin (IL)-1
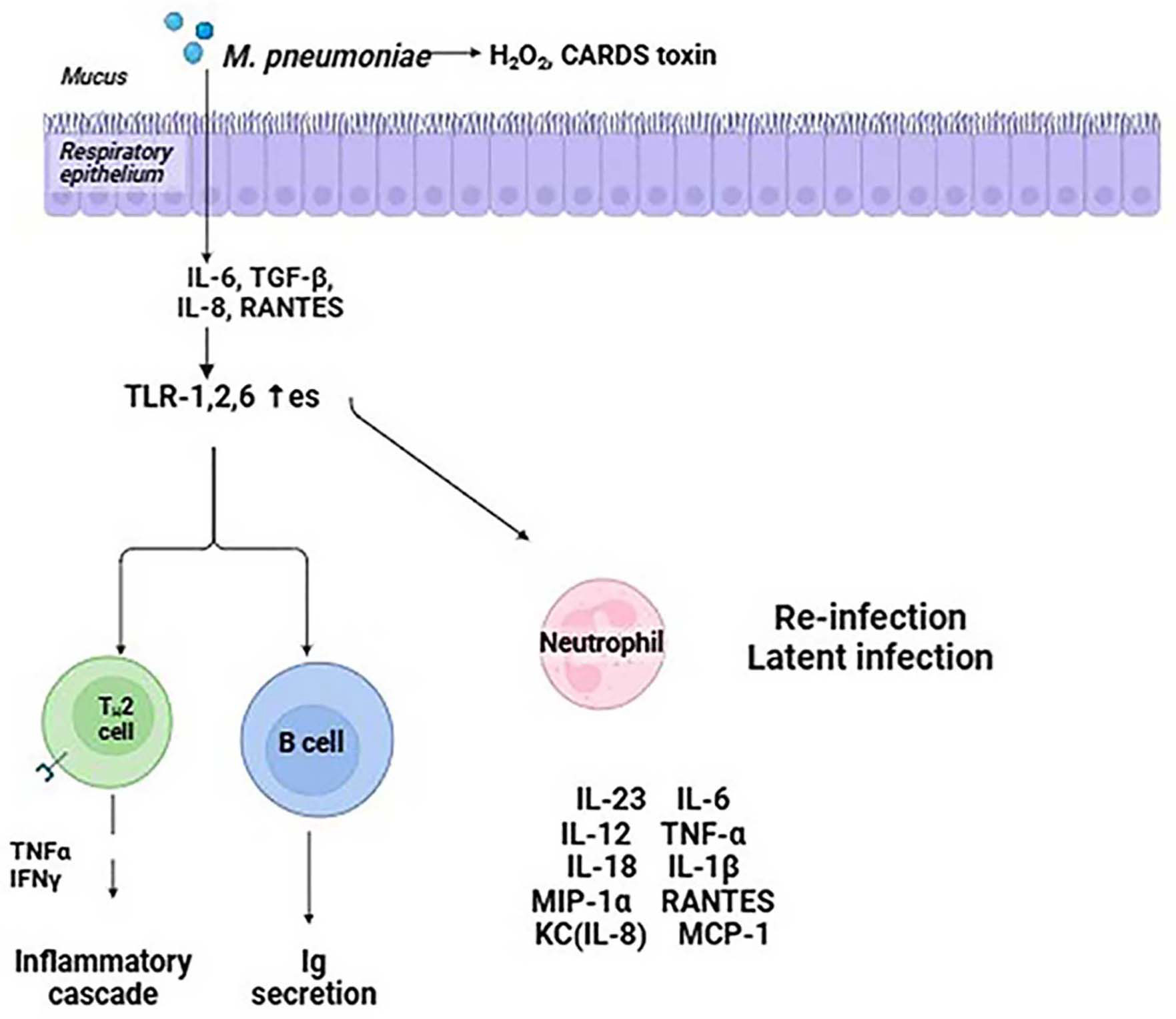 Fig. 1.
Fig. 1.
