






1 Group for Molecular Oncology, Institute for Medical Research, National Institute of the Republic of Serbia, University of Belgrade, 11129 Belgrade, Serbia
2 Integrative Center for Biology and Applied Chemistry (CIBQA), Bernardo O’Higgins University, 8370993 Santiago, Chile
Abstract
Myeloid-derived suppressor cells (MDSCs) are believed to be key promoters of tumor development and are recognized as a hallmark of cancer cells’ ability to evade the immune system evasion. MDSC levels often increase in peripheral blood and the tumor microenvironment (TME). These cells exert immunosuppressive functions, weakening the anticancer immune surveillance system, in part by repressing T-cell immunity. Moreover, MDSCs may promote tumor progression and interact with cancer cells, increasing MDSC expansion and favoring an immunotolerant TME. This review analyzes the primary roles of MDSCs in cancer and T-cell immunity, discusses the urgent need to develop effective MDSC-targeted therapies, and highlights the potential synergistic combination of MDSC targeting with chimeric antigen receptors and immune checkpoint inhibitors.
Keywords
- MDSC
- T-cell lymphocyte immunosuppression
- tumor progression
- cancer
- myeloid-derived suppressor cells
The tumor microenvironment (TME) might favor tumor growth and metastasis. In this context, myeloid-derived suppressor cells (MDSCs) have attracted attention because they may be able to generate an immunosuppressive cancer microenvironment. Their involvement in cancer-related inflammation and immunosuppressive functions principally, but not exclusively consists of repressing T-cell immunity [1, 2]. Moreover, MDSCs are actively involved in various other aspects of tumor progression. They facilitate extracellular matrix (ECM) reorganization, induce cancer-associated epithelial-to-mesenchymal transitions (EMTs), and form pre-metastatic niches [3, 4, 5]. Cancer cells also release cytokines, chemokines, and growth factors that recruit, activate, and expand MDSCs. These cells also actively interact with lymphocytes, and other myeloid cells collaborate to create a TME that supports immune responses and disease progression [6, 7, 8, 9].
Various therapeutic strategies have been devised to intervene in MDSC activity, expansion, and migration to restore or boost the immune system’s anticancer functions. As a result, it is imperative to target the cancer-associated functions of MDSCs. This review aims to explore the complex activities of MDSCs in tumor immunology within the TME. We also analyze the current data, focusing on MDSC targeting in combination with immunotherapies. These combined therapies have the potential to substantially improve the effectiveness of current and emerging oncotherapies, providing reassurance about the progress in cancer treatment and ultimately significantly improving patients’ quality of life.
MDSCs constitute diverse precursor subsets in which the expression of surface biomarkers typically found in differentiated myeloid cells has not been observed. They possess phenotypical features similar to granulocytic and monocytic cells [5, 10, 11]. In the 1970s, it was demonstrated that tumor progression frequently leads to an abnormal increase in the number of immature myeloid cells [4]. These immature myeloid cells were initially identified in mice xenograft models and then in patients with head and neck squamous cell cancer (HNSC) [12]. The name “myeloid-derived suppressor cells” was postulated by Gabrilovich et al. in 2007 [13]. These cells are a significant feature of cancer development and a prominent way for cancer cells to avoid being detected by the immune system, as they exert immunosuppressive effects [12, 14].
MDSCs exhibit a baseline suppressive activity, and they can also migrate to other organs and differentiate into mature immune cells to support normal immune functions [15]. However, under pathological conditions such as infection, tissue damage, or cancer, emergency myelopoiesis is triggered in response to inflammatory factors. These events might trigger MDSC proliferation and increase their immunosuppressive abilities, serving as a defensive cellular process intended to restrain excessive tissue injury resulting from unresolved immune reactions [16, 17, 18].
MDSCs are categorized into the following subsets: polymorphonuclear MDSCs (PMN-MDSCs) (formerly known as granulocytic MDSCs), which are characterized as CD11b+, CD33+, CD15+, CD66b+, or CD14–, and monocytic MDSCs (M-MDSCs), which are classified as CD11b+, CD14+, CD15–, or human leukocyte antigen – DR isotype (HLA-DR)-/low. There are also early-stage MDSCs (eMDSCs), comprising very immature progenitors, classified as HLA-DR–, CD33+, CD14–, or CD15– [10, 19]. In contrast, mouse MDSC counterparts are classified as granulocyte antigen (Gr)-1+ (which shares a common epitope with lymphocyte antigen (Ly) 6C and Ly6G) or CD11b+. These cells can be classified as PMN-MDSCs, characterized as CD11b+, Ly6G+, Ly6Clow, and M-MDSCs, and identified as CD11b+, Ly6G–, and Ly6Chi [19] (Fig. 1).
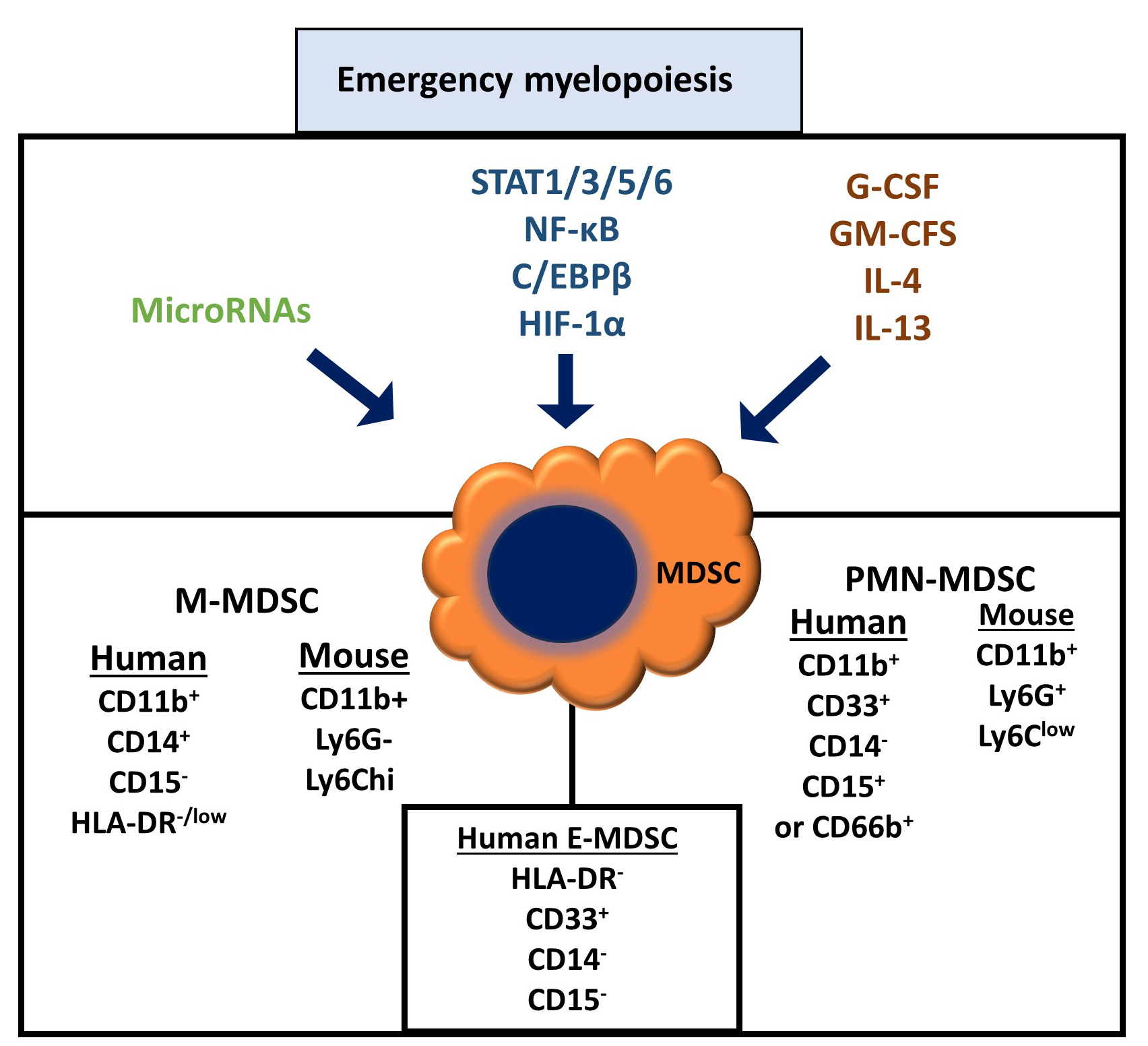 Fig. 1.
