




 , Csaba Horváth 1,†, Izabela Jarabicová 1, Petra Majerová 2, Dominika Olešová 2,3, M. Saadeh Suleiman 4, Adriana Adameová 1,5,*
, Csaba Horváth 1,†, Izabela Jarabicová 1, Petra Majerová 2, Dominika Olešová 2,3, M. Saadeh Suleiman 4, Adriana Adameová 1,5,*
1 Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University in Bratislava, 83232 Bratislava, Slovakia
2 Institute of Neuroimmunology, Slovak Academy of Sciences, 84104 Bratislava, Slovakia
3 Institute of Experimental Endocrinology, Biomedical Research Center, Slovak Academy of Sciences, 84104 Bratislava, Slovakia
4 Faculty of Health Sciences, Bristol Heart Institute, The Bristol Medical School, University of Bristol, BS8 1TH Bristol, UK
5 Centre of Experimental Medicine, Institute for Heart Research, Slovak Academy of Sciences, 81438 Bratislava, Slovakia
†These authors contributed equally.
Abstract
Regulated forms of necrosis-like cell death (e.g., necroptosis) have been shown to contribute to cardiac ischemia/reperfusion (I/R) injury. However, pro-inflammatory necroptosis is unlikely to be involved during early reperfusion and little is known about the associated molecular changes. Thus, this study aimed to provide an in-depth protein screening with a particular focus on pro-pyroptotic and mitochondrial damage-related pathways.
Langendorff-perfused rat hearts were subjected to 30-minute global ischemia followed by 10-minute reperfusion. Liquid chromatography coupled with mass spectrometry (LC-MS/MS) and immunoblotting techniques were used to study the complex cardiac proteome. In addition, calcium-induced mitochondrial swelling and lactate dehydrogenase (LDH) release were examined to assess mitochondrial stress and necrosis phenotype, respectively.
Approximately 160 proteins linked to cell death signaling, cellular metabolism, and post-translational modifications were significantly differentially expressed in I/R hearts compared to controls. Conventional proteins of pyroptosis, either of canonical or non-canonical signaling, were not affected during the short reperfusion. Notably, this type of I/R was associated with increased expression of p25 cleaved form of poly [ADP-ribose] polymerase 1 (PARP1 p25) and mature apoptosis-inducing factor (AIF), alongside nitrosative stress and mitochondrial swelling. Conversely, a receptor-interacting protein kinase 3 (RIP3) inhibitor (GSK′872, 250 nM) reversed mitochondrial swelling and plasma membrane rupture and mitigated the increase in the expression of PARP1 p25 and AIF.
This study shows for the first time that necrosis-like injury during early I/R of the isolated heart is associated with mitochondrial events, rather than pro-inflammatory pyroptotic cell death. Furthermore, the inhibition of RIP3 mitigates this injury independent of targeting pro-inflammatory signaling.
Keywords
- cell death
- inflammation
- myocardial ischemia/reperfusion
- pyroptosis
- receptor-interacting protein kinase 3 inhibition
Myocardial ischemia/reperfusion (I/R) injury is a complex of various cellular, molecular, and metabolic changes that trigger events such as calcium overload, adenosine triphosphate (ATP) depletion, overproduction of reactive oxygen species and protein synthesis suppression some of which can trigger the opening of the mitochondrial permeability transition pore [1, 2]. Collectively, they result in the death of cardiac cells, impair heart function and result in the development of heart failure over time [3].
Although early research indicated only apoptosis and spontaneous necrosis to be involved in myocardial I/R injury, recent findings highlight the role of programmed necrosis-like cell death modes, such as necroptosis, pyroptosis, ferroptosis, parthanatos, and neutrophil extracellular traps-(NET)osis [1, 4, 5]. Although all these regulated necrosis cell death forms differ in the activation and execution mechanisms, their common feature is the rupture of the plasma membrane [6].