Cellular pathways implicated in inflammation and cellular
transformation are impacted by Mycoplasma pneumoniae. H2O2,
hydrogen peroxide; CARDS, community-acquired respiratory distress syndrome; TLR,
Toll-like receptor; IL, interleukin; TGF-
With its ability to produce H2S, which damages blood cells, the HapE found in M. pneumoniae serves as a possible virulence factor. HapE’s generation of H2S causes phagocytes to produce pro-inflammatory substances, which in turn intensifies inflammatory reactions [60]. Additionally, HapE uses ATP-sensitive K+ (KATP) channels to contribute to inflammatory reactions [61]. When cysteine is broken down within the KATP channel complex, more H2S is produced, which exacerbates inflammation.
The immunoglobulin-binding protein of Mycoplasma (IbpM), sometimes referred to as mpn400, binds firmly to a variety of immunoglobulins generated by the host, including IgM, IgA, and IgG. The importance of IbpM as a virulence factor was demonstrated by a prior investigation that revealed strains of M. pneumoniae lacking IbpM show a minor decrease in cytotoxicity [62].
When M. pneumoniae adheres to host cells, it enters the cells and
releases superoxide and hydrogen peroxide radicals, leading to oxidative stress
in the respiratory tract epithelial cells. Furthermore, M. pneumoniae
does not have catalase or superoxide dismutase, thus the radicals it generates
can prevent the host cell’s catalase action. As a result, there is less peroxide
breakdown, making host cells more susceptible to damage from oxygen molecules
[63]. It has been documented that M. pneumoniae’s hydrogen peroxide can
control the detachment of infected cells, promoting the bacterial infection’s
persistence [64]. L-
Reactive oxygen species (ROS) are generated by nicotinamide adenine dinucleotide phosphate oxidase and are a component of the host’s non-specific immunological defense against invasive pathogens [67]. ROS have two roles: they target proteins, lipids, carbohydrates, and nucleic acids in M. pneumoniae cells, acting as direct antibacterial agents and significantly impairing these biological components. At the same time, ROS are crucial signals for innate immunity signaling, which activates the immune system to fight off infections. A recent investigation found that mpn668 encodes a protective antioxidant enzyme in