Fig. 1.
Main features of myeloid-derived suppressor cell generation.
Under pathological inflammation, an emergency myelopoiesis is initiated that
generates several molecules converging in generating myeloid-derived suppressor
cells (MDSCs), such as inflammatory cytokines and microRNAs, and the contribution
of transcription factors necessary to display MDSC characteristics. As the figure
indicates, these MDSCs expressed a series of cell surface proteins, allowing them
to differentiate their immunophenotype from mature macrophages and neutrophils.
For more details, see the text. STAT, signal transducer and activator of
transcription; NF-
MDSCs’ increased frequency and functionality are dependent on various biomolecules such as cytokines, growth factors, signal transduction pathways, transcription factors, and microRNAs (miRs) [1] (Fig. 1). For example, the induction of inducible nitric oxide synthetase (iNOS) and arginase-1 (ARG1) through signal transducer and activator of transcription (STAT)1 may positively affect MDSC function [20], whereas STAT3 promotes reactive oxygen species (ROS) in MDSCs [21].
Granulocyte colony-stimulating factor (G-CSF) and granulocyte–monocyte
colony-stimulating factor (GM-CSF) induce the downregulation of interferon
regulatory factor (IRF)-8 through STAT3 and STAT5 pathways in MDSCs, whereas
IRF-8-deficient mice generate MDSCs. Notably, advanced M-MDSCs obtained from
human malignant melanoma express increased levels of STAT3, and inhibition of the
latter significantly reduces MDSC-mediated immunosuppression [22]. Conversely,
mice with IRF-8 overexpression exhibit reduced MDSC accumulation [23].
Furthermore, interleukin (IL)-4 and IL-13 upregulate ARG1 expression and
transforming growth factor (TGF)-
The activation of nuclear factor (NF)-
Furthermore, hypoxia-inducible factor (HIF)-1
In addition, when mouse myeloid cells harbor an Src homology 2 (SH2) domain-containing inositol-5-phosphatase 1 (SHIP1) gene deletion, the frequency of MDSCs increases. This increase is partially due to elevated levels of G-CSF. Additionally, in vivo, splenocytes exhibit reduced SHIP-1 expression, associated with MDSC growth and immunosuppression activity, thus facilitating pancreatic cancer progression [29, 30].
Cancer involves a complex interplay between several cell types, including neoplastic cells, fibroblasts, mesenchymal cells, and innate and adaptive immune cells. This interaction significantly affects tumor growth, progression, and metastasis [31, 32, 33]. MDSCs are present in high numbers within the TME and promote cancer development by enhancing cancer cell expansion, neoangiogenesis, and tumor malignancy. Moreover, MDSCs support cancer development by decreasing immunosurveillance, facilitating TME rearrangement, and promoting pre-metastatic niche generation [3, 34, 35].
MDSCs may support the development of primary tumors through immune and
non-immune processes. One non-immunological process by which MDSCs support tumor
progression induces cancer cells to undergo a cancer-associated EMT [36]. This
cellular program increases tumor progression and cancer cell aggressivity.
Soluble factors secreted by MDSCs, including TGF-
Furthermore, it has been shown that, in vivo, the decrease in PMN-MDSCs frequency is related to an EMT reduction within tumors [37]. In addition, M-MDSCs derived from pancreatic tumors can trigger the expansion of cancer stem cells expressing aldehyde dehydrogenase 1 (ALDH1) [38]. MDSCs also enhance NANOG and cellular myelocytoma protein (c-MYC) (stemness biomarkers) in epithelial ovarian cancer (EOC) cells in vitro. Furthermore, these cells can induce the increased production of colony-stimulating factor 2 (CSF2) via EOC cells, which can further stimulate cancer cell stemness by activating STAT3 signaling in an autocrine manner [39].
It is worth noting that MDSCs significantly promote cell migration and invasion by modifying the ECM, matrix stiffness, and basement membrane [40]. Specifically, the ECM is primarily affected by proteinases produced and secreted by transformed malignant cells’ MDSCs, such as matrix metalloproteinases (MMPs). In particular, MDSCs express MMP2, MMP8, MMP9, MMP13, and MMP14, contributing to ECM degradation and enhancing cancer cell motility, invasiveness, and metastasis [41].
Furthermore, PMN-MDSCs enhance melanoma and liver cancer cell migration through the endothelial layer by interacting with integrin (MAC-1)- ntercellular adhesion molecule (ICAM)-1 expressed in endothelial cells. In addition, MDSCs can transdifferentiate into VEGFR2+ endothelial cells, contributing to tumor endothelial development [42]. The dysfunctional tumor vasculature formed from this process can facilitate tumor cell intravasation and extravasation [35]. Neutrophils and inflammatory monocytes also promote the extravasation of cancer cells from blood vessels [35, 43].
MDSCs interact with and regulate immune system functions; the ability to inhibit T-cells is an essential hallmark of MDSCs [44, 45] (Fig. 2).
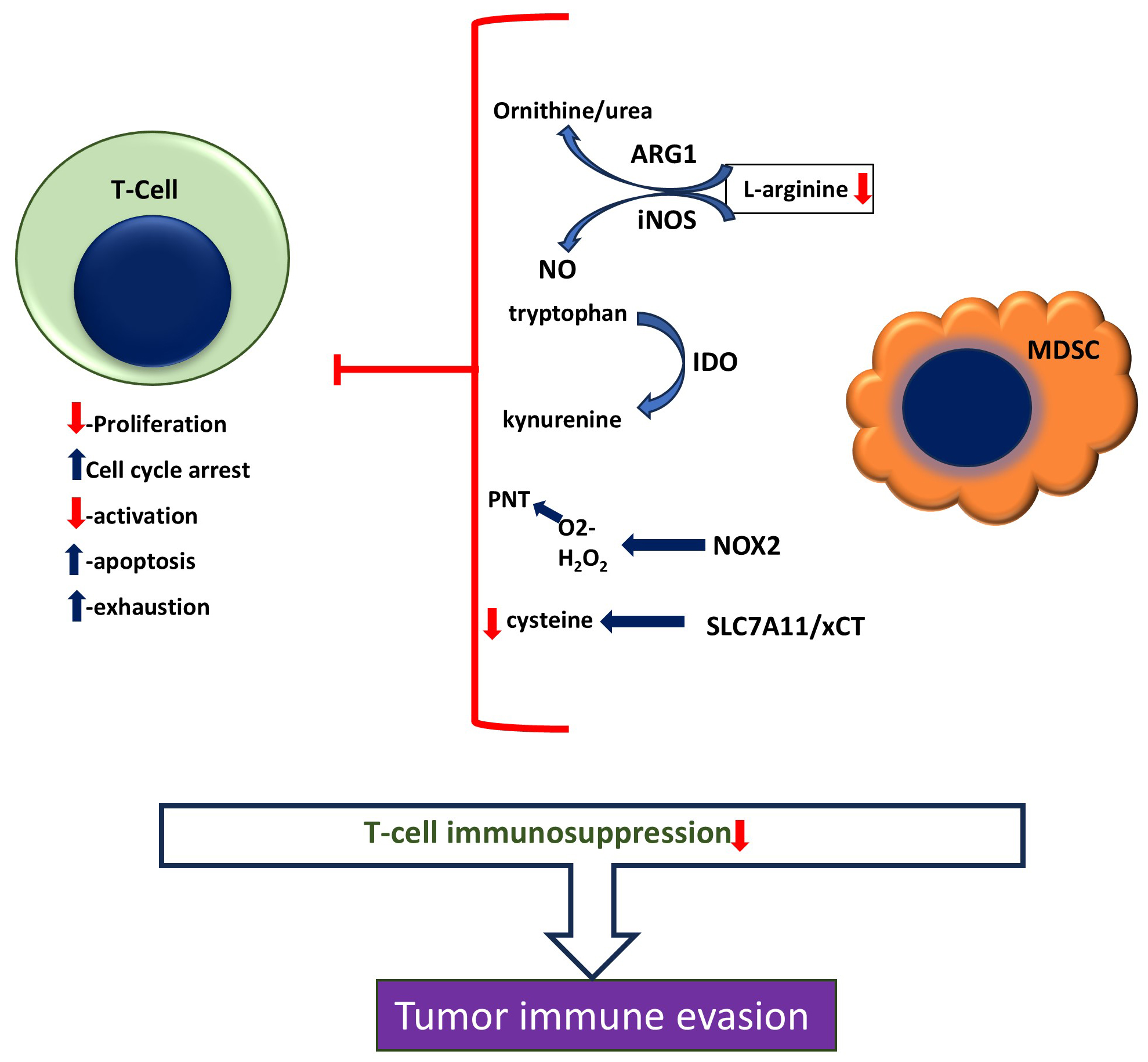 Fig. 2.
