




1 Department of Immunology, St Jude Children’s Research Hospital, Memphis, TN 38105, USA
Abstract
Ubiquitin ligases play pivotal roles in the regulation of NLR family pyrin domain containing 3 (NLRP3) inflammasome activation, a critical process in innate immunity and inflammatory responses. This review explores the intricate mechanisms by which various E3 ubiquitin ligases exert both positive and negative influences on NLRP3 inflammasome activity through diverse post-translational modifications. Negative regulation of NLRP3 inflammasome assembly is mediated by several E3 ligases, including F-box and leucine-rich repeat protein 2 (FBXL2), tripartite motif-containing protein 31 (TRIM31), and Casitas B-lineage lymphoma b (Cbl-b), which induce K48-linked ubiquitination of NLRP3, targeting it for proteasomal degradation. Membrane-associated RING-CH 7 (MARCH7) similarly promotes K48-linked ubiquitination leading to autophagic degradation, while RING finger protein (RNF125) induces K63-linked ubiquitination to modulate NLRP3 function. Ariadne homolog 2 (ARIH2) targets the nucleotide-binding domain (NBD) domain of NLRP3, inhibiting its activation, and tripartite motif-containing protein (TRIM65) employs dual K48 and K63-linked ubiquitination to suppress inflammasome assembly. Conversely, Pellino2 exemplifies a positive regulator, promoting NLRP3 inflammasome activation through K63-linked ubiquitination. Additionally, ubiquitin ligases influence other components critical for inflammasome function. TNF receptor-associated factor 3 (TRAF3) mediates K63 polyubiquitination of apoptosis-associated speck-like protein containing a CARD (ASC), facilitating its degradation, while E3 ligases regulate caspase-1 activation and DEAH-box helicase 33 (DHX33)-NLRP3 complex formation through specific ubiquitination events. Beyond direct inflammasome regulation, ubiquitin ligases impact broader innate immune signaling pathways, modulating pattern-recognition receptor responses and dendritic cell maturation. Furthermore, they intricately control NOD1/NOD2 signaling through K63-linked polyubiquitination of receptor-interacting protein 2 (RIP2), crucial for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein kinase (MAPK) activation. Furthermore, we explore how various pathogens, including bacteria, viruses, and parasites, have evolved sophisticated strategies to hijack the host ubiquitination machinery, manipulating NLRP3 inflammasome activation to evade immune responses. This comprehensive analysis provides insights into the molecular mechanisms underlying inflammasome regulation and their implications for inflammatory diseases, offering potential avenues for therapeutic interventions targeting the NLRP3 inflammasome. In conclusion, ubiquitin ligases emerge as key regulators of NLRP3 inflammasome activation, exhibiting a complex array of functions that finely tune immune responses. Understanding these regulatory mechanisms not only sheds light on fundamental aspects of inflammation but also offers potential therapeutic avenues for inflammatory disorders and infectious diseases.
Keywords
- ubiquitin ligases
- NLRP3 inflammasome
- innate immunity
- inflammation
- post-translational modifications
Inflammasomes are sophisticated multiprotein complexes that play a crucial role in innate immunity, acting as critical sensors and integrators of danger signals [1, 2, 3]. These complexes are essential for the body’s initial defense against both pathogenic microorganisms pathogen-associated molecular patterns (PAMPs) and sterile incursions damage-associated molecular patterns (DAMPs) such as trauma, cancer, ischemia, and metabolic perturbations [4, 5]. Typically, an inflammasome consists of three main components: a sensor protein (e.g., NOD-like receptors (NLR) family members or Pyrin and HIN domain-containing (PYHIN) proteins), an adaptor protein (usually ASC - apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD), and an effector protein (typically pro-caspase-1) [6, 7, 8].
Inflammasome activation is a tightly regulated process involving two distinct
steps. The initial step, known as priming, involves the upregulation of
inflammasome components and pro-forms of inflammatory cytokines, typically
through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-
Several types of inflammasomes have been identified, each responding to specific stimuli and playing distinct roles in immune responses [14, 15, 16, 17]. The NLR family pyrin domain containing 3 (NLRP3) inflammasome, for instance, is activated by a diverse range of stimuli including ATP, pore-forming toxins, crystalline substances, and cellular stress signals, and is involved in responses to a wide array of pathogens and sterile inflammatory conditions [18]. The NLR family pyrin domain containing 1 (NLRP1) inflammasome, activated by Bacillus anthracis lethal toxin and muramyl dipeptide, plays a significant role in anthrax infection and certain autoimmune disorders [6, 19]. The NLR family caspase activation and recruitment domain (CARD) domain-containing protein (NLRC4) inflammasome, often in conjunction with Neuronal Apoptosis Inhibitory Protein (NAIP) proteins, detects bacterial flagellin and components of type III secretion systems, making it critical for responding to intracellular bacterial pathogens [20, 21]. The absent in melanoma 2 (AIM2) inflammasome responds to cytosolic double-stranded DNA, playing an important role in host defense against DNA viruses and intracellular bacteria [22, 23]. Additionally, the Pyrin inflammasome, activated by bacterial toxins that modify Ras homolog gene family member A (RhoA) GTPases, is involved in autoinflammatory diseases and responses to certain bacterial infections [24, 25].
Other inflammasomes, including Interferon-Inducible Protein 16 (IFI16), NLRP6,
and NLRP7, have unique activators and roles in immune responses [17, 26, 27, 28]. The
activation of inflammasomes results in the proteolytic activation of inflammatory
caspases, leading to the maturation and secretion of pro-inflammatory cytokines
IL-1
While inflammasome activation is critical for host defense, it requires tight regulation to prevent excessive inflammation and tissue damage [32, 33]. Dysregulation of inflammasome activity has been associated with various inflammatory disorders, including autoinflammatory diseases, cardiometabolic diseases, infections, cancer, and neurological disorders [34, 35]. In the context of intracerebral hemorrhage (ICH), edaravone has emerged as a promising neuroprotective agent due to its multifaceted mechanisms of action [36, 37, 38]. As a potent free radical scavenger, neutralizes reactive oxygen species generated from hemoglobin breakdown, potentially mitigating oxidative stress-induced neuronal damage [39]. Additionally, edaravone exhibits anti-inflammatory properties by suppressing microglial activation and reducing pro-inflammatory cytokine production [40]. Preclinical study demonstrated edaravone’s ability to preserve blood-brain barrier integrity, inhibit neuronal apoptosis in the perihematomal region, and attenuate early brain injury following ICH. In animal models, edaravone treatment has been associated with reduced brain edema, decreased neurological deficits, and improved long-term functional outcomes [41]. While clinical evidence remains limited, a small randomized controlled trial showed promising results, with early edaravone administration linked to reduced hematoma growth and improved functional outcomes in ICH patients [38, 42]. The diversity of inflammasome types allows for a nuanced and targeted immune response to a wide range of potential threats [43]. Understanding the specific activation mechanisms and regulatory pathways of each inflammasome type is crucial for developing targeted therapeutic strategies for various inflammatory and infectious diseases [44]. Ongoing research continues to unravel the complex interplay between different inflammasomes and their roles in health and disease.
The NLRP3 inflammasome is a cornerstone of the innate immune system, serving as a sophisticated sentinel that detects a wide array of pathogenic threats and cellular stresses [10]. This multiprotein complex, comprising the NLRP3 sensor protein, the ASC adaptor, and pro-caspase 1, orchestrates the initiation and amplification of inflammatory responses crucial for host defense [45].
The NLRP3 protein itself is an architectural marvel, featuring three distinct domains: a C-terminal leucine-rich repeat (LRR) domain involved in ligand sensing, a central NACHT domain that facilitates oligomerization, and an N-terminal pyrin (PYD) domain responsible for protein-protein interactions during inflammasome assembly [46]. This intricate structure allows NLRP3 to function as a molecular switch, transitioning from an inactive to an active state in response to cellular danger signals [47].
Activation of the NLRP3 inflammasome is a meticulously regulated process that
typically unfolds in two stages: priming and activation. The priming phase,
initiated by pathogen-associated molecular patterns (PAMPs) or damage-associated
molecular patterns (DAMPs), activates NF-
The activation phase, triggered by a remarkably diverse array of stimuli ranging from extracellular ATP and pore-forming toxins to particulate matter and various pathogens, initiates the assembly of the inflammasome complex [1]. While the precise mechanism by which NLRP3 recognizes such a broad spectrum of activators remains a subject of intense research, emerging evidence suggests that NLRP3 likely senses common cellular perturbations induced by these stimuli rather than directly binding to them [55]. One proposed mechanism involves the disassembly of the trans-Golgi network (TGN), which serves as a cellular stress indicator that can recruit and activate NLRP3 [47, 56].