Pyroptosis can proceed through canonical (inflammasome-related), non-canonical
(inflammasome-independent), and alternative axes [7]. Upon the initiation of
canonical pyroptotic death, the protein expression of nucleotide-binding domain
leucine-rich-containing family (NLRC), pyrin domain-containing-3 (NLRP3), NLRC4
or absent in melanoma 2 (AIM2) increases. Their increase leads to their
oligomerisation and assembly with apoptosis-associated speck-like protein (ASC)
and pro-caspase-1, finally forming the NLRP3, NLRC4 or AIM2 inflammasome,
respectively. Additionally, activated caspase-1 cleaves gasdermin D (GSDMD)
releasing its cytotoxic N-terminal (GSDMD-NT) fragment which can oligomerize and
translocate to the plasma membrane causing disruption to the membrane. Caspase-1
also leads to the production of interleukin-1
In an earlier study, we showed that necrosis-like cell death is likely to be responsible for the damage during 10-minute reperfusion in the isolated Langendorff-perfused rat hearts [10]. Because necroptosis has not been proven as a predominant form of regulated necrosis under such conditions, we aimed to further explore potential pyroptotic mechanisms. We evaluated canonical, non-canonical and alternative signaling pathways of pyroptosis and possible mitochondrial cell death-associated events that might be involved in promoting both these processes. In addition, we carried out a detailed liquid chromatography coupled with mass spectrometry (LC-MS/MS) protein screening to provide a more comprehensive protein profile and to uncover other potential mechanisms involved.
Approval for the study was granted by the Animal Welfare Ethical Review Board at
the University of Bristol (ethical approval number UB/15/017). Procedures adhered
to the Guide for the Care and Use of Laboratory Animals issued by the US National
Institutes of Health (Guide, NRC 2011) and complied with the UK’s Animals Act of
1986 [11]. Adult male Wistar rats (14–16 weeks old; 250–260 g) were provided by
Charles River Laboratories (Oxford, UK). The animals were acclimatised in a
controlled environment (23
Heart tissues were homogenised in a lysis buffer comprising 200 mM Tris, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM sodium orthovanadate (Na3VO4), 20 mM sodium fluoride (NaF), 0.5% Triton X-100 (pH 7.4) (Sigma-Aldrich, MO, USA), and a complete protease inhibitor cocktail (Roche, Mannheim, Germany). Protein precipitate was achieved overnight using 80% ice-cold acetone (Sigma-Aldrich, MO, USA). Following centrifugation, the pellets were solubilised in 8 M urea (Sigma-Aldrich, MO, USA). Protein concentrations were quantified using the Bio-Rad protein assay (Bio-Rad Laboratories GmbH, Colbe, Germany). A total of 100 µg of protein was reduced with 10 mM dithiothreitol (Sigma-Aldrich, MO, USA) in 100 mM ammonium bicarbonate (Sigma-Aldrich, MO, USA) at 37 °C for one hour. Alkylation was conducted using 15 mM iodoacetamide (Sigma-Aldrich, MO, USA) in 100 mM ammonium bicarbonate, protected from light for 30 minutes. Trypsin digestion (Promega, Wisconsin, USA) was performed at an enzyme-to-substrate ratio of 1:100 for overnight incubation at 37 °C. The resulting peptide mixtures (100 ng) were separated by Acquity M-Class Ultra-High-Performance Liquid Chromatography (Waters, Milford, MA, USA), utilising a nanoEase Symmetry C18 trap column (25 mm length, 180 µm diameter, 5 µm particle size) (Waters, Milford, MA, USA) for desalting before transferring to a nanoEase High Strength Silica T3 C18 analytical column (100 mm length, 75 µm diameter, 1.8 µm particle size). A 90-minute gradient of acetonitrile (5% to 35%) (Sigma-Aldrich, MO, USA) with 0.1% formic acid (Sigma-Aldrich, MO, USA) was applied at a flow rate of 300 nL/minute. The analysis was performed using a Synapt G2-Si quadrupole time-of-flight mass spectrometer with ion mobility (Waters, Milford, MA, USA). Data were captured in a data-independent fashion while maintaining a spectral acquisition rate of one second. Peak detection and processing were executed using Progenesis QI 4.0 software (Waters, Milford, MA, USA). Calibration and normalisation of peak intensities were based on median distribution [12].