Fig. 2.
MDSC’s Immunosuppressive mechanisms. MDSCs potently suppress T-cells’ function and activity by expressing and releasing inhibitory proteins and molecules. ARG1, arginase-1; iNOS, inducible nitric oxide synthase; NO, nitric oxide; IDO, indoleamine 2,3-dioxygenase; PNT, Peroxynitrite; NOX2, NADPH oxidase 2; SLC7A11/xCT, cystine/glutamate transporter.
MDSCs exert T-cell immunosuppressive functions via a plethora of cellular and
molecular mechanisms that include oxidative stress, cell metabolism,
proliferation inhibition, apoptosis induction, and the inhibition of cell
migration. Specifically, the L-arginine (Arg) catabolism can impact T-cell
growth. When L-arginine is converted to ornithine and urea by ARG1 or used as a
precursor for nitric oxide (NO) production by iNOS, the amount of Arg in the
surrounding environment decreases. This depletion of Arg causes T-cell
proliferation to be inhibited by cell cycle arrest at G0/G1 by
inhibiting the expression of CD3
Moreover, STAT3 signaling promotes NADPH oxidase 2 (NOX2) expression in MDSCs,
which further increases levels of ROS, such as superoxide anions (O2–)
and hydrogen peroxide (H2O2) [21]. These ROS can combine with the free
radical NO– to generate reactive nitrogen species such as peroxynitrite
(PNT, ONOO–). PNT causes T-cell cell death via the tyrosine nitration of the
T-cell receptor (TCR), leading to the inhibition of antigen peptide–major histocompatibility complex (MHC) dimers
bound with CD8+ T-cells due to changes in the TCR-CD8 complex, hindering
antigen-specific stimulation [49]. On the other hand, H2O2 produced by
MDSCs inhibits TCR
Furthermore, MDSCs contribute to T-cell immunosuppression through indoleamine 2,3-dioxygenase (IDO) expression. IDO is a crucial cytosolic enzyme that controls the degradation of tryptophan (Trp) through the kynurenine pathway, and the degradation of Trp generates kynurenine, kynurenic acid, and 3-hydroxykynurenine. Additionally, Trp reduction inhibits the mechanistic target of rapamycin complex 1 (mTORC1), inhibits T-cell cell cycle progress, and induces cell death. Kynurenine is toxic to lymphocytes, provoking the exhaustion of CD4+ T-cells [51, 52, 53]. Meanwhile, MDSCs sequester and reduce cysteine levels through the cystine/glutamate transporter (SLC7A11/xCT) [54], thus decreasing cysteine availability. Therefore, the suppression of T-cell activation by MDSCs promotes apoptosis by reducing the availability of intratumoral cysteine (Cys) levels; this is because T-cells cannot produce Cys independently and rely on the Cys provided by dendritic cells. Moreover, Baumann et al. [55] showed that MDSCs produce and release high levels of the toxic metabolite methylglyoxal, which can induce T-cell paralysis and inhibit their function through direct cell-to-cell transfer.
MDSCs also impede T-cells’ motility, limiting their ability to infiltrate inflammation sites. MMP17, expressed by MDSCs, cleaves the cell adhesion molecule L-selectin/CD62L ectodomain, decreasing T-cell-associated L-selectin. Likewise, MDSCs can reduce the expression of T-cell-associated L-selectin [26, 56, 57]. Consequently, the number of T-cells homing to peripheral lymph nodes and tumors is reduced.
Finally, non-small-cell lung cancer (NSCLC)-derived MDSCs produce TGF-
Additionally, activated T-cells can trigger the death of MDSCs due to a reciprocal inhibitory relationship. This occurs because activated T-cells generate and release the Fas ligand, which binds to the Fas death receptor on MDSCs, thereby regulating the frequency of circulating MDSCs [60].
Over the years, many research labs have focused on uncovering the critical cellular and molecular processes implicated in MDSCs’ function in cancer. This research has led to progress in basic and clinical approaches to targeting MDSCs’ growth and activities, with the aim of reducing tumor development and progression. MDSCs contribute to numerous events in cancer progression. While their immunosuppressive function is a significant factor in tumor progression, they also play a role in cancer cell stemness, local invasion, angiogenesis, and cancer EMT. Therefore, MDSCs are an essential target for antitumor therapeutic strategies. Numerous research efforts have focused on impeding their expansion, reducing their frequency, and revoking MDSCs’ immune-suppressing capabilities [61, 62].
Existing approaches that target MDSCs have four main strategies: (1) Inhibiting
their immunosuppressive function. This is achieved by blocking key transcription
factors, such as STAT3, or inhibiting critical immunosuppressive enzymes, such as
cyclooxygenase-2, ARG1, and phosphodiesterase-5 [63, 64, 65]. (2) Reducing MDSCs’
frequency within the TME and in circulation, which is accomplished using
chemotherapeutic agents, including gemcitabine, paclitaxel, and 5-fluorouracil
(5-FU) [66, 67, 68]. However, some small chemical molecules reduce MDSC levels in some
cancers. For instance, the tyrosine kinase inhibitor (TKI) sunitinib
significantly reduces MDSCs in renal cell carcinoma patients. At the same time,
in NSCLC, the liver-X nuclear receptor (LXR) agonists GW3965 and RGX-104 reduce
MDSC viability through the increased secretion of Apolipoprotein E and through
binding to MDSC-associated low-density lipoprotein receptor-related protein 8
[69, 70]. Moreover, the monoclonal antibody gemtuzumab ozogamicin, which targets
CD33, reduces the frequency of MDSCs in acute myeloid leukemia (AML) patients
[71]. (3) Inhibition of MDSC migration and recruitment to the TME. This is achieved, for example, by neutralizing chemotactic cytokine receptors (CCR)5 ligands via a fusion protein mCCR5-Ig, which can reduce the migration of MDSCs toward tumors [72]. (4) Inducing MDSCs towards
mature myeloid cells lacking immunosuppressive activity within the TME. This
approach has been successfully demonstrated using all-trans retinoic acid (ATRA).