M. pneumoniae [68]. This enzyme breaks down hydroperoxide, which may lessen the oxidative damage the host generates. Furthermore, neutrophils quickly gather at the infection site after an M. pneumoniae infection due to chemokine chemotaxis. As a result of their increased phagocytic activity, infections are successfully eradicated by neutrophil extracellular traps (NETs) and the release of several bactericidal chemicals. Extracellular nucleases produced by M. pneumoniae can break down NETs. Interestingly, the magnesium-dependent nuclease produced by M. pneumoniae mpn491 is an important extracellular nuclease that increases the pathogen’s survival rate and helps it evade the host’s immune response by breaking down NETs, which further damages the host [68].
The lipoprotein components of the M. pneumoniae membrane, which are
produced on the surface as lipid-associated membrane proteins (LAMPs), are
essential for the host immunological inflammatory response [69]. It has been
discovered that LAMPs attach to monocytes and macrophages’ TLRs 1, 2, and 6 as
well as CD-14. These cells then activate downstream IL-1R-associated signal
molecules, which activate NF-
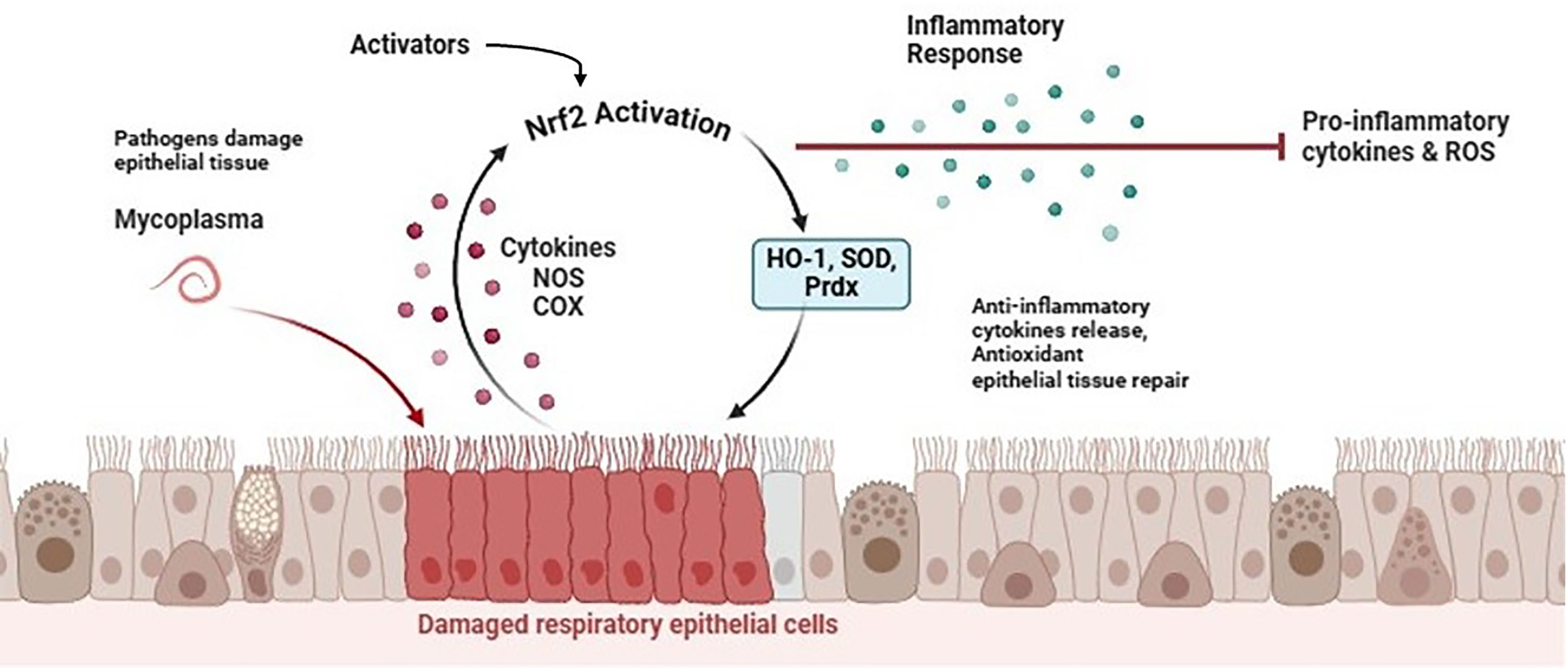 Fig. 2.
Fig. 2.
Progression of Mycoplasma-induced infections and role of Nrf2 signaling pathway. Nrf2, erythroid 2-related factor 2; HO-1, heme oxygenase-1; SOD, superoxide dismutase; Prdx, peroxiredoxin; NOS, nitric oxide synthase; ROS, reactive oxygen species; COX, cyclooxygenase. Created with Biorender.com.
Macrophage-activating lipopeptide-2 (MALP-2), derived from the NH2 terminal of the mycoplasmal lipoprotein, is commonly used as the pathogen-associated molecular pattern (PAMP) for Mycoplasma studies because it shares many pro-inflammatory properties with membrane lipoproteins in nearly all Mycoplasmas [72]. In an earlier report, it was shown that MALP-2 might activate HO-1 monocyte expression to control the overexpression of cyclooxygenase 2 [73]. In THP-1 cells, MALP-2 reportedly increased HO-1 enzyme activity and stimulated HO-1 mRNA and protein expression [73]. However, HO-1 expression was dramatically reduced by mitogen-activated protein kinase (MAPK) inhibitors SB203580, PD98059, and SP600125. This implies that MAPK pathways might be an upstream signal required for the production of HO-1 that is dependent on MALP-2.