Upon activation, NIMA-related kinase 7 (NEK7) emerges as a critical player, binding to NLRP3 and inducing a conformational change that disrupts its inactive double-ring structure [57]. This structural rearrangement exposes the PYD domains, allowing the NACHT domain to oligomerize [58]. The exposed PYD domain then recruits the adaptor molecule ASC, forming a remarkable filamentous structure known as the ASC pyroptosome or “speck” [59]. This speck serves as a molecular platform, with the caspase recruitment domain (CARD) of ASC binding to pro-caspase-1 and facilitating its conversion to active caspase-1 through proximity-induced autoproteolysis [60, 61].
Activated caspase-1 functions as the effector arm of the inflammasome,
processing the pro-forms of IL-1
The NLRP3 inflammasome is tightly regulated through multiple mechanisms to maintain immune homeostasis. Autophagy plays a crucial role in limiting NLRP3 activation by promoting its degradation [65]. MicroRNAs, particularly miR-223, negatively regulate NLRP3 expression by targeting its mRNA [50, 65]. Anti-inflammatory cytokines like IL-10 suppress NLRP3 activity [45], while endogenous inhibitors such as CARD8 and pyrin-only proteins provide additional control [66, 67]. Species-specific differences in NLRP3 activation exist between humans and mice, highlighting the importance of careful interpretation in translational research [68]. Recent structural biology insights have revealed conformational changes during NLRP3 activation, enhancing our understanding of its molecular mechanisms [67]. Cellular metabolism, especially glycolysis and mitochondrial function, significantly influences NLRP3 activation [47]. NLRP3 function varies across different tissue contexts, with distinct roles in immune cells versus epithelial cells [69]. From an evolutionary perspective, NLRP3 is conserved across species, suggesting its fundamental importance in innate immunity [70].
Complementing this canonical pathway, NLRP3 inflammasome activation can also proceed through a non-canonical route mediated by caspase-4 and caspase-5 in humans, or caspase-11 in mice [71, 72]. This pathway is particularly relevant in the context of intracellular LPS detection from Gram-negative bacteria, further expanding the surveillance capacity of the inflammasome system [50, 73].
The exquisite regulation of NLRP3 inflammasome activation is paramount for maintaining immune homeostasis [66]. Dysregulation of this process has been implicated in a spectrum of inflammatory disorders, ranging from autoinflammatory diseases to metabolic syndromes and neurodegenerative conditions [74]. This central role in health and disease has positioned the NLRP3 inflammasome as a prime target for therapeutic interventions, with ongoing research exploring various strategies to modulate its activity in inflammatory diseases [75].
Post-translational modifications (PTMs) play a pivotal role in regulating the activity of the NLRP3 inflammasome, a critical component of innate immunity responsible for detecting cellular stress and pathogen invasion [76, 77]. These PTMs exert precise control over NLRP3 function, influencing its activation, assembly into the inflammasome complex, and subsequent cytokine secretion [56, 78].
Phosphorylation plays a critical and multifaceted role in regulating NLRP3 inflammasome activation, with different phosphorylation sites exerting diverse and sometimes opposing effects [79]. At Ser295 (human)/Ser293 (mouse), phosphorylation by protein kinase D (PKD) at the Golgi apparatus promotes activation. In contrast, phosphorylation by protein kinase A (PKA) inhibits activation by suppressing the ATPase activity of the NLRP3 NACHT domain. At Ser5 (human)/Ser3 (mouse), phosphorylation by AKT has a dual role: it impedes inflammasome activation by inhibiting NLRP3 oligomerization and prevents proteasome-mediated degradation of NLRP3 during lipopolysaccharide (LPS) priming. Dephosphorylation of this site by protein phosphatase 2A (PP2A) enhances activation by promoting NLRP3-ASC interaction [45, 80, 81]. Additionally, phosphorylation at Ser198 (human)/Ser194 (mouse) by c-Jun N-terminal kinase 1 (JNK1) facilitates NLRP3 homo-oligomerization and is essential for inflammasome activation [82]. In contrast, enhanced tyrosine phosphorylation at Tyr861 negatively regulates inflammasome activation through autophagy induction, with protein tyrosine phosphatase nonreceptor 22 (PTPN22) promoting activation by dephosphorylating this site [77, 83].
Ubiquitination plays a crucial role in regulating NLRP3 inflammasome function through various linkage types, with the balance between ubiquitination and deubiquitination being essential for controlling inflammasome activation [84]. K48-linked ubiquitination targets NLRP3 for proteasomal degradation, inhibiting inflammasome activation [85]. Several E3 ubiquitin ligases mediate this process, including TRIM31, which promotes K48-linked polyubiquitination of NLRP3 [45]. Other E3 ligases such as MARCH7, ARIH2, and FBXL2 also attenuate NLRP3 inflammasome activation through protein degradation [86]. K63-linked ubiquitination, present in resting macrophages, has been associated with NLRP3 regulation. The E3 ubiquitin ligase TRIM65 can induce both K48- and K63-linked ubiquitination of NLRP3, with a higher efficiency for K63-linked ubiquitination. Deubiquitination enhances inflammasome activation, with BRCA1/BRCA2-containing complex 3 (BRCC3), a component of the BRCC36 isopeptidase complex (BRISC), acting as a positive regulator by promoting deubiquitination of the Leucine Rich Repeat (LRR) region of NLRP3 [86]. The USP1-Associated Factor 1/Ubiquitin-Specific Peptidase 1 UAF1/USP1 deubiquitinase complex specifically removes K48-linked ubiquitination of NLRP3, stabilizing its expression and facilitating inflammasome activation [84].
Other post-translational modifications (PTMs) intricately regulate NLRP3
inflammasome activity through diverse mechanisms. SUMOylation, mediated by the E3
ligase MAPL (MUL1), inhibits NLRP3 by preventing its oligomerization and
interaction with ASC, thereby blocking inflammasome assembly and activation [45, 87, 88]. S-nitrosylation at cysteine residues, such as Cys3 and Cys394, by nitric
oxide (NO) donors, impedes NLRP3 oligomerization and inflammasome formation [89].
ADP-ribosylation mediated by Poly (ADP-ribose) polymerase 1 (PARP1) suppresses
NLRP3 activation by disrupting its interaction with NIMA-related kinase 7 (NEK7), which is crucial for inflammasome assembly [90]. Acetylation at lysine
residues by p300/CBP enhances NLRP3 activation, promoting its interaction with
ASC and facilitating IL-1
The intricate regulation of the NLRP3 inflammasome is heavily influenced by various post-translational modifications (PTMs), with ubiquitination playing a pivotal role [93]. Understanding these modifications is crucial for developing targeted therapies to modulate inflammasome activity in inflammatory and immune-related diseases [78]. Ubiquitin ligases, through the addition of ubiquitin molecules, exert precise control over NLRP3’s stability, localization, and activity, ultimately modulating inflammasome activation [94]. These modifications can either tag NLRP3 for degradation or facilitate its assembly and activation, thus balancing the immune response to prevent both insufficient and excessive inflammation [56]. Understanding the specific ubiquitin ligases involved in these processes is crucial for comprehending the broader regulatory mechanisms at play. Each ubiquitin ligase, including TRIM31, MARCH7, FBXL2, Pellino2, and TRAF6, contributes uniquely to the modulation of NLRP3 activity through distinct ubiquitination pathways [1, 77]. Below, we delve into the detailed roles of these key ubiquitin ligases and their impact on NLRP3 inflammasome regulation (Fig. 1) created using Biorender (https://biorender.com/).