In tumor-bearing mice models, ATRA induces differentiation, reduces the increase
in MDSCs in BALB/C mice bearing cervical tumors, and increases the intratumoral
frequency of CD8+ T-cells [73]. Similarly, 1
Increasing evidence indicates a link between the frequency of MDSCs and cancer patients’ outcomes. This suggests that MDSCs could be valuable biomarkers for tumor development [76]. Several cancer types, including lung, colon, uterus, cervix, bladder, and thyroid gland cancers, and advanced non-small-cell lung cancer (NSCLC), have been linked to elevated levels of circulating MDSCs. Furthermore, MDSCs’ elevated frequency is inversely correlated with clinical responses and associated with reduced overall survival and tumor recurrence [77, 78, 79, 80].
Targeting MDSCs is an emerging and promising approach to enhancing immune cells’ anticancer functions and further ameliorating the effectiveness of cancer immunotherapy. Cancer immunotherapy is a rapidly growing area in cancer treatment and is considered the fourth type of antitumor therapy, following surgery, radiotherapy, and chemotherapy. This innovative treatment involves regulating immunity to activate its natural defenses against cancer or using biological compounds to prevent and suppress tumor growth [81]. However, excessive MDSC depletion or inhibition of their immunosuppressive functions can have significant side effects. This suggests that carefully managing the levels of regulatory cells can be critical to enhancing the effectiveness of cancer immunotherapy [82].
An immunosuppressive tumor stroma is a significant obstacle to the effectiveness of chimeric antigen receptor T-cells (CAR-T-cells). Several studies support this statement, suggesting that an immunosuppressive TME can impede CAR-T-cell immunotherapies (Table 1, Ref. [71, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93]) [94, 95]. Patients with a higher myeloid cell frequency tend to exhibit worse results in CAR-T-cell therapy. This highlights the need to reduce the quantity of these cells or their inhibitory role to enhance CAR-T-cells’ effectiveness against tumors (Fig. 3). Therefore, MDSCs inhibit CAR-T-cells’ gene transfer, activation, killing activities, and cytokine expression. A promising solution to this problem is the proposed combination of CAR-T-cell therapy with ATRA to reduce MDSC numbers. In this sense, the co-administration of ATRA with CAR-T-cells engineered to express a first-generation specific disialoganglioside (GD2) led to improved antitumoral activities in a sarcoma xenograft model in vivo [83]. This approach, along with the use of anti-CD33 gemtuzumab ozogamicin, has demonstrated potential in targeting MDSCs and enhancing the activity of anti-GD2-/mesothelin-/EGFRvIII-CAR-T-cells [71]. Moreover, in neuroblastoma patients, the success of a third-generation GD2-specific CART strategy is inversely correlated with the levels of peripheral blood PMN-MDSCs [96].
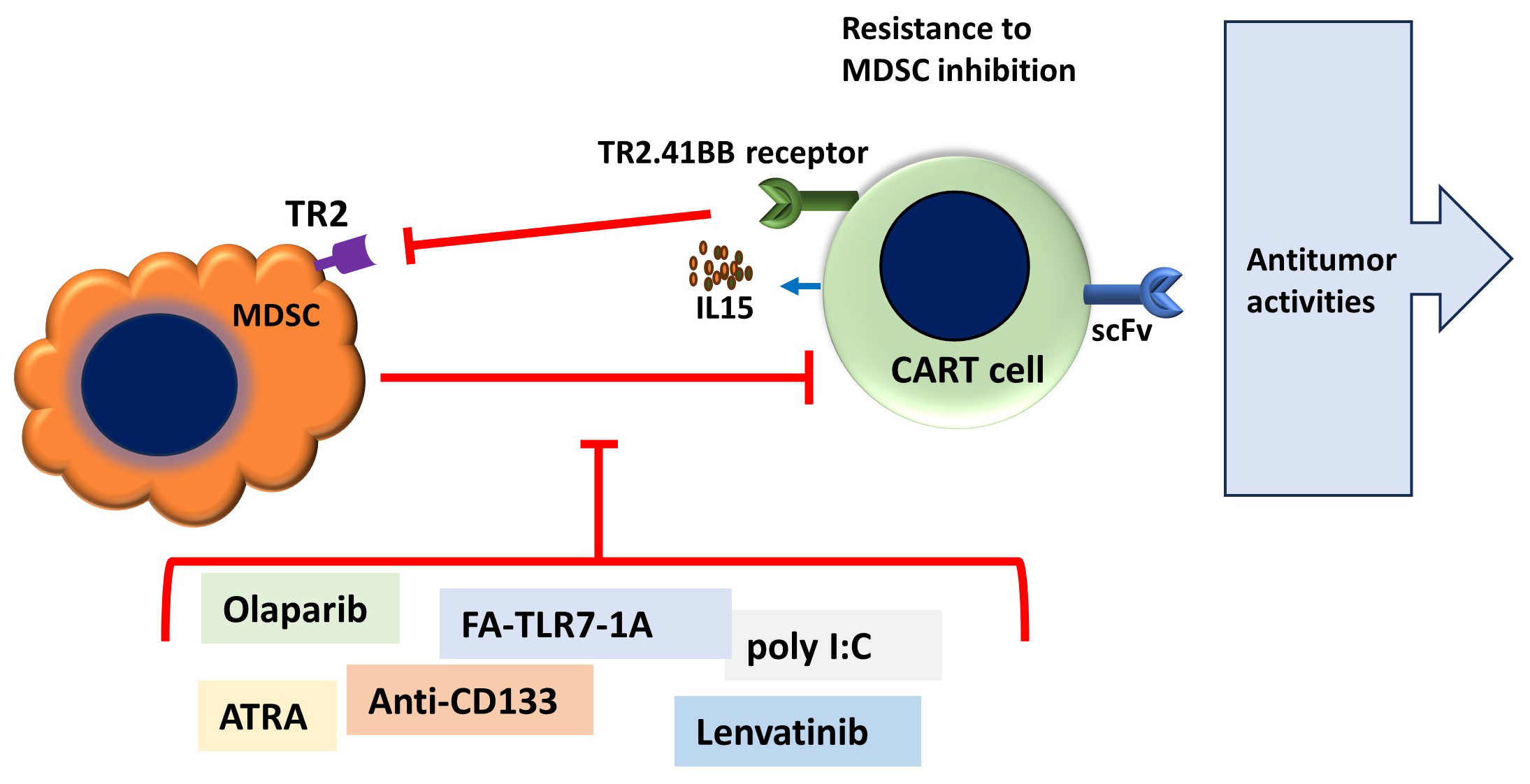 Fig. 3.
Fig. 3.