The activity of lactate dehydrogenase (LDH) was assessed by analysing the
effluent collected from both groups during the entirety of the 10-minute
reperfusion [10]. Samples were mixed with a buffer containing 100 mM
triethanolamine and 100 µM reduced beta-nicotinamide adenine dinucleotide
(
The mitochondrial fraction was isolated in accordance with previously
established protocols [10]. Ventricular tissue was placed in chilled sucrose
buffer consisting of 300 mM sucrose, 10 mM Tris-HCl, 1 mM ethylene glycol
bis (2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) (pH = 7.2)
(Sigma-Aldrich, MO, USA) and thoroughly homogenized. The resulting homogenate was
quickly diluted with an isolation buffer containing 5 mg/mL bovine serum albumin
(Sigma-Aldrich, MO, USA) and centrifuged at 2000
For immunoblotting assessment, left ventricular tissues were prepared using
sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western
blotting techniques as previously documented [10]. Briefly, after
electrophoresis, proteins were transferred onto polyvinylidene fluoride (PVDF)
membranes (Immobilon-P, Merck Millipore) and incubated with various primary
antibodies, including apoptosis-inducing factor (AIF) (#4642, Cell Signaling
Technology, Danvers, MA, USA), AIM2 (ab119791, Abcam, Cambridge, UK), caspase-1
(ab179515, Abcam, Cambridge, UK), Caspase-11 (#14340, Cell Signaling Technology,
Danvers, MA, USA), Caspase-3 (#9662, Cell Signaling Technology, Danvers, MA,
USA), Caspase-8 (#4790, Cell Signaling Technology, Danvers, MA, USA), CypD (#
78247, Cell Signaling Technology, Danvers, MA, USA), GSDMC (ab225635, Abcam,
Cambridge, UK), GSDMD (#46451, Cell Signaling Technology, Danvers, MA, USA),
GSDME (#88874, Cell Signaling Technology, Danvers, MA, USA), IL-1
Data are reported as means
In the case of the proteomic analysis, significance (p
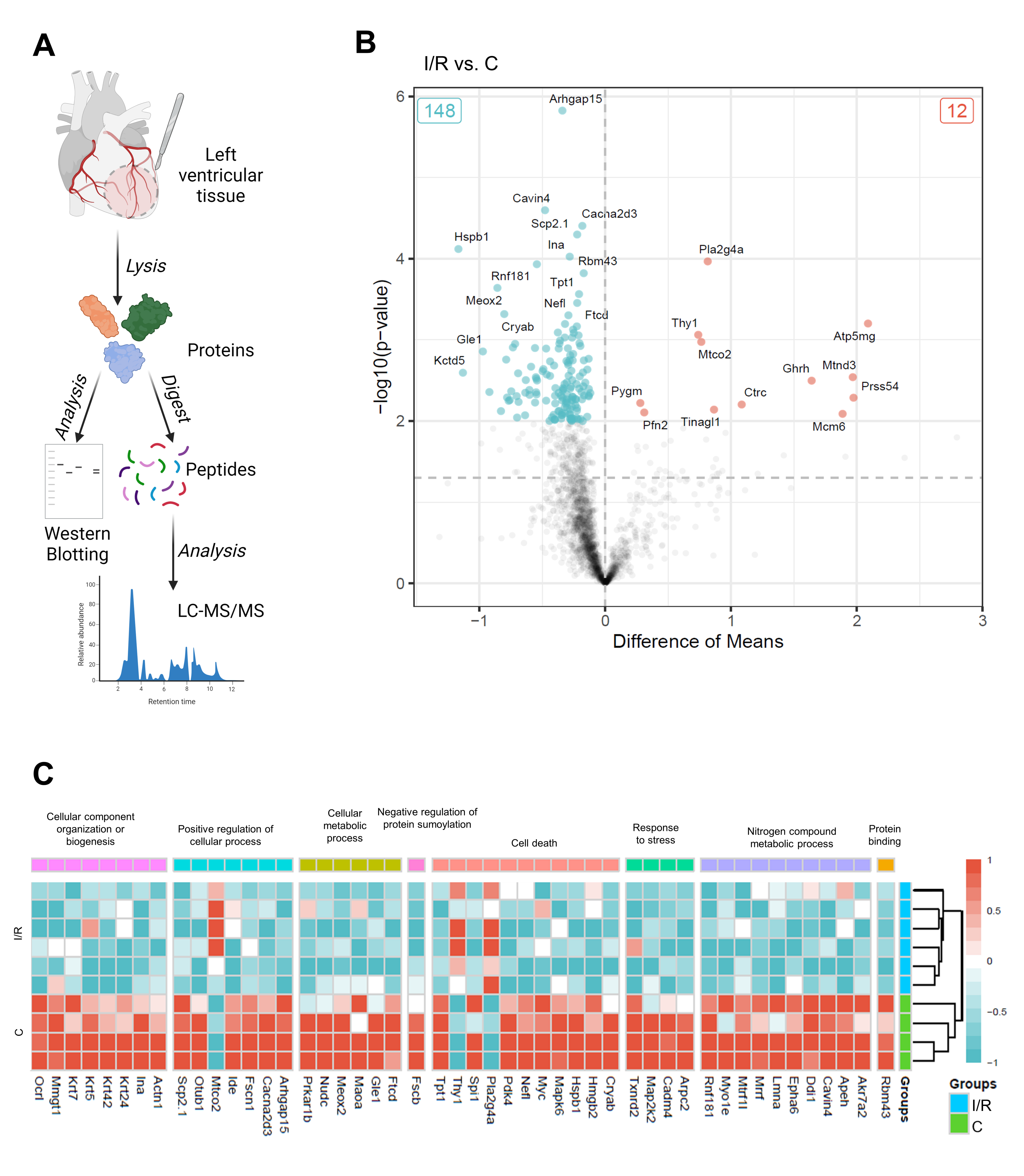 Fig. 1.