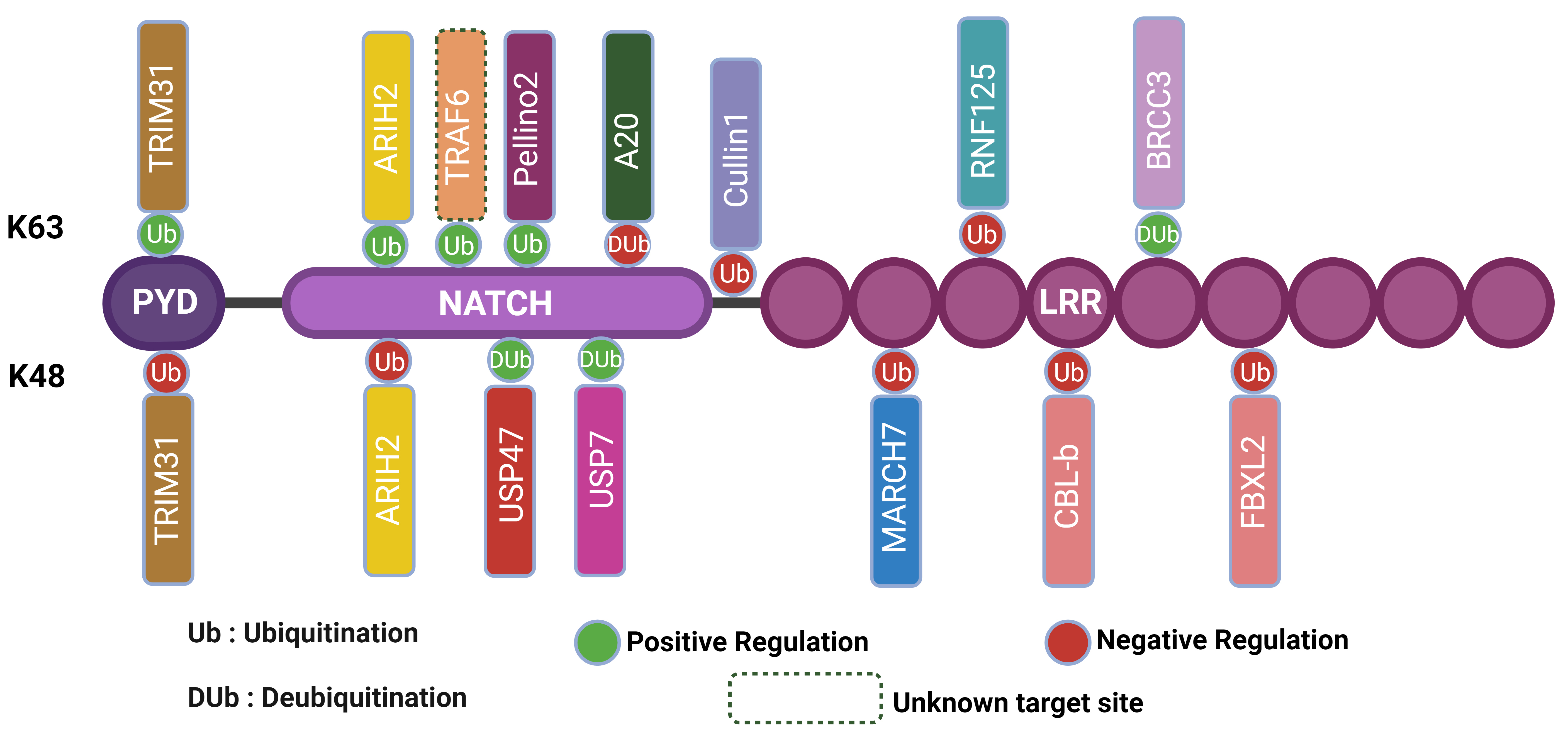 Fig. 1.
Fig. 1.
Regulation of NLRP3 inflammasome activation through ubiquitination and deubiquitination mechanisms. The figure illustrates the regulation of the NLRP3 inflammasome through ubiquitination and deubiquitination mechanisms. The NLRP3 inflammasome consists of the (Pyrin Domain) PYD, Nucleotide-binding domain, NATCH, and Leucine-Rich Repeat (LRR) domains. Positive regulation occurs via K63 ubiquitination by TRIM31 and ARIH2, promoting inflammasome activation, while negative regulation is mediated by K48 ubiquitination by the same proteins, leading to inhibition. Deubiquitinating enzymes such as USP47, USP7, and BRCC3 remove ubiquitin, thereby influencing inflammasome activity. Additionally, other regulatory proteins, including TRAF6, Pellino2, A20, Cullin1, RNF125, MARCH7, CBL-b, and FBXL2, modulate inflammasome activity through various ubiquitination pathways. The legend indicates ubiquitination (Ub), deubiquitination (DUb), positive regulation (green circles), negative regulation (red circles), and unknown target sites (dashed lines). This figure highlights the complex interplay of post-translational modifications in regulating NLRP3 inflammasome activation. Created using BioRender.com. NLRP3, NLR family pyrin domain containing 3; BRCC3, BRCA1/BRCA2-containing complex subunit 3; TRIM31, Tripartite Motif Containing 31; ARIH2, Ariadne RBR E3 Ubiquitin Protein Ligase 2; TRAF6, TNF Receptor-Associated Factor 6; RNF125, Ring Finger Protein 125; MARCH7, Membrane-Associated Ring-CH-Type Finger 7; CBL-b, Casitas B-Lineage Lymphoma-b; FBXL2, F-Box And Leucine Rich Repeat Protein 2.
Understanding the negative regulation of the NLRP3 inflammasome is crucial for comprehending how the body maintains immune homeostasis and prevents excessive inflammation. Several ubiquitin ligases act as key negative regulators, mediating the ubiquitination and subsequent proteasomal degradation of NLRP3, thus limiting its activation. These ubiquitin ligases ensure that the NLRP3 inflammasome is tightly controlled, preventing unwarranted inflammatory responses that can lead to chronic inflammation and autoimmune diseases [94].
TRIM31, a member of the TRIM protein family with E3 ubiquitin ligase activity,
intricately regulates NLRP3 inflammasome activation through multifaceted
mechanisms [95]. It directly binds to NLRP3 via its PYD domain, facilitating both
K48-linked ubiquitination for proteasomal degradation and K63-linked
ubiquitination that enhances NLRP3 oligomerization and assembly with ASC, thereby
promoting inflammasome activation [85]. This dual regulatory role of TRIM31 is
pivotal in modulating immune responses: it negatively regulates NLRP3 by
promoting degradation to prevent hyperinflammation, while positively influencing
inflammatory responses through assembly promotion. Experimental evidence
demonstrates that TRIM31 overexpression decreases NLRP3 levels and IL-1
Membrane-Associated Ring-CH-Type Finger 7 (MARCH7) is a member of the MARCH family of E3 ubiquitin ligases, characterized by the presence of a Really Interesting New Gene-Cysteine/Histidine-rich (RING-CH) domain essential for their ubiquitin ligase activity [96]. MARCH7 specifically targets the NLRP3 protein for ubiquitination, catalyzing the addition of K48-linked polyubiquitin chains to NLRP3, typically signaling it for degradation [97]. Unlike other E3 ligases that promote proteasomal degradation, MARCH7 mediates the autophagic degradation of NLRP3 [98]. This distinct autophagic pathway helps regulate NLRP3 levels and activity. MARCH7 binds to the Leucine-Rich Repeat (LRR) and NACHT domains of NLRP3, which are critical for its function in inflammasome formation. This specific interaction does not affect other inflammasome components such as ASC or Pro-Caspase-1, highlighting MARCH7’s targeted regulation [66].