Main features of combined adoptive therapy using chimeric antigen receptors and targeting of myeloid-derived suppressor cells. MDSC strongly reduces the effectiveness of chimeric antigen receptors T-cells (CART) immunotherapies, which may be overturned by combined therapy to reduce MDSC immunosuppression or reduce MDSC frequency, as well as double genetic CART manipulation to target cancer cells and MDSC simultaneously, such as IL15 secretion and TR2 agonist antibody DS-8273a co-expression. ScFv, single chain variable fragment; TR2, TNF-related apoptosis-inducing ligand receptor 2; ATRA, all-trans retinoic acid; Poly I:C, Polyinosinic-polycytidylic acid; FA-TLR7-1A, a combined compound of Toll-like receptor (TLR)7 agonist to folic acid.
| Chimeric antigen receptors T-cells (CART) type | Combined therapy | Cancer type, model | Outcomes | Ref. |
| First-generation disialoganglioside (GD2)-specific CAR T-cells | All-trans retinoic acid (ATRA) | Sarcoma xenografts model in vivo | MDSC depletion; improvement of antitumoral activities | [83] |
| Anti-GD2-/mesothelin-/EGFRvIII-CAR-T-cells | Gemtuzumab ozogamicin (Anti-CD33) | Colon and pancreatic carcinoma, Neuroblastoma, Mesothelioma in vitro | MDSC reduction; restores T-cell immunity | [71] |
| Murine EGFRvIII specific CAR-T cells | Polyinosinic-polycytidylic acid (poly I:C), MDSC depletion with anti-Gr1 | Orthotopic breast tumors mouse model | Improves CAR T-cells Antitumor Effects | [84] |
| EGFRvIII-targeted CAR (806-28Z CAR) T-cells | Olaparib, a poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor | Immunocompetent mouse models of breast cancer | MDSC migration inhibition; CAR T-cell antitumor activityimprovement | [85] |
| CAR T-cells with null mutations of CD28 subdomains | none | B-ALL mouse model, spontaneous lymphoid tumor cell line (Eµ-ALL cells)/C57BL/6 mice | Resistance to MDSC-mediated immunosuppression; increased CAR T-cell antigen sensitivity and survival | [86, 87] |
| CAR.MUC1.TR2.41BB T and CAR.HER2.TR2.41BB T cells | none | Cell-derived xenograft (CDX) mouse model | MDSC elimination; enhancing CAR-T in vivo expansion and antitumor efficacy | [88] |
| Second-generation CAR targeting human carbonic anhydrase IX (CAIX-CAR) | Multi-kinase inhibitor Lenvatinib | Murine renal cancer model | Decrease in MDSCs frequency; T-cell immunity recovery | [89] |
| CD19-CAR T-cells | Folic acid and TLR7 agonist (FA-TLR7-1A) MDSC reprogramming into proinflammatory M1 macrophages | CD19-expressing 4T1 tumors in immune-competent mice model | MDSC reduced levels; enhances CAR T-cell activation and tumor infiltration | [90] |
| IL13R |
none | Human and syngeneic murine models of glioblastoma | MDSC depletion; mice cancer models survival improvement | [91] |
| Dual-targeted CAR-T (123NL CAR-T) cell targeting CD123 and NKG2DL | none | Acute myeloid leukemia | Amelioration of the immunosuppressive environment by simultaneously acute myeloid leukemia (AML) and MDSC targeting | [92] |
| Murine epithelial cell adhesion molecule (EpCAM)-targeting third-generation CAR (EpCAM28.BBz) | Hypofractionated radiotherapy and Cysteine-X-cysteine motif chemokine receptor 2 (CXCR2) blockade | Immunocompetent mice bearing triple-negative breast cancer | Inhibition of MDSC tumor microenvironment (TME) infiltration; enhancement of CAR T-cells trafficking into TME and antitumor efficacy | [93] |
Polyinosinic-polycytidylic acid (poly I:C), a compound that binds to Toll-like receptor (TLR) 3, significantly enhances CAR-T-cells’ antigen-specific responses and their antitumor activity in vivo. Poly I:C, combined with anti-Gr1 antibodies to reduce MDSC numbers, has remarkably improved CAR-T-cell therapy efficacy in vivo [84]. In addition, poly I:C has been found to reduce MDSC frequency in the peripheral blood and spleen, ameliorating CAR-T-cell proliferation and cytolytic functions. These findings regarding poly I:C provide new therapeutical options for future strategies based on CAR-T-cells.
Olaparib, a poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, suppresses MDSC
migration, promoting CD8+ T-cell viability in mouse models of breast cancer.
Furthermore, olaparib has been shown to significantly increase the antitumor
efficacy of EGFRvIII-targeted CAR (806-28Z CAR) T-cell therapy in vivo.
Mechanistically, olaparib reduces cancer-associated fibroblast stromal
cell-derived factor (SDF)1
A novel chimeric costimulatory receptor (TR2.41BB), which encodes the single-chain variable fragment (scFv) of the TNF-related apoptosis-inducing ligand receptor 2 (TR2) agonist antibody DS-8273a, followed by a 41BB endodomain, was developed to target TME-associated MDSCs. MDSC-associated TR2 induces MDSC death via the induction of apoptosis through binding with the soluble tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [88, 97]. Moreover, the co-expression of the TR2.41BB receptor in targeted CAR-T-cells enhances their toxicity against orthotopic tumors in breast cancer models. Additionally, targeting MDSCs through TRAIL receptor 2 (TR2) enhances the efficacy of CART therapy in breast cancer models [88, 98].
The potential of combining Lenvatinib, a tyrosine kinase inhibitor, with a second-generation CAR targeting human carbonic anhydrase IX (CAIX-CAR) is a promising avenue in cancer treatment. This CAR comprises a mouse anti-human CAIX scFv, a mouse 4–1BB costimulatory domain, and a CD3 zeta intracellular activation domain. By reducing the number of MDSCs, Lenvatinib significantly improves T-cell anticancer activities, and combining it with these CART cells significantly inhibits tumorigenesis and improves mice survival in ovarian cancer models [89].
Luo et al. [90] detailed one interesting approach to reducing the immunosuppressive milieu within the TME using co-administration of FA-Toll-like receptor (TLR)7-1A using a murine CD19-expressing 4T1 cell clone as a model. This receptor is a combined compound of a TLR7 agonist (TLR7a) to folic acid and can reprogram MDSCs into a proinflammatory M1 macrophage phenotype [99]. CD19-CAR-T-cells effectively improved tumor eradication in vivo without evidence of toxicity. Moreover, the authors claim that MDSC polarization, via FA-TLR7-1A, towards an inflammatory phenotype may increase lymphocyte recruitment and activation within the TME, thus facilitating CAR-T-cells’ infiltration into the tumor mass [90, 99].
Recently, CAR-T-cells expressing IL-15 were shown to provoke MDSC depletion and
release of immunosuppressive molecules in vitro. Specifically, the
authors of that study aimed to target MDSCs in both humans and syngeneic murine
models of glioblastoma (GBM). Murine T-cells expressing IL13R
Furthermore, dual-targeted CAR-T (123NL CAR-T) cells targeting CD123 and natural killer group 2D (NKG2DL), both expressed in acute myeloid leukemia (AML) and MDSC cells, demonstrated efficacy in eradicating AML cells and improving selectivity concerning immunosuppressive cells. One study highlighted that dual-target CAR-T-cells are more effective than single-target CAR-T-cells at eliminating neoplastic cells and ameliorating the immunosuppressive environment via simultaneous MDSC targeting. In addition, 123NL CAR-T constructs harboring a highly compact marker/suicide gene, RQR8, which binds targeting epitopes of CD34 and CD20 antigens and lends CAR-T-cells sensitivity to rituximab to eliminate 123NL CAR-T-cells when not needed, working as a switch-off system [92].
A reduced TME-infiltrating capacity affects the efficacy of CAR-T therapy in solid tumors [100]. Although hypofractionated radiotherapy (HFRT) can improve leukocyte tumor infiltration abilities by reshaping the tumor stroma, it also induces MDSC expansion and activity within the TME, highlighting the need to block MDSCs when CAR-T therapies are combined with HFRT [101, 102]. This inhibits these immunosuppressive cells’ intratumoral infiltration via the CXCR2 blockade, significantly enhancing the trafficking towards tumors and the effectiveness of CAR-T-cell therapy [93].
MDSCs may inhibit T-cell activation and cell death by interacting with immune
checkpoint proteins through cell-to-cell communication. For instance, programmed
death (PD)-1/PD-ligan(L)1 interaction is crucial in MDSC-mediated
immunosuppression. Specifically, interferon-gamma (IFN-
Several approaches targeting MDSCs in combination with ICIs have been demonstrated to improve immunotherapy (Table 2, Ref. [110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123]). For instance, immunotherapies using ICIs have been successful in numerous patients by blocking the interaction between immune checkpoints, including PD-1/PD-L1 and CTLA-4, and promoting T-cell activity [124] (Fig. 4). Several investigations have demonstrated the critical functions of immune checkpoint proteins in the immunosuppression induced by MDSCs. Indeed, T-cells become anergic and undergo cell death when MDSC-associated PD-L1 binds to PD-1 on their cell surface.