Fig. 1.
Liquid chromatography with mass spectrometry (LC-MS/MS) proteome
analysis. (A) Schematic representation of proteomic workflow; (B) Volcano plots
comparing protein levels between C and I/R groups (significantly differentially
expressed proteins were defined with
In the first 10 minutes of reperfusion, the LDH activity in the effluent was significantly increased, indicating a necrosis-like phenotype of cardiac damage. Likewise, the maximal extent of mitochondrial swelling as a marker of mitochondrial damage (sensitivity to Ca2+) was higher due to I/R. However, RIP3 inhibition significantly decreased LDH release and inhibited I/R-induced mitochondrial swelling (Table 1).
| Group | Maximal mitochondrial swelling [A520 |
LDH release [AUC] |
| C | 44.21 |
83.68 |
| I/R | 78.24 |
1148 |
| I/R + GSK′872 | 44.18 |
637.4 |
Control (C), untreated ischemic/reperfused hearts (I/R), and
receptor-interacting protein kinase 3 (RIP3) inhibitor-treated hearts
(I/R+GSK′872). AUC, area under the curve. Data are presented as mean
A liquid chromatography coupled with mass spectrometry (LC-MS/MS)-based
untargeted proteomics approach identified 160 significantly (p
| Protein | Full protein name | Molecular functions | Biological process and related pathways | UniProt ID |
| Cryab | Alpha-crystallin B chain | Chaperone-like activity, small heat shock protein. | Negative regulation of reactive oxygen species metabolic process, response to hypoxia, programmed cell death - negative regulation of apoptotic processes. | P23928 |
| HMGB2 | High mobility group protein B2 | Damaged DNA binding. | Inflammatory response to antigenic stimulus, positive regulation of innate immune response. | P52925 |
| Hspb1 | Heat shock protein beta-1 | Molecular chaperone, small heat shock protein, cellular component of cardiac myofibril. | Intracellular signal transduction, negative regulation of cellular response to oxidative stress, positive regulation of IL-1 |
P42930 |
| Mapk6 | Mitogen-activated protein kinase 6 | Serine/threonine-protein kinase; transferase. | Intracellular signal transduction. | P27704 |
| Myc | Myc proto-oncogene protein | DNA-binding; glycoprotein; transcription regulation. | Cellular response to cytokine stimulus, cellular response to hypoxia, glucose metabolism process, inner mitochondrial membrane organization, positive regulation of mitochondrial membrane potential, cellular response to hypoxia, immune processes. | P09416 |