Furthermore, Nrf2 translocation was also triggered by MALP-2 and the levels of
MALP-2-mediated HO-1 expression were reduced in THP-1 cells when Nrf2 expression
was silenced. Additionally, THP-1 cells’ levels of cyclooxygenase 2 (COX-2)
protein expression were elevated in response to MALP-2, and transfected with HO-1
siRNAs markedly raised COX-2 accumulation. According to another study,
phosphoinositide 3-kinases (PI3K)/Akt, ROS, and Nrf2-dependent HO-1 expression
influence the inhibitory action of sulforaphane on the release of several
pro-inflammatory mediators in MALP-2-stimulated monocytes [74]. Sulforaphane
induced Nrf2 to move from the cytoplasm to the nucleus, however, HO-1 expression
was markedly suppressed by short interfering RNA-mediated Nrf2 knockdown. The
pharmacological inhibitors LY294002 and N-acetylcysteine (NAC) also demonstrated
that PI3K/Akt and ROS were implicated in Sulforaphane-induced Nrf2 activation and
HO-1 expression. Additionally, animals treated with sulforaphane showed reduced
lung inflammation and pro-inflammatory cytokine release, as well as
NF-
In a previous report, the authors examined H2S’s anti-inflammatory
properties in an in vitro model of proinflammatory
Mycoplasma-infected macrophage microbe with the ability to initiate the
rapid enrolment of a significant number of macrophages particularly in the
airways and lungs [19]. Additionally, the research demonstrated that exogenous
H2S can prevent NF-
LncRNAs, or long non-coding RNAs, are crucial for controlling respiratory conditions including pneumonia [77]. In many inflammatory disorders, the growth arrest-specific 5 (GAS5) lncRNA plays an essential role. It has been shown that GAS5-silencing reduces the viability and exacerbates the inflammatory damage of chondrocytes produced by LPS [78]. Significantly, children with M. pneumoniae pneumonia have increased expression of miR.222.3p [79]. A study examined the expression of GAS5, miR-222-3p, and tissue inhibitor of metalloproteinases-3 (TIMP3) in M. pneumoniae pneumonia [80]. GAS5 and TIMP3 expression was found to be downregulated in THP-1 cells stimulated by LAMP. GAS5 was found to interact directly with miR-222-3p, which in turn targets TIMP3. The stimulating effect on cell viability and the suppression of inflammation generated by GAS5-overexpression in LAMP-induced THP-1 cells was reversed by miR-222-3p upregulation or TIMP3-knockdown. There is supportive evidence that showed that blocking miR-222-3p reduces inflammation via activation of Nrf2/HO-1 signaling, which in turn attenuates oxidative stress [81]. However, the specific regulatory association between GAS5, miR.222.3p, and Nrf2 signaling pathway remains to be elucidated in M. pneumoniae pneumonia.
M. hyopneumoniae, causes swine enzootic pneumonia, a chronic respiratory disease. A study used M. hyopneumoniae strain J to infect swine epithelial NPTr cells to detect mRNAs and miRNAs that were differently expressed [82]. Genes linked to redox homeostasis and antioxidant defense, which are known to be controlled by the transcription factor Nrf2 in similar species, had up-regulated mRNAs. Since they found that miRNAs anticipated to target antioxidant genes were down-regulated and miRNAs targeting ciliary and cytoskeleton genes were up-regulated, the bioinformatic analysis indicated a relationship between changes in miRNA and mRNA levels.
Treating chronic lung disorders linked to M. pneumoniae poses several difficulties that need to be resolved to guarantee the best possible outcomes for patients and avoid long-term issues. These difficulties include the necessity for long-term management plans, possible adverse effects, and antibiotic resistance. Since macrolides and fluoroquinolones have historically been the cornerstones of treatment for M. pneumoniae, the bacterium has become resistant to several widely used antibiotics [83]. The extensive and frequently careless use of antibiotics, along with M. pneumoniae’s capacity to develop resistance through mutations and horizontal gene transfer, have been connected to the rise of antibiotic-resistant strains [84]. High-level resistance to this family of antibiotics can be conferred by mutations in the 23S rRNA gene, which is the primary mechanism of macrolide resistance in M. pneumoniae [85]. The genes parC and parE encode DNA topoisomerase IV subunits, and mutations in these genes are frequently linked to fluoroquinolone resistance [86]. The genome of M. hominis contains two rRNA operons, the full sequences of which are not yet known, whereas M. pneumoniae only has one copy of 23S rRNA [87]. Genetic changes in DNA gyrase (GyrA and GyrB) and/or the topoisomerase IV complex (parC and parE) are linked to bacterial resistance to fluoroquinolones [88]. M. hominis’s resistance to 16-membered macrolides may be caused by changes in the ribosomal proteins L4 and L22, as well as in domain II or V of 23S rRNA [88]. Both M. fermentans, another erythromycin-resistant Mycoplasma, and M. hominis had a G2057A transition in their 23S rRNA sequence in comparison to that of M. pneumoniae [87]. Antibiotic-resistant M. pneumoniae strains are becoming more common, which makes it difficult to effectively treat chronic lung diseases because they reduce the number of available treatments and can result in treatment failures or the need for alternative, possibly less effective, or more toxic agents [89]. Chronic lung disease patients frequently need to take several drugs at the same time to treat their underlying problems, thus it is important to carefully assess the possibility of drug interactions and cumulative toxicity [90]. Long-term management techniques might be necessary in cases with chronic lung disorders made worse by M. pneumoniae infection to manage symptoms, stop the disease from getting worse, and reduce the chance of recurring infections or exacerbations [91]. Various supportive therapies, including bronchodilators, corticosteroids, and antibiotics, may be used in these regimens, depending on the severity of the disease and the underlying condition [92].