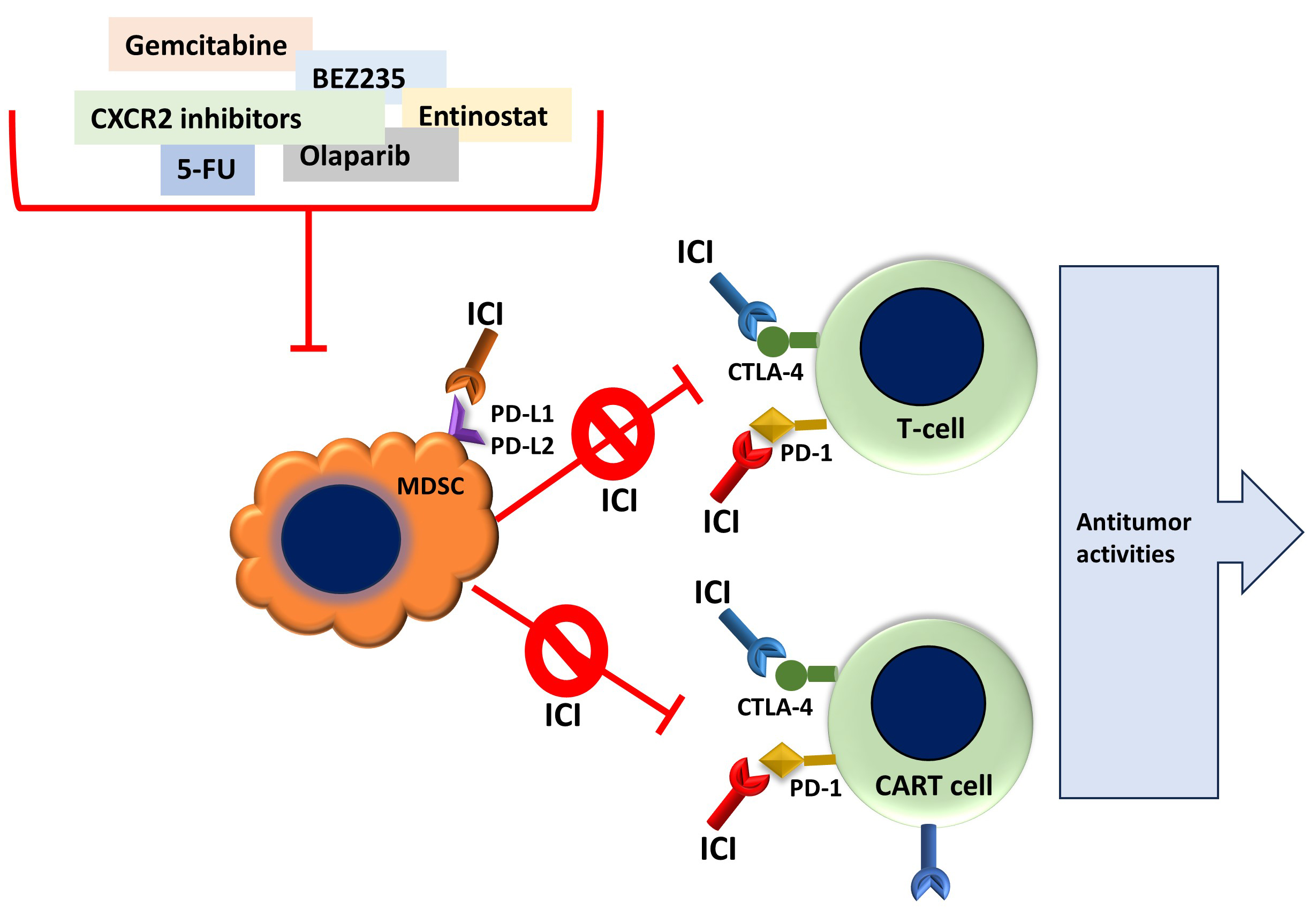 Fig. 4.
Fig. 4.
Main features of combined Immune Checkpoint Inhibitors therapy and the targeting of MDSCs. MDSCs may exert cancer immunosuppression by interacting with T-cells via binding immune checkpoint proteins, such as PD-L1 and PD-L2. Therefore, combining several anticancer agents with immune checkpoint inhibitors (ICI) may subvert MDSCs immunosuppression, ameliorate T-cell anti-cancer immunity, and improve CART immunotherapy. PD-L1/PD-L2, programmed death Ligand-1/-2; PD-1, programmed death receptor-1; CTLA-4, cytotoxic T-lymphocyte antigen 4; 5-FU, 5-fluorouracil; CXCR2, chemokine receptor 2.
| Immune checkpoint inhibitor | Combined therapy | Cancer type, models | Outcomes | Ref. |
| Anti-programmed death (PD)-1-nivolumab, cytotoxic T-lymphocyte-associated protein (CTLA)-4 with ipilimumab | none | Advanced melanoma | MDSC frequency reduction; sustained long-term overall survival at five years was observed in patients receiving nivolumab and ipilimumab or only nivolumab | [110, 111] |
| Anti-PD-1 | Chemotactic cytokine receptor (CCR)2 inhibitor, ChemoCentryx (CCX)872, to reduce tumor trafficking | Mouse glioma model | MDSC TME trafficking and number reduction; increased anti-PD-1 effectiveness and significantly increased overall mouse survival | [112] |
| Anti-PD1 and anti-CTLA4 antibodies | Multikinase inhibitors cabozantinib and BEZ235 | Prostate cancer mouse model | Reduction in the TME frequency and immunosuppression; reduced resistance to immune checkpoint inhibitors (ICIs) | [113] |
| Anti-PD-1, anti-CTLA-4 | Class I histone deacetylase, entinostat | Murine breast cancer, and Lung and renal cell carcinoma | Reduced MDSC TME trafficking and reprogramming to macrophages; increased sensitivity to ICIs therapies by tumor growth inhibition and promoting animal survival | [114, 115] |
| Anti-PD-L1 | Gemcitabine, checkpoint kinase 1 inhibitor SRA737 | Mesothelioma, small-cell lung cancer | Reduced TME MDSC number; enhances antitumor activities, significantly prolongs mouse survival, and overcomes the resistance to ICI in patients | [116, 117] |
| Anti-PD-L1 | 5-fluoro-uracil | Renal cell carcinoma xenograft tumor-bearing mice | Increased CD8+ T cells and MDSC ratio; better animal survival time and survival improvement | [118] |
| Anti-PD-1 | PARP-1 inhibitor olaparib | Syngeneic colon cancer animal models | Reduced MDSC suppressive function; tumor reduction and improved anti-PD-1 sensitivity | [119] |
| Anti-PD-1 | Triggering receptor expressed on myeloid cell 1 (TREM1) inhibitor VJDT | Syngeneic murine melanoma (B16F10) and fibrosarcoma (MCA205)/normal and Trem1deficient mice model | MDSC reduction; ameliorate immunosuppressive TME and improve efficacy of anti-PD-1 immunotherapy | [120] |
| Anti-PD-L1 | Bruton’s tyrosine kinase (BTK) ibrutinib | Mouse models of breast cancer and melanoma | Reduced MDSCs’ frequency; enhancement of anti-PD-L1 effectiveness | [121, 122] |
| Anti-PD-1 | none | Orthotopic esophageal cancer and G-protein coupled receptor (GPR)84−/− mice mouse models | MDSC decreased TME frequency; GPR84 inhibition ameliorates CD8+ T cell dysfunction and improves anti-PD-1 therapy | [123] |
In advanced melanoma, patients responding to nivolumab, an anti-PD-1 antibody
treatment, showed decreased M-MDSC levels, while non-responders did not
experience any changes [NCT01844505]. In vitro experiments revealed that
CD8+ T-cells actively secrete IFN-
In a liver cancer model, the increase in MDSCs within the TME was found to be reliant on GM-CSF derived from the tumor. Moreover, GM-CSF, via STAT3, promotes PD-L1 expression, thus enhancing MDSC–PD-1 bonds on CAR-T-cells and, as a result, suppressing their activity. At the same time, the PD-L1 blockade restores CAR-T-cell function and effectiveness in this model. Thus, combining CAR-T-cell and ICI therapy to neutralize MDSC-mediated immunosuppression has emerged as a promising clinical approach [126, 127].