By promoting the degradation of NLRP3, MARCH7 acts as a negative regulator,
suppressing the formation and activation of the NLRP3 inflammasome [66]. This
process reduces NLRP3 protein levels, preventing unnecessary or excessive
inflammasome activation and maintaining cellular homeostasis under basal
conditions. This process is intricately linked to the dopamine D1 receptor
pathway, providing a fascinating connection between neurotransmitter signaling
and innate immunity [50]. By promoting NLRP3 degradation in response to
dopaminergic stimulation, MARCH7 contributes to the anti-inflammatory effects of
dopamine, effectively inhibiting NLRP3 inflammasome activation. This
dopamine-MARCH7-NLRP3 axis represents a sophisticated regulatory mechanism that
highlights the intricate crosstalk between the nervous system and inflammatory
processes [99]. A Study showed that overexpression of MARCH7 decreases NLRP3
protein levels and IL-1
Ring Finger Protein 125 (RNF125) is an E3 ubiquitin ligase that plays a crucial
role in the regulation of the NLRP3 inflammasome. RNF125 initiates K63-linked
polyubiquitination of the NLRP3 leucine-rich repeat (LRR) domain, a modification
that facilitates the recruitment of another E3 ubiquitin ligase, Cbl-b [100]. By
targeting NLRP3 for K63-linked ubiquitination, RNF125 sets the stage for a
two-step process that ultimately controls NLRP3 protein levels and inflammasome
activity [100]. This mechanism creates a negative feedback loop to control
inflammation, particularly in the context of viral infections where type I
interferons are prominently induced. By reducing NLRP3 protein levels, RNF125
suppresses inflammasome assembly and activation, leading to decreased production
of pro-inflammatory cytokines like IL-1
Casitas B-lineage Lymphoma-b (Cbl-b) is another E3 ubiquitin ligase involved in the regulation of the NLRP3 inflammasome [100]. Cbl-b specifically targets the LRR domain of NLRP3 for K48-linked polyubiquitination, also leading to proteasomal degradation. By targeting NLRP3 for degradation, Cbl-b acts as a negative regulator, preventing the excessive activation of the inflammasome [100]. This regulation is critical for balancing immune responses and avoiding hyperinflammation. Research indicates that Cbl-b deficiency leads to increased NLRP3 levels and heightened inflammasome activity, contributing to exacerbated inflammatory conditions [45]. The role of Cbl-b in inflammasome regulation underscores the importance of ubiquitin-mediated pathways in controlling innate immune responses. Furthermore, the negative regulation of NLRP3 inflammasome activation by Cbl has been associated with the inhibition of glycolysis through GLUT1-dependent mechanisms [101]. By inhibiting Cbl, there is an increase in GLUT1 expression, leading to enhanced glycolytic capacity and subsequent NLRP3 inflammasome activation [101]. This demonstrates the diverse pathways through which proteins like Cbl modulate NLRP3 activity.
F-box and Leucine-rich repeat protein 2 (FBXL2) is a critical regulator of the
NLRP3 inflammasome, functioning as part of the Skp1-Cullin-F-box (SCF) E3
ubiquitin ligase complex [102]. This complex consists of Skp1, Cullin1, Rbx1, and
FBXL2 as the F-box protein that determines substrate specificity. FBXL2 directly
interacts with NLRP3 through its leucine-rich repeats (LRRs), specifically
targeting Lysine689 of NLRP3 for ubiquitination [77, 103]. It facilitates the
addition of K48-linked polyubiquitin chains to NLRP3, marking it for proteasomal
degradation by the 26S proteasome [104]. This process effectively reduces
cellular levels of NLRP3, thereby inhibiting inflammasome activation and
decreasing production of pro-inflammatory cytokines like IL-1
FBXL2’s activity is modulated by various cellular signals, including JNK1-mediated phosphorylation, which enhances its ability to target NLRP3 [10]. Interestingly, FBXL2 itself is regulated by another F-box protein, FBXO3, creating a regulatory loop where TLR signaling can indirectly increase NLRP3 levels by reducing FBXL2-mediated degradation [105]. This mechanism helps maintain homeostasis under basal conditions and plays a crucial role in resolving inflammation after the initial immune response.
Experimental evidence underscores FBXL2’s importance, showing that its overexpression decreases NLRP3 levels and reduces inflammasome activation, while its knockdown has the opposite effect [103]. Dysregulation of FBXL2 could potentially contribute to excessive NLRP3 activation in various inflammatory disorders, making it a promising therapeutic target [45].
Ariadne RBR E3 Ubiquitin Protein Ligase 2 (ARIH2), a member of the Ring-Between-Ring (RBR) family of E3 ubiquitin ligases, plays a pivotal role in modulating NLRP3 inflammasome activation through its distinctive ubiquitin ligase activity [106, 107]. It specifically targets NLRP3 for ubiquitination, catalyzing both K48-linked and K63-linked polyubiquitination. While K48-linked ubiquitination typically marks proteins for proteasomal degradation, the exact consequences of ARIH2-mediated ubiquitination on NLRP3 function are still under investigation [108]. This dual mode of ubiquitination by ARIH2 highlights its nuanced regulatory role in fine-tuning inflammasome activity, potentially impacting the formation and activation of the NLRP3 complex [109]. This mechanism is crucial during the priming phase of NLRP3 activation, influencing the expression and assembly of inflammasome components in response to inflammatory cues.
ARIH2’s interaction with NLRP3 is pivotal in macrophages, where it negatively
regulates inflammasome activation. By modifying NLRP3 through ubiquitination,
ARIH2 likely modulates its stability or activity, thereby affecting downstream
inflammatory responses mediated by IL-1
Cullin1, as an essential component of the Skp1-Cullin1-F-box (SCF) E3 ubiquitin ligase complex, plays a critical role in regulating NLRP3 inflammasome activation through its distinct mechanism. Structurally, Cullin1 acts as a scaffold within the SCF complex, facilitating the assembly of Skp1, an F-box protein, and a RING protein (typically Rbx1). This complex formation is crucial for its function as an E3 ubiquitin ligase. Interaction studies have revealed that Cullin1 directly interacts with the PYD domain of NLRP3 [45, 110]. This interaction facilitates the specific ubiquitination of NLRP3, particularly through K63-linked polyubiquitin chains [111]. Unlike K48-linked ubiquitination, which typically targets proteins for proteasomal degradation, K63-linked ubiquitination serves primarily as a signaling mechanism involved in protein-protein interactions and cellular signaling pathways.
Specifically, Cullin1 promotes K63-linked ubiquitination at lysine 689 (K689) of NLRP3. This modification plays a pivotal role in preventing NLRP3 inflammasome assembly and activation under basal conditions [112]. By ubiquitinating NLRP3 in this manner, Cullin1 interferes with the assembly of the NLRP3 inflammasome complex or its responsiveness to activating signals, thereby maintaining NLRP3 in an inactive state [13]. This regulatory role of Cullin1 is distinct from other E3 ligases such as TRIM31 or FBXL2, which primarily induce proteasomal degradation of NLRP3 [105]. The non-degradative nature of K63-linked ubiquitination by Cullin1 suggests a mechanism for finely tuning NLRP3 activity without the need for protein degradation, enabling rapid activation or inactivation as needed by the cell [77]. Understanding the intricate regulatory mechanisms involving Cullin1 and NLRP3 provides insights into how cells tightly control inflammatory responses. This knowledge could potentially lead to new therapeutic strategies for managing NLRP3-associated inflammatory disorders by modulating Cullin1 activity or its interaction with NLRP3 to influence inflammasome activation appropriately.
While most ubiquitin ligases negatively regulate NLRP3 inflammasome activation, some promote its activation. These positive regulators play crucial roles in ensuring a timely and adequate inflammatory response, highlighting the complex and balanced nature of NLRP3 regulation [113]. Understanding the role of positive regulators in inflammasome dynamics and their implications for immune responses and disease is crucial to a better understanding of inflammasome dynamics.
Pellino2, a member of the Pellino family of E3 ubiquitin ligases, plays a
crucial role in regulating the NLRP3 inflammasome [114]. Unlike many other E3
ligases that negatively regulate NLRP3, Pellino2 acts as a positive regulator,
specifically during the priming phase of inflammasome activation [115]. It
contains a RING-like domain that confers its ubiquitin ligase activity, allowing
it to catalyze the addition of K63-linked polyubiquitin chains to NLRP3 [93, 114]. The K63-linked ubiquitination facilitated by Pellino2 is crucial for NLRP3
inflammasome activation. This type of ubiquitination, unlike K48-linked
ubiquitination which typically targets proteins for degradation, is involved in
regulating protein interactions and signaling pathways [13]. By adding these
ubiquitin chains, Pellino2 enhances NLRP3’s ability to oligomerize and assemble
into the inflammasome complex. This promotes the activation of NLRP3, leading to
the cleavage of pro-caspase-1 and the subsequent maturation and secretion of
pro-inflammatory cytokines such as IL-1
Pellino2’s activity is particularly important in response to pathogenic infections and other danger signals, ensuring a rapid and robust activation of the NLRP3 inflammasome [114]. Its expression and activity are regulated by various signaling pathways, including those activated by Toll-like receptors (TLRs) and other pattern recognition receptors (PRRs). This regulation ensures that Pellino2 activity is tightly controlled and context dependent [115].