| Nefl | Neurofilament light polypeptide | Direct protein sequencing, glycoprotein. | Protein polymerization. | P19527 |
| Pdk4 | [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 4, mitochondrial | Cellular component of mitochondrial matrix, protein kinase activity. | Cellular response to starvation, reactive oxygen species metabolic process, regulation of glucose metabolic process, regulation of pH. | O54937 |
| Pla2g4a | Phospholipase A2 | Membrane lipid remodelling and biosynthesis of lipid mediators of the inflammatory response. | Response to calcium ion. | F7EZZ6 |
| Spi1 | Transcription factor PU.1 | DNA-binding; transcription regulation. | Immune system processes, programmed cell death. | Q6BDS1 |
| Thy1 | Thy-1 membrane glycoprotein | Cell surface glycoprotein, protein kinase binding. | Cytoskeleton organization, negative regulation of protein kinase activity, positive regulation of release of sequestered calcium ion into cytosol, receptor clustering, immune system processes, programmed cell death, cell-cell signalling, positive regulation of release of sequestered calcium ion into cytosol. | P01830 |
| Tpt1 | Translationally controlled tumor protein | Calcium ion binding, DNA-binding transcription factor binding. | Programmed cell death - negative regulation of intrinsic apoptotic signaling pathway in response to DNA damage. | P63029 |
The table represents the most significantly altered proteins between the control
and ischemia/reperfusion groups. Data was acquired from the UniProt database
[17]. IL-1
The protein expression of the main inflammasome components, NLRP3, absent in
melanoma 2 (AIM2) and NLRC4, was unaffected, indicating no involvement in acute
I/R injury. Furthermore, there were no changes in the expression of the main
pro-pyroptotic caspases, caspase-1 and caspase-11, nor in the expression of
caspases associated with both pyroptotic and apoptotic signalling, caspase-8 and
pro-caspase 3. The active caspase-3 was fully absent. With the exception of the
I/R-mediated upregulation in the total GSDMC level, there were no changes in the
expression of other investigated gasdermins, namely GSDMD and GSDME, including
their active forms. We also found no I/R-mediated changes in the cleavage of
pyroptosis-related IL-1
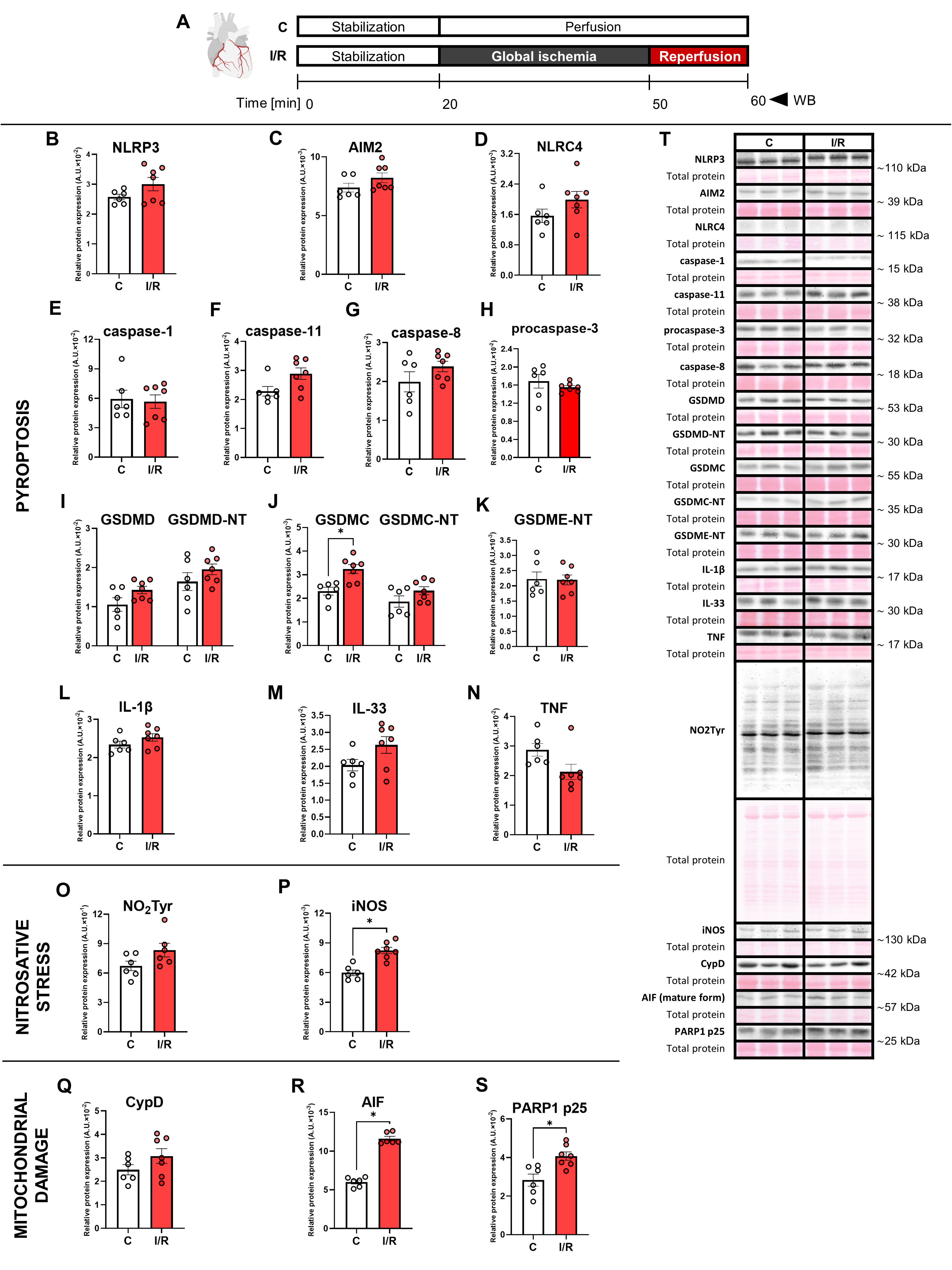 Fig. 2.