PMN-MDSCs’ infiltration into the TME depends on chemokine (C-X-C motif) ligands/CXCR2. CXCR2+ PMN-MDSCs upregulate the expression of T-cell immune checkpoints, such as CTLA-4 and LAG3, leading to T-cell anergy. Preclinical investigations have indicated that targeting CXCR2 could reduce the immunosuppressive activity of MDSCs, consequently enhancing the effectiveness of anti-PD-1 therapy in rhabdomyosarcoma, pancreatic ductal adenocarcinoma, and colorectal cancer [128, 129, 130]. Recently, it has been reported that CD300ld, a single-pass transmembrane protein that belongs to the CD300 family, which is mainly expressed in myeloid cells, is upregulated in PMN-MDSCs, facilitates PMN-MDSC infiltration within the TME, and improves their immunosuppressive functions. In this sense, CD300ld knockout leads to reduced tumor development in combination with anti-PD1 therapy [131, 132]. Moreover, CCR2 and CCR5 are critical in recruiting M-MDSCs to the TME, with MDSCs-CCR5+ demonstrating better immunosuppressive properties within tumor sites. Cells lacking CCR2 or the use of ChemoCentryx (CCX)872 have been shown to improve the effectiveness of anti-PD-1 therapy in a mouse glioma model. CCX872 works by inhibiting MDSC trafficking, provoking MDSC accumulation in the bone marrow, and reducing their frequency within the TME [112].
Furthermore, the phosphoinositide 3-kinase (PI3K)
Moreover, entinostat, an oral class I histone deacetylase, ameliorates ICIs’ responses in different tumor models with immunosuppressive TMEs. Combined with the methyltransferase inhibitor 5-azacytidine, it reduces MDSC trafficking and reprograms MDSCs towards an interstitial macrophage-like phenotype. Entinostat improves sensitivity to ICIs by significantly alleviating MDSC-induced immunosuppression in murine breast cancer, pancreatic tumor models, and lung and renal cell carcinoma [114, 115, 135].
Overcoming resistance to ICIs in mesothelioma patients, gemcitabine combined with the oral checkpoint kinase 1 (CHK1) inhibitor SRA737 enhances
anti-PD-L1 effectiveness in small-cell lung cancer. Gemcitabine-improved ICI
treatment is associated with a decreased immunosuppressive TME due to a reduction
in MDSCs and Tregs levels. At the same time, it increases the M1/M2 macrophage
ratio [116, 117, 136]. Furthermore, the pyrimidine analog 5-FU, in combination with
ICIs against PD-L1, produces a better survival time and survival improvement in
renal cell carcinoma xenograft tumor-bearing mice; these effects, along with
increased IL-2, IFN-
Anti-PARP-1 metronomic therapy is a prolonged administration of low doses of chemotherapy that can sustain active levels of drugs in plasma to increase drug tolerability [137]. When combined with olaparib in colon cancer, this therapy enhances the efficacy of anti-PD-1 therapy. PARP-1 inhibition reduces the suppressive function of MDSCs without influencing intratumoral migration or differentiation, which increases tumor-associated T-cell function. Combining anti-PD-1 immunotherapy with metronomic PARP-1 inhibition reduces MDSC activity, ameliorates antitumor activities, and enhances the survival of animals in colon cancer models [119].
Targeting the triggering receptor expressed on myeloid cell 1 (TREM1), a cell surface receptor member of the immunoglobulin superfamily that is categorized as an amplifier of myeloid cell immune responses [138], demonstrated efficacy in reducing MDSC immunosuppression and expansion in cytotoxic CD8+ T-cells, and also increased PD-1 expression. These effects on MDSC occurred in parallel with the enhanced antitumorigenic effect of anti-PD-1 immunotherapy, likely due to a decrease in MDSC frequency within the TME and an amelioration of T-cell exhaustion. Therefore, TREM1 is an excellent novel candidate for reprogramming immunosuppressive TMEs and sensitizing and enhancing ICI immunotherapies [120].
Interestingly, Bruton’s tyrosine kinase (BTK), expressed in MDSCs, seems to play a crucial role in their function, marking BTK as a potential target for regulating MDSC activities [139]. For instance, BTK inhibition via ibrutinib recovered T-cell activation via MDSCs, likely by impairing NOX2 expression, NO production, cell migration, and in vitro MDSC generation. Moreover, BTK inhibition reduced MDSCs’ frequency in mouse breast cancer and melanoma models and significantly enhanced anti-PD-L1’s effectiveness [121, 122].
Additionally, the free fatty acid G-protein coupled receptor (GPR) 84, also known as EX33, and a member of the G protein-coupled receptor (GPCR) superfamily, influence the differentiation and function of bone marrow cells and leukocytes. Moreover, increased GPR84 expression in stem cells promotes AML cell proliferation, migration, and disease progression [140, 141]. GPR84 expression is increased in MDSCs in patients with esophageal cancer and preclinical samples of tumor mouse models. Moreover, the frequency of GPR84+MDSC increases in patients who are resistant to anti-PD-1 treatment, which also seems to negatively impact the overall survival of patients under ICI treatment. GPR84 inhibition effectively ameliorated CD8+ T-cell dysfunction and enhanced anti-PD-1 treatment in orthotopic esophageal cancer and GPR84-/- mice mouse models [123].
Interestingly, there is a correlation between MDSC frequency and ICI clinical outcomes, as is observed during chemotherapy, indicating that MDSCs may be valuable biomarkers for immunotherapy responses in cancer patients [142].
Tumor stroma barriers impede CAR-T-cells’ therapeutic effectiveness via various
means, such as by competing for metabolic resources, presenting obstacles to
successful TME infiltration, by high levels of immune suppressive cytokines, and
through different cell types. MDSCs adversely impact CAR-T-cell-based therapies
due to their inherent immunosuppressive ability to directly inhibit effector
T-cells. Therefore, there is an urgent need to develop clinical strategies that
combine CAR-T-cells and anti-MDSC therapies. In this context, several
investigations have addressed novel approaches that combine ICIs with CAR-T-cell
strategies, which may synergistically improve cancer immunotherapies. Preclinical
research has demonstrated that combined CAR-T-cell and anti-PD-1 treatment
reduces MDSC frequency within the TME [143]. Although several ongoing clinical
trials address combined ICI and CAR-T-cell strategies, such as [NCT03179007] in
solid tumors and [NCT02862028] in NCLC, simultaneous MDSC interventions
and analyses have not been performed. There are limited numbers of approved
CAR-T-cell therapies. Examples of these therapies include
ABECMA®, BREYANZI®, CARVYKTI™,
KYMRIAH™, TECARTUS™, and YESCARTA™ [144]. However, an
open-label, phase I clinical trial study [NCT00372437] has recruited patients to
determine the safety and efficacy of CD4-CAR-T-cells (CD4CAR). These cells are
engineered to recognize and eliminate CD4+ cells in chronic myelomonocytic
leukemia (CMML) patients [145]. CD4 is linked to CD28, 4-1BB, and CD3
ICIs have become highly effective approaches for combatting T-cell exhaustion. They involve antibodies that block either an immune receptor or its ligand. Drugs targeting PD-L1 (atezolizumab, durvalumab, and avelumab), PD-1 (pembrolizumab, nivolumab), and CTLA-4 (ipilimumab and tremelimumab), used in combination with CAR-T-cell strategies, have shown increased therapeutic effectiveness in several patients [148, 149, 150]. Moreover, ICI immunotherapy has opened up a broad interest in developing clinical trials and patient treatments. For instance, Ozbay et al. [151] and Arshad et al. [152] provide comprehensive reviews of clinical trials investigating the use of molecules and biomolecules to inhibit MDSC suppressive functions, depletion, cell migration inhibition, and the induction of cell differentiation.