Experimental evidence has shown that Pellino2 deficiency results in impaired
NLRP3 inflammasome activation and reduced levels of IL-1
Given its role in promoting NLRP3 inflammasome activation, dysregulation of Pellino2 could contribute to inflammatory diseases characterized by excessive or chronic inflammation. Targeting Pellino2 activity might offer a novel approach to modulating NLRP3 inflammasome activation, with potential applications in boosting immune responses or reducing pathological inflammation in various disorders.
Tumor Necrosis Factor Receptor-Associated Factor 6 (TRAF6) is a crucial adaptor protein and E3 ubiquitin ligase that plays a key role in regulating the activation of the NLRP3 inflammasome, a multiprotein complex central to inflammatory responses [118]. TRAF6 directly interacts with NLRP3, catalyzing the addition of K63-linked ubiquitin chains, which promotes signaling processes rather than protein degradation [118, 119]. Unlike some E3 ligases that negatively regulate NLRP3, TRAF6 enhances its activation by promoting oligomerization and assembly of the inflammasome complex [77].
TRAF6 is involved in multiple aspects of NLRP3 regulation. It participates in
the priming step by mediating NF-
The TRAF6-mediated activation of the NLRP3 inflammasome is particularly important in response to microbial infections and danger signals. TRAF6’s activity and expression are regulated by upstream signaling molecules and receptors, including Toll-like receptors (TLRs) and interleukin-1 receptor (IL-1R), which recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) [45]. Experimental evidence shows that knockdown or inhibition of TRAF6 impairs NLRP3 inflammasome activation, while its overexpression enhances inflammasome activity and cytokine production [123].
The positive regulation of NLRP3 inflammasome by TRAF6 is crucial for immune responses to certain pathogens and plays a role in the development of autoimmune diseases [123]. Dysregulation of TRAF6 can contribute to various inflammatory and autoimmune conditions, such as rheumatoid arthritis, atherosclerosis, and neuroinflammatory diseases [124]. Understanding the TRAF6-NLRP3 interaction holds significant therapeutic potential, as modulating TRAF6 activity could help control NLRP3-mediated inflammation in various disease contexts [125].
While ubiquitin ligases play a crucial role in modulating NLRP3 inflammasome activity through the addition of ubiquitin chains, the removal of these chains is equally important for the precise regulation of this complex [126]. Deubiquitinating enzymes (DUBs) are specialized proteases that cleave ubiquitin moieties from substrate proteins, thereby reversing the actions of ubiquitin ligases. In the context of the NLRP3 inflammasome, DUBs can either enhance or suppress inflammasome activation by removing specific types of ubiquitin modifications [77, 105]. The dynamic interplay between ubiquitination and deubiquitination ensures a balanced and timely response to inflammatory signals, maintaining immune homeostasis and preventing excessive inflammation [127]. In the following sections, we will discuss key deubiquitinating enzymes involved in the regulation of the NLRP3 inflammasome and their mechanisms of action.
BRCA1/BRCA2-containing complex subunit 3 (BRCC3) is a crucial deubiquitinating enzyme that plays a significant role in regulating the NLRP3 inflammasome, a key component of the innate immune response [128]. As a member of the JAB1/MPN/Mov34 Metalloenzyme (JAMM) family of zinc-dependent metalloproteases, BRCC3 specifically cleaves K63-linked polyubiquitin chains from NLRP3, acting as a positive regulator of inflammasome activation [129]. This function is particularly important in the context of inflammatory responses and immune regulation.
BRCC3 is a component of the BRCC36 isopeptidase complex (BRISC), which is
involved in various cellular processes, including DNA repair and maintenance of
genomic stability [130]. In its role regulating the NLRP3 inflammasome, BRCC3
directly interacts with NLRP3, removing K63-linked ubiquitin chains from its LRR
region [129]. This deubiquitination event is crucial for NLRP3 assembly and
activation, facilitating the formation of the inflammasome complex and subsequent
activation of caspase 1. The process leads to the production and secretion of
pro-inflammatory cytokines such as IL-1
The timing of BRCC3’s action is specific, occurring after the priming step but before or during the activation step of NLRP3 inflammasome formation [48]. This mechanism ensures that NLRP3 activation is timely and appropriately scaled, preventing unwarranted inflammation while allowing for an effective immune response when needed. BRCC3’s activity has been observed in various cell types, including macrophages and dendritic cells, underscoring its importance in diverse immune contexts [131].
The regulation of NLRP3 by BRCC3 has significant implications for both physiological and pathological processes [47]. Proper functioning of BRCC3 is essential for the immune system’s ability to respond to infections and tissue damage. However, dysregulation of BRCC3 activity can lead to either insufficient or excessive inflammasome activation, potentially contributing to a range of inflammatory disorders [128]. Conditions such as autoinflammatory syndromes, chronic inflammatory diseases, and certain cancers have been associated with aberrant NLRP3 activation, highlighting the importance of BRCC3 in maintaining immune homeostasis [1].
The therapeutic potential of targeting BRCC3 and its regulatory pathways is significant. Modulating BRCC3 activity could be beneficial in treating diseases characterized by excessive inflammasome activation, such as gout, atherosclerosis, and autoimmune disorders. Inhibiting BRCC3 could potentially dampen NLRP3 inflammasome activation in conditions with excessive inflammation. Conversely, enhancing BRCC3 activity might improve immune responses in situations where NLRP3 activation is deficient [78].
Ubiquitin-specific peptidase 7 (USP7), also known as Herpesvirus-associated ubiquitin-specific protease (HAUSP), is a crucial deubiquitinating enzyme that plays a significant role in regulating the NLRP3 inflammasome [132]. As a member of the ubiquitin-specific protease (USP) family, USP7 removes ubiquitin moieties from substrate proteins, rescuing them from proteasomal degradation or altering their functional state [133]. This cysteine protease has diverse cellular functions, including roles in DNA damage response, apoptosis, and immune regulation.
In the context of NLRP3 inflammasome regulation, USP7 directly interacts with NLRP3 and modulates its stability and activity through deubiquitination [134]. It primarily removes K48-linked polyubiquitin chains, which are typically associated with targeting proteins for proteasomal degradation. By deubiquitinating NLRP3, USP7 stabilizes the protein and enhances its accumulation within the cell. This mechanism counteracts the effects of various E3 ubiquitin ligases, such as TRIM31, MARCH7, and FBXL2, which target NLRP3 for degradation [134].
USP7 acts as a positive regulator of the NLRP3 inflammasome [135]. By preventing
the degradation of NLRP3, USP7 increases the availability of this protein,
thereby promoting the formation and activation of the inflammasome complex [135].
This leads to the subsequent activation of caspase-1 and the maturation and
secretion of pro-inflammatory cytokines such as IL-1
The activity of USP7 in regulating NLRP3 has been observed in various cell types, including macrophages and other immune cells [13]. USP7 displays specificity for NLRP3 among various inflammasome components, underscoring its targeted regulatory role. It does not significantly affect other key inflammasome proteins like ASC or Pro-caspase-1, focusing its activity primarily on NLRP3 [137].
Experimental evidence supports USP7’s role in NLRP3 regulation. A study showed that the inhibition or knockdown of USP7 leads to decreased NLRP3 protein levels and reduced inflammasome activation [47]. Conversely, overexpression of USP7 results in increased NLRP3 stability and enhanced inflammasome activity, corroborating its role as a positive regulator.
The activity and expression of USP7 itself are subject to regulation, which can influence its effect on the NLRP3 inflammasome. Various factors, including post-translational modifications and protein-protein interactions, regulate USP7’s activity. These regulatory mechanisms can indirectly affect NLRP3 inflammasome activation [135].
Given its role in promoting NLRP3 activation, dysregulation of USP7 could contribute to inflammatory disorders characterized by excessive or chronic NLRP3 activation. Conditions such as autoinflammatory diseases, metabolic disorders, and certain infections might be influenced by alterations in USP7 activity [138]. This makes USP7 a potential therapeutic target for managing NLRP3-related inflammatory diseases. Modulating USP7 activity, possibly using USP7 inhibitors, could be a strategy to control NLRP3-mediated inflammation [139].
USP7 functions within a broader network of NLRP3 regulators, including other deubiquitinating enzymes and ubiquitin ligases. Understanding how USP7 interacts with these other factors is crucial for a comprehensive view of NLRP3 regulation. The balance between ubiquitination by E3 ligases and deubiquitination by DUBs like USP7 allows for fine-tuning of NLRP3 levels and activity, which is crucial for maintaining appropriate inflammatory responses while preventing excessive inflammation [77].