On the other hand, there is disagreement over the best long-term management strategies and the best duration and regimens for antibiotic therapy in chronic lung disorders linked to M. pneumoniae [93]. Long-term or recurrent antibiotic courses may raise the likelihood of antibiotic resistance, side effects, and respiratory microbiome disturbance, all of which could have negative consequences [94]. These elements, along with M. pneumoniae’s long-term effects on respiratory health, make treating chronic lung diseases especially difficult due to its antibiotic resistance.
One essential innate immune response that produces a protective host response is inflammation. Nevertheless, proinflammatory factors, ROS, NO, and other bioactive molecules may be released following a prolonged infection, which might distort the host’s reaction to “hyperinflammation” and cause excessive tissue damage. Therefore, some sort of negative regulatory mechanism must be in place to control an excessive inflammatory response.
Certain transcription factors and cellular signaling pathways influence
inflammatory responses [95]. According to Sundaresan et al. [96], the
traditional NF-
Even though there is increasing evidence that M. pneumoniae infections are linked to chronic lung disorders, more research is still needed in a few areas to further our knowledge and create efficient therapeutic approaches. Several pathogenic factors contribute to the development of linked diseases. The development of medications that target the common pathogenic mechanism of extrapulmonary and intrapulmonary infections may aid in the prevention of M. pneumoniae infections. Further studies are needed to develop a complete treatment plan that incorporates immunomodulators, and antibiotics for problems brought on by M. pneumoniae. Furthermore, investigating other approaches to treating macrolide antibiotics may alleviate the clinical signs of therapy failure. Patients who were infected with other prevalent respiratory pathogens also had M. pneumoniae infections. The emergence and spread of antibiotic-resistant M. pneumoniae strains must be tracked, and ongoing research and surveillance are required to look into the processes behind resistance and possible countermeasures. To tackle the increasing problem of antibiotic resistance, it should also be a top priority to discover new antimicrobial drugs with unique modes of action. In this regard, Nrf2 controls the expression of hundreds of genes that code for proteins with anti-inflammatory, antioxidant, drug-metabolizing, and other homeostatic properties. In this article, we have highlighted the role of the Nrf2 pathway in Mycoplasma infections and some of the ongoing challenges in the therapeutic management of Mycoplasma that need to be addressed. Thus, the aforementioned research suggests that targeting Nrf2 activation as a therapeutic target is probably going to be beneficial. Additionally, it is currently unknown if long-term pharmacological Nrf2 activation could change the risk of inflammation in situations that are susceptible to M. pneumoniae infection; this could be affected by the intervention’s timing about the disease’s course.
NAATs, nucleic acid amplification tests; Nrf2, erythroid 2-related factor 2; bZIP, basic-region leucine zipper; RBX1, ring box protein-1; GST, glutathione-S-transferase; ARE, antioxidant response element; HO-1, heme oxygenase-1; LPS, lipopolysaccharide; LPs, lipoproteins; MALP-2, macrophage-activating lipopeptide-2; IbpM, immunoglobulin-binding protein; ROS, reactive oxygen species; NETs, neutrophil extracellular traps; LAMPs, lipid-associated membrane proteins; AP-1, activating protein 1; PGE2, prostaglandin E2; NO, nitric oxide; PAMP, pathogen-associated molecular pattern; COX-2, cyclooxygenase 2; PI3K, phosphoinositide 3-kinases; CORM-2, carbon monoxide releasing molecule 2; lncRNA, long non-coding RNA; NF-
SS, NS, OK, EN and NZ, conceptualization, writing, and editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors acknowledge GLA University, India for providing support.
This study was supported by the RUDN University Strategic Academic Leadership Program.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.