Fig. 2.
Analysis of inflammasomes and signalling pathways leading to
pyroptosis, nitrosative stress and mitochondrial damage in left ventricles
lysates. (A) Schematic representation of the experimental protocol; Immunoblot
quantification of (B) NLR family pyrin domain containing 3 (NLRP3); (C) Absent in
melanoma 2 (AIM2); (D) NLR family CARD domain-containing 4 (NLRC4); (E)
Caspase-1; (F) Caspase-11; (G) Caspase-8; (H) Procaspase-3; (I) Gasdermin D
(GSDMD) and N-terminal GSDMD (GSDMD-NT); (J) Gasdermin C (GSDMC) and N-terminal
GSDMC (GSDMC-NT); (K) N-terminmal gasdermin E (GSDME-NT); (L) Interleukin-1 beta
(IL-1
Because mitochondrial damage is associated with nitrogen species production and uncoupled nitric oxide synthase [18], we evaluated the protein tyrosine nitration and the expression of inducible NO synthase (iNOS). Although protein tyrosine nitration was unchanged, iNOS expression was significantly increased due to I/R (Fig. 2).
There were no changes in the expression of cyclophilin D (CypD), a significant regulator of mitochondrial permeability transition pore (mPTP) opening [19], among the groups. However, an increase in mPTP sensitivity to Ca2+ (cell swelling) is an important factor in mediating cell death. The levels of the active 57-kDa fragment of apoptosis-inducing factor (AIF), known to be translocated within mitochondria [20] and the p25 fragment of poly (adenosine diphosphate-ribose) polymerase 1 (PARP1) were significantly upregulated in the I/R group (Fig. 2).
Since RIP3 inhibition is able to ameliorate the I/R-mediated increase in both the maximal extent of mitochondrial swelling and LDH release (Table 1), we next investigated the associated underlying mechanisms. We found that the RIP3 inhibitor GSK′872 suppressed the I/R-induced increase in the active fragment of AIF and PARP1 p25 expression despite no effect on the CypD levels (Fig. 3).