Several clinical trials have addressed the combined targeting of MDSC and ICI treatment. For instance, one phase II clinical trial was initiated to determine whether MDSC depletion via gemcitabine increased nivolumab efficacy and reduced immunosuppression in stage IIIB NSCLC [NCT03302247]. Gemcitabine efficacy reduces MDSC frequency in cancer [66] and has been used in a combined treatment with anti-PD-L1 in preclinical studies [116, 117]. Patients should have stage IV disease and stage IIIb disease that is not responsive to current chemotherapy. The primary outcome measures consider reducing the peripheral blood MDSC frequency due to the gemcitabine treatment and increases in T-cell activity. This study was prematurely terminated due to insufficient data collected for analysis. Along similar lines, a recent phase IV clinical trial [NCT04331626], not yet recruiting, aims to determine the safety and efficacy of half-dose gemcitabine with nivolumab in NSCLC patients. Gemcitabine efficacy reduces MDSC frequency in cancer [66] and has been used in a combined treatment with anti-PD-L1 in preclinical studies [116, 117]. This clinical trial will measure the disease control rate, disease-free progression, and overall patient survival.
Clinical trials addressing MDSC differentiation with ATRA and ICI treatments were conducted. A randomized phase II clinical trial has assessed the safety and efficacy of combined ATRA (VESANOID) and anti-CTLA-4 (Ipilimumab) in melanoma patients [NCT02403778]. Outcome measures include the tolerability of combined VESANOID and Ipilimumab, the peripheral blood MDSC frequency, and the T-cell immunosuppression activity of MDSCs. Patients were followed for evidence of disease progression. This clinical trial has been completed, and the main findings indicate that ATRA does not increase the advanced melanoma frequency of grade 3 or 4 adverse events, demonstrating the safety of combining it with the standard Ipilimumab therapy and substantially reducing circulating MDSC numbers. Thus, MDSC targeting may increase the efficacy of immunotherapeutic strategies in advanced-stage melanoma patients [153].
Similarly, a phase Ib/II clinical trial evaluated the safety and effectiveness of VESANOID (ATRA) combined with anti-PD1 (pembrolizumab) in stage IV melanoma patients [NCT03200847]. This study aimed to determine the safety and toxicity of VESANOID and pembrolizumab to establish its maximum tolerated dose; at the same time, it examined antitumor effects by determining MDSC frequency in the peripheral blood and the progression-free survival of unresectable stage III or stage IV melanoma patients. The published results of this study indicate that the combined treatment was well tolerated by the patients, allowing the authors to recommend a phase II dose of 150 mg/m2 ATRA + 200 mg Q3W pembrolizumab. Furthermore, 50% of patients underwent a complete response, with an overall response rate of 71%, a one-year overall survival of 80%, and 20.3 months of progression-free survival. The combined treatment effectively reduced the frequency of circulating MDSCs and appears to be a promising strategy for improving the effectiveness of anti-PD-1 therapy [150].
A recently recruiting phase I dose-escalation trial [NCT03161431] aims to determine whether the newly oral SX-682 drug, a bioavailable, potent allosteric inhibitor of CXCR1 and CXCR2, can target cancer by reducing the tumor chemoattraction of MDSCs in melanoma patients. Chemokines and their receptors are vital for recruiting MDSCs within the TME and constitute promising targets for immunotherapies [72]. The preliminary results indicate that SX-682, combined with pembrolizumab, has a proven tolerable safety profile and shows activity in metastatic melanoma patients [154].
Furthermore, a phase II single-center open-label trial of the combination of ATRA and pembrolizumab for relapsed or refractory Hodgkin lymphoma or B-Non-Hodgkin lymphoma treatment is recruiting patients [NCT06484920]. In total, 24 patients are expected to be enrolled. They will be treated with a 200 mg dose of pembrolizumab every three weeks (Q3W) and 150 mg/m2 of ATRA orally for the three days around each of the first four infusions of pembrolizumab (day –1, day 0, day +1). Outcome measures include the overall response rate, the complete remission rate, the MDSC frequency, and in vitro immunosuppressive activity. Since this trial is still recruiting patients, no results have been published. A recent, not-yet-recruiting, single-arm, open-label, prospective trial [NCT06371274] aims to determine the safety and efficacy of ATRA and anti-PD-1 (toripalimab) in triple-negative breast cancer (TNBC) patients. This study expects to enroll 32 subjects who failed prior standard or second-line therapies. These subjects will be administered 150 mg/m2 of ATRA orally twice a day for three consecutive days and receive an intravenous infusion of 240 mg on day one of each cycle, with cycles repeated every three weeks. The outcomes of this clinical trial will be studied by determining the objective response rate, progression-free survival, duration of response, and overall survival. Peripheral blood MDSCs will be analyzed to determine their frequency, subsets, and surface biomarkers to evaluate their association with the combined ATRA and toripalimab treatment response.
PI3K was reported to enhance intratumoral infiltration of MDSCs, promoting an
immunosuppressive TME [133]. An ongoing phase I/Ib clinical trial [NCT02637531]
aims to target PI3K with the IPI-549 (Eganelisib) inhibitor in combination with
nivolumab in advanced solid tumors, including NSCL, melanoma, squamous cell
cancer of the head and neck, TNBC, adrenocortical carcinoma, and mesothelioma.
These patients should have high-circulating MDSCs (i.e.,
In recent years, the significance of understanding the cellular and molecular processes that cause immunosuppression in the TME has been highlighted. MDSCs—by directly regulating immune responses, influencing T-cell, NK, and DC roles, and promoting Treg induction—may facilitate immunological tolerance, providing a conducive environment for cancer cells to evade immune clearance. This review highlights the crucial aspects of the diverse functions of MDSCs in promoting and maintaining an immunosuppressive TME, which hinders effective immunosurveillance.
In summary, MDSCs are crucial for developing an immunosuppressive TME, promoting cancer cell evasion from immune clearance. As such, MDSCs are promising targets for improving immunotherapy strategies, with many clinical trials aiming to eliminate MDSCs to improve chemo- and immunotherapies. Therefore, determining the complex functions of MDSCs in cancer immunology is critical for generating combined clinical therapies that will increase the effectiveness of immunotherapy. Combining these strategies with ICIs may restrain tumor development and malignancy by reducing tumor aggressivity and immune resistance.
During the preparation of this work, the author used Grammarly, an AI-powered writing assistant, to check spelling and grammar. After using this tool, the author reviewed and edited the content as needed and took full responsibility for the publication’s content.
JFS designed the research study, performed the literature research, analyzed the data, wrote the manuscript, and read and approved the final manuscript.
Not applicable.
I apologize to those colleagues whose work, although relevant to the issues dealt with within this review, has not been included due to space limitations. I also express our thankfulness for the support provided to JFS by the Visiting Professor Program of the UBO.
This research received no external funding.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.