USP47, a member of the ubiquitin-specific protease family, plays a crucial role in regulating the NLRP3 inflammasome, a key component of the innate immune response. This deubiquitinating enzyme acts by removing ubiquitin moieties from NLRP3, preventing its proteasomal degradation and increasing its stability within the cell [140]. Through this mechanism, USP47 functions as a positive regulator of NLRP3 inflammasome activation, enhancing the availability of this key component and promoting its assembly into the active inflammasome complex [135].
The deubiquitinating activity of USP47 facilitates efficient inflammasome assembly by allowing NLRP3 to interact with the adaptor protein ASC and Pro-caspase 1 [45]. This process is essential for a robust inflammatory response, enabling cells to effectively respond to pathogenic infections and other danger signals [141]. USP47 displays specificity for NLRP3 among various inflammasome components, underscoring its targeted regulatory role [21]. Its activity is particularly significant in contexts where enhanced inflammasome activation is required, such as during microbial infections, tissue damage, or sterile inflammation.
Experimental study has demonstrated that inhibition or knockdown of USP47 leads to reduced NLRP3 protein levels and diminished inflammasome activation, while overexpression results in increased NLRP3 stability and enhanced inflammasome activity [47]. These findings suggest that USP47 is critical for maintaining appropriate levels of NLRP3 to ensure effective inflammasome responses. Given its role in promoting NLRP3 activation, dysregulation of USP47 could contribute to inflammatory disorders characterized by excessive or chronic NLRP3 activation, such as autoinflammatory diseases, metabolic disorders, and certain infections [142].
Targeting USP47 offers a promising therapeutic approach for managing NLRP3-related inflammatory diseases. Inhibiting USP47 might help reduce excessive inflammation in conditions where NLRP3 hyperactivation is detrimental, while enhancing its activity could boost NLRP3 responses when a stronger immune response is needed [135]. USP47 functions within a complex network of NLRP3 regulators, including other deubiquitinating enzymes and ubiquitin ligases.
A20, also known as Tumor Necrosis Factor Alpha-Induced Protein 3 (TNFAIP3), is a pivotal anti-inflammatory molecule that plays a crucial role in regulating immune and inflammatory responses, including the NLRP3 inflammasome [143]. As a ubiquitin-editing enzyme with both deubiquitinating (DUB) and E3 ubiquitin ligase activities, A20 possesses a unique dual functionality that allows it to modulate protein ubiquitination status in complex ways, making it a versatile regulator of various signaling pathways [144].
In the context of NLRP3 inflammasome regulation, A20 primarily functions as a negative regulator, helping to prevent excessive or prolonged inflammatory responses. Its mechanism of action involves the removal of K63-linked polyubiquitin chains from NLRP3 and potentially other inflammasome components [143]. This deubiquitination is crucial for controlling the activation and assembly of the NLRP3 inflammasome, as it interferes with the recruitment of downstream signaling molecules necessary for inflammasome assembly. By maintaining NLRP3 in an inactive state under normal conditions, A20 prevents unwarranted inflammasome activation and excessive inflammation [145].
The importance of A20 in regulating the NLRP3 inflammasome is underscored by an
experimental study showing that the absence or inhibition of A20 leads to
enhanced NLRP3 inflammasome activation and increased production of
pro-inflammatory cytokines such as IL-1
A20’s regulatory effects extend beyond the NLRP3 inflammasome, as it also
modulates other signaling pathways involved in immune responses, such as
NF-
The impact of A20 on NLRP3 inflammasome activation may vary depending on the cell type and specific inflammatory context. This cell type-specific effect adds another layer of complexity to its regulatory role and highlights the need for further research to fully understand the nuances of A20’s function in different cellular environments [150].
Given its central role in regulating inflammation, A20 represents a potential therapeutic target for various inflammatory and autoimmune diseases. Enhancing A20 activity could be beneficial in conditions characterized by excessive NLRP3 activation, such as rheumatoid arthritis, systemic lupus erythematosus, and certain autoinflammatory syndromes [151]. Therapeutic strategies aimed at modulating A20 function may help restore immune homeostasis and alleviate disease symptoms. By controlling the ubiquitination status of NLRP3 and other signaling molecules, A20 helps maintain immune homeostasis and prevent chronic inflammatory responses [152]. Understanding the mechanisms by which A20 regulates the NLRP3 inflammasome and developing strategies to modulate its activity holds significant therapeutic potential for managing inflammatory and autoimmune diseases.
The ubiquitination of NLRP3 plays a critical role in regulating its activation and degradation [45]. Ubiquitination involves the attachment of ubiquitin molecules to lysine residues on NLRP3, which can lead to different cellular outcomes depending on the type of ubiquitin linkage [66]. Specifically, K48-linked ubiquitination of NLRP3 typically targets it for proteasomal degradation. E3 ubiquitin ligases such as TRIM31 and MARCH7 have been shown to mediate K48-linked polyubiquitination of NLRP3, promoting its degradation via the proteasome and thereby inhibiting inflammasome activation [85, 153]. On the other hand, K63-linked ubiquitination is associated with autophagic degradation [77]. This process involves the interaction of ubiquitinated NLRP3 with autophagic adaptors like p62, which facilitate its delivery to the autophagosome for degradation [98, 154]. The balance between these ubiquitination pathways ensures that NLRP3 levels are tightly regulated, preventing excessive inflammasome activation that could lead to pathological inflammation [54].
Pathogens have evolved sophisticated strategies to manipulate host cellular pathways to evade immune responses and establish infections. One such strategy involves hijacking the host ubiquitination pathway to regulate NLRP3 inflammasome activation [155]. Ubiquitination, a post-translational modification where ubiquitin proteins are attached to target proteins, plays a crucial role in controlling the stability, localization, and activity of many cellular proteins, including those involved in immune responses [156]. Below Table 1 (Ref. [94, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179, 180, 181, 182, 183, 184, 185, 186, 187, 188]) depicts various pathogen strategies for modulating ubiquitination to influence inflammasome activation.
| Pathogen type | Pathogen | Effector proteins | Ubiquitination mechanism and effect | Mechanism of action | Target pathway/Sensor |
| Bacterial pathogens | Mycobacterium tuberculosis | PtpA | Deubiquitinase activity | Removes K63-linked ubiquitin chains from NLRP3, inhibiting activation and reducing IL-1 |
NLRP3 [157] |
| Zmp1 | Interferes with ubiquitination | Interferes with K63-linked ubiquitination of pro-IL-1 |
Pro-IL-1 | ||
| Yersinia species | YopJ/YopP | Inhibits ubiquitination | Inhibits NF-κB and MAPK pathways, interfering with NLRP3 priming and ubiquitination of TRAF proteins. | NF-κB, MAPK [159] | |
| YopM | Recruits deubiquitinase USP7 | Recruits USP7 to deubiquitinate NLRP3, leading to its destabilization and degradation. | NLRP3 [160] | ||
| Shigella flexneri | IpaH proteins (e.g., IpaH7.8) | E3 ubiquitin ligase activity | Targets NLRP3 inflammasome components for degradation, enhancing inflammasome activation. | GLMN [161] | |
| Listeria monocytogenes | LLO | Modulates ubiquitination pathways | Induces K+ efflux, modulating NLRP3 activation and manipulating host ubiquitination pathways. | NLRP3 [162] | |
| InlC | Interacts with ubiquitination-related signaling | Interacts with IKK |
NF-κB [94] | ||
| Brucella abortus | TcpB | Interferes with ubiquitination-related signaling | Mimics TIRAP/Mal, interfering with TLR signaling pathways and reducing NLRP3 inflammasome activation. | TLR signaling [163] | |
| Pseudomonas aeruginosa | Various effectors | Disrupt host ubiquitination processes | Disrupt host cell signaling and ubiquitination processes, indirectly affecting NLRP3 inflammasome activation. | NLRP3 [164, 165] | |
| Legionella pneumophila | LubX | E3 ubiquitin ligase activity | Targets host kinase CLK1, affecting cellular processes including inflammasome regulation. | NLRP3 [166] | |
| Salmonella enterica | SseL | Deubiquitinase activity | Removes K63-linked ubiquitin chains from host proteins, potentially affecting NLRP3 inflammasome activation. | NLRP3 [167] | |
| Francisella tularensis | Indirect modulation of ubiquitination pathways | Indirect modulation of ubiquitination pathways | Prostaglandin E2 production inhibits NLRP3 inflammasome activation by interfering with potassium efflux. | NLRP3 [168] | |
| Rickettsia species | Sca proteins | Interacts with host ubiquitin ligases | Interact with host ubiquitin ligases, modulating cellular processes including inflammasome regulation. | NLRP3 [169] | |
| Coxiella burnetii | Dot/Icm type IV secretion system | Injects effector proteins to modulate host ubiquitination pathways | Injects effector proteins to modulate host cell processes including ubiquitination pathways. | NLRP3 [170] | |
| Streptococcus pyogenes | SpeB, Streptolysin O | Degrades ubiquitination-related host proteins | Degrades host proteins involved in inflammasome activation, modulating NLRP3 inflammasome activation. | NLRP3 [171, 172] | |
| Porphyromonas gingivalis | Gingipains | Degrades ubiquitination-related host proteins | Degrade multiple host proteins, potentially affecting NLRP3 components. | NLRP3 [173, 174] | |
| Neisseria gonorrhoeae | Opa proteins | Modulates ubiquitination-related signaling | Interact with host receptors and modulate signaling pathways. | NLRP3 [175] | |
| Borrelia burgdorferi | OspA, OspB | Potentially modulates ubiquitination-related signaling | Interact with host cells, potentially modulating inflammasome activation. | NLRP3 [176, 177] | |
| Viral Pathogens | Influenza A Virus | NS1 | Interacts with TRIM25, an E3 ubiquitin ligase | Inhibits RIG-I ubiquitination and subsequent antiviral signaling. | RIG-I [177] |
| Hepatitis C Virus | NS3/4A | Cleaves MAVS and interacts with Parkin, affecting ubiquitination pathways | Cleaves MAVS and interacts with Parkin, affecting ubiquitination pathways. | MAVS [178] | |
| Dengue Virus | NS3, NS2B3 | Cleaves ubiquitination-related proteins STING and cGAS | Cleaves STING and targets cGAS for degradation, disrupting innate immune signaling. | STING, cGAS [179] | |
| Human Papillomavirus | E6 | Recruits E6AP, an E3 ubiquitin ligase | Recruits E6AP, targeting p53 for degradation. | p53 [180] | |
| Epstein-Barr Virus | BPLF1 | Deubiquitinase activity | Deubiquitinates TRAF6 and NEMO, suppressing NF-κB signaling. | TRAF6, NEMO [181] | |
| Kaposi’s Sarcoma-associated Herpesvirus | ORF64, K5 | Viral deubiquitinase and E3 ubiquitin ligase activity | Suppresses RIG-I-mediated signaling and targets host immune proteins for degradation. | RIG-I [182] | |
| Enterovirus 71 | 3C protease | Cleaves ubiquitination-related protein TRIM25 | Cleaves TRIM25, inhibiting RIG-I-mediated antiviral signaling. | TRIM25 [183] | |
| Human | pUL26, pUL48 | Interferes with ubiquitination-related NF-κB activation and acts as a deubiquitinase | Interferes with NF-κB activation and acts as a deubiquitinase. | NF-κB [184] | |
| Cytomegalovirus | |||||
| Herpes Simplex Virus | ICP0 | E3 ubiquitin ligase activity | Targets various host proteins for degradation. | NLRP3 [185] | |
| Measles Virus | V protein | Interacts with multiple host factors affecting ubiquitination processes | Interacts with multiple host factors affecting ubiquitination processes. | NLRP3 [186] | |
| Nipah Virus | W protein | Inhibits ubiquitination-related RIG-I-like receptor signaling | Inhibits RIG-I-like receptor signaling. | RIG-I-like receptors [187] | |
| Crimean-Congo Hemorrhagic Fever Virus | L protein | Deubiquitinase activity | [188] |
PtpA, protein tyrosine phosphatase A; Zmp1, zinc metalloproteinase 1; NLRP3,
NOD-like receptor family pyrin domain containing 3; IL-1
Intracellular bacteria have evolved diverse strategies to manipulate the host
ubiquitination pathway and regulate NLRP3 inflammasome activation (Fig. 2) [189, 190]. Mycobacterium tuberculosis employs protein tyrosine phosphatase A (PtpA) as
a deubiquitinase to remove K63-linked ubiquitin chains from NLRP3, inhibiting its
activation, reducing IL-1
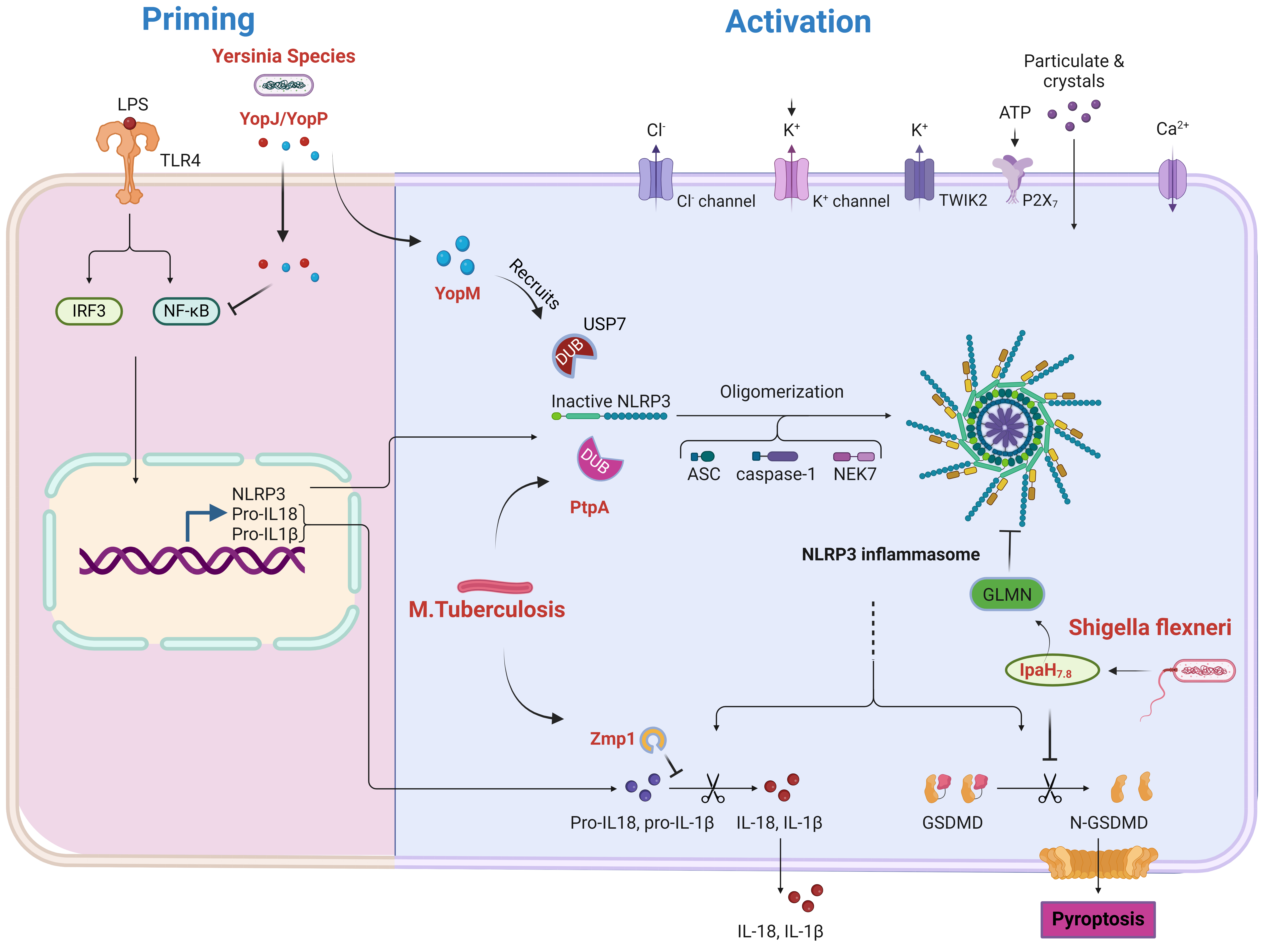 Fig. 2.
Fig. 2.
Modulation of NLRP3 inflammasome priming and activation by
bacterial pathogens. This figure depicts how bacterial pathogens influence NLRP3
inflammasome priming and activation. During priming, lipopolysaccharide (LPS) triggers TLR4, leading
to NF-
Other bacterial pathogens like Pseudomonas aeruginosa, use various effector proteins to disrupt host cell signaling and ubiquitination processes indirectly affecting NLRP3 inflammasome activation [164, 165]. Legionella pneumophila, an intracellular bacterium uses its type IV secretion system to inject effector proteins like LubX, E3 ubiquitin ligase that targets the host kinase clock homolog 1 (CLK1), affecting various cellular processes including inflammasome regulation [205]. Salmonella enterica uses different effector protein like Salmonella secreted effector L (SseL), deubiquitinase that removes K63-linked ubiquitin chains from various host proteins, potentially affecting NLRP3 inflammasome activation [206]. Francisella tularensis, a highly virulent bacterium induces prostaglandin E2 production, which inhibits NLRP3 inflammasome activation by interfering with potassium efflux [207].
Surface Cell Antigen (Sca) proteins from Rickettsia species, an obligate intracellular bacteria interact with host ubiquitin ligases, modulating cellular processes including inflammasome regulation [208]. Coxiella burnetiid, intracellular bacterium injects effector proteins via its Dot/Icm type IV secretion system to modulate host cell processes including ubiquitination pathways [209]. Streptococcus pyogenes uses Streptococcal Pyrogenic Exotoxin B (SpeB), cysteine protease that degrades host proteins, including those involved in inflammasome activation and Streptolysin O, pore-forming toxin that can modulate the NLRP3 inflammasome activation [210]. Porphyromonas gingivalis employs gingipains to degrade multiple host proteins, potentially affecting NLRP3 components [173]. Neisseria gonorrhoeae uses Opa proteins to interact with host receptors and modulate signaling pathways [211]. Borrelia burgdorferi’s, the causative agent of Lyme disease uses outer surface proteins like outer secreted protein A (OspA) and outer secreted protein B (OspB) proteins to interact with host cells, potentially modulating inflammasome activation [212].
Viruses have also developed sophisticated mechanisms to manipulate host
ubiquitination and NLRP3 inflammasome activation [213]. Influenza A Virus uses
its NS1 protein to interact with TRIM25, an E3 ubiquitin ligase, inhibiting the
ubiquitination of RIG-I and subsequent activation of antiviral signaling pathways
indirectly affecting the NLRP3 inflammasome activation [214]. Human
Papillomavirus employs its E6 oncoprotein to recruit the cellular E3 ubiquitin
ligase E6AP, targeting p53 for degradation [180]. Epstein-Barr Virus’s BamHI
fragment P leftward open reading frame 1 (BPLF1) deubiquitinates TRAF6 and
NF-kappa-B Essential Modulator (NEMO), suppressing NF-
Enterovirus 71 (EV71) virus, responsible for hand, foot, and mouth disease uses
its 3C protease to cleave TRIM25, inhibiting RIG-I-mediated antiviral signaling
[183]. Hepatitis C Virus employs non-structural proteins 3 and 4A (NS3/4A)
protease to cleave mitochondrial antiviral signaling protein (MAVS) and its core
protein interacts with Parkin. Dengue Virus uses NS3 protease to cleave
stimulator of interferon genes (STING) and Non-Structural Protein 2B and 3
(NS2B3) to target cyclic GMP-AMP synthase (cGAS) for degradation [215]. Human
Cytomegalovirus’s protein UL26 (pUL26) interferes with NF-
The NLRP3 inflammasome plays a crucial role in the innate immune response
against various parasitic infections [45, 78, 220]. Protozoan parasites such as
Plasmodium, Toxoplasma gondii, Leishmania species, and Trypanosoma cruzi, as well
as helminth parasites like Schistosoma mansoni and Heligmosomoides polygyrus
manipulate host ubiquitination pathways to regulate NLRP3 inflammasome activation
[220, 221, 222]. In Plasmodium infections, hemozoin and parasite DNA have been
identified as potent NLRP3 activators, contributing to both protective immunity
and immunopathology in malaria [223, 224]. Leishmania species use Glycoprotein
63 (GP63), a surface protease, to cleave and inactivate multiple host
proteins involved in innate immune signaling [225, 226]. Similarly, Toxoplasma
gondii employs sophisticated mechanisms to modulate host cellular processes,
including the regulation of the NLRP3 inflammasome [227, 228]. The parasite
secretes Granule Protein 16 (GRA16), a dense granule protein that
interacts with the host deubiquitinase Herpesvirus-Associated Ubiquitin-Specific
Protease/ Ubiquitin-Specific Peptidase 7 (HAUSP/USP7), potentially affecting p53
stability and indirectly influencing NLRP3 inflammasome activation [227, 229].
This interaction highlights the complex interplay between T. gondii and host
cellular pathways [224]. Concurrently, T. gondii activates the NLRP3 inflammasome
through mechanisms involving potassium efflux and reactive oxygen species (ROS)
production. The resulting NLRP3-dependent IL-1
These diverse strategies employed by various pathogens highlight the complex interplay between host defense mechanisms and pathogen evasion tactics, centered around the manipulation of the ubiquitination system and NLRP3 inflammasome activation [189].
The regulation of the NLRP3 inflammasome through ubiquitin ligases and deubiquitinating enzymes represents a sophisticated and tightly regulated system essential for maintaining immune balance [105]. This review has elucidated the complex network of both positive and negative regulators that modulate NLRP3 inflammasome activity through diverse ubiquitination and deubiquitination processes. The roles of E3 ubiquitin ligases, such as TRIM31, MARCH7, FBXL2, and Pellino2, alongside deubiquitinating enzymes like BRCC3, USP7, and A20, highlight the intricate nature of this regulatory system [105]. These enzymes collectively influence NLRP3 protein levels, stability, and activation, thereby affecting the magnitude and duration of inflammatory responses. In the context of intracerebral hemorrhage (ICH), where excessive inflammation contributes to secondary brain injury, understanding these regulatory mechanisms offers insights into potential therapeutic strategies [36]. Edaravone, for instance, may modulate the activity of specific ubiquitin ligases, potentially attenuating inflammasome-driven inflammation and providing a novel approach to managing ICH [42].
Furthermore, pathogens’ ability to exploit and manipulate the host’s ubiquitination machinery to evade immune detection adds another layer of complexity. Various bacteria, viruses, and parasites have developed advanced strategies to disrupt NLRP3 inflammasome activation by targeting key components of the ubiquitination pathway [191]. This interplay exemplifies the evolutionary battle between host defense mechanisms and pathogen evasion tactics, revolving around post-translational modifications.
Understanding these regulatory mechanisms offers valuable insights into the fundamental aspects of inflammation and presents potential therapeutic opportunities for inflammatory and infectious diseases. The delicate balance between NLRP3 inflammasome activation and inhibition, governed by ubiquitination and deubiquitination, presents both challenges and prospects for targeted interventions.
However, several questions remain for future investigation:
1. How do distinct ubiquitination patterns on NLRP3 influence its function and
activation kinetics? 2. What are the specific molecular mechanisms by which pathogens disrupt host
ubiquitination machinery to modulate NLRP3 inflammasome activation? 3. How do cell type-specific variations in ubiquitin ligase and deubiquitinase
expression impact NLRP3 inflammasome regulation across different tissues and
disease states? 4. Can targeting specific ubiquitin ligases or deubiquitinases provide effective
and selective treatments for inflammatory diseases while preserving overall
immune function? 5. How do environmental factors and cellular stress affect the activity of
ubiquitin ligases and deubiquitinases in the context of NLRP3 inflammasome
regulation? 6. What is the role of interactions between ubiquitination and other
post-translational modifications in fine-tuning NLRP3 inflammasome activation?
Addressing these questions will enhance our understanding of NLRP3 inflammasome regulation and could lead to innovative therapeutic strategies for managing inflammatory disorders and combating infectious diseases.
During the preparation of this work the author used ChatGPT in order to check spell and grammar. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.
SB was responsible for the conception of ideas presented, writing, and the entire preparation of this manuscript.
Not applicable.
The author acknowledges many investigators in the field whose primary data could not be cited in this review because of space limitations. Figures were created with BioRender.com.
This research received no external funding.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.