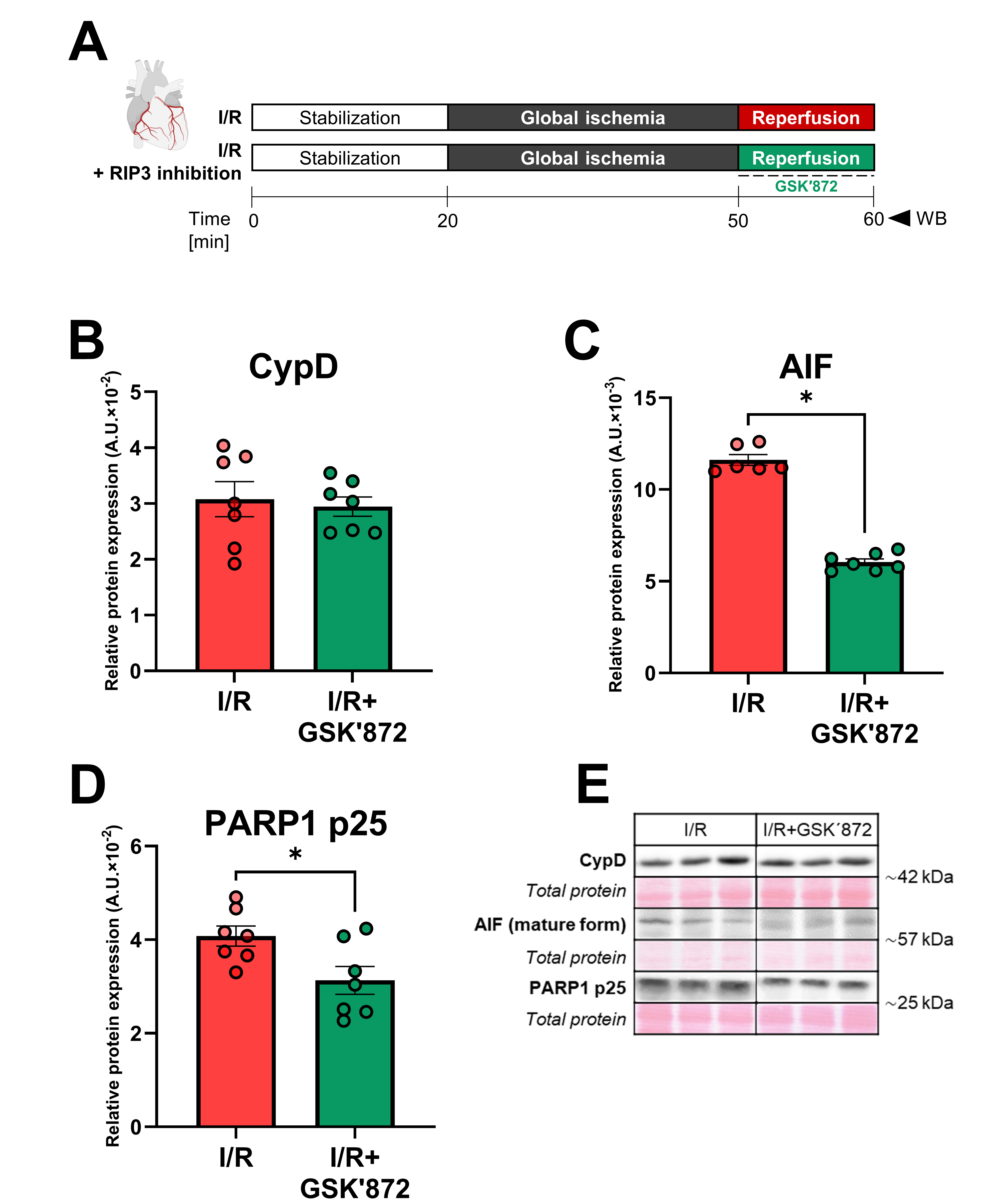 Fig. 3.
Fig. 3.
Analysis of molecules associated with mitochondrial damage in
left ventricles and the effect of RIP3 inhibition. (A) Schematic representation
of experimental protocol. Immunoblot quantification of (B) CypD, (C) AIF (mature
form), (D) PARP1 p25; (E) Representative immunoblots and total protein staining.
C, control group; I/R, ischemia/reperfusion group; I/R+GSK′872,
ischemia/reperfusion + RIP3 inhibition group. Data are presented as mean
This study aimed to provide in-depth protein analysis with a particular focus on pyroptosis and mitochondria-related damage measured during early reperfusion following global ischemia. LC-MS/MS-based proteomic screening has revealed 160 differentially expressed proteins due to 30-minute ischemia and 10-minute reperfusion. There were changes in proteins involved in the signaling of cell death, cellular metabolism, and post-translational modifications. Since the activity of LDH, arguing for the pro-necrotic plasma membrane disruption was increased, we were primarily interested in proteins mediating necrosis-like cell death. However, this detailed untargeted protein approach did not indicate alterations in the most common proteins of necrosis-like regulated forms of cell death. Instead, there were changes in the regulators of other signaling pathways that are linked to cell death. Further blotting analysis showed no changes in the expression of pyroptosis-mediating proteins (Fig. 4). Indeed, neither inflammasome-dependent nor independent signalling of pyroptosis was activated. On the other hand, unlike control hearts, the I/R hearts exhibited increased mitochondrial swelling and upregulated protein expression of AIF and PARP1 p25. Additionally, even though mitochondrial and nitrosative stress were shown to be intertwined, protein tyrosine nitration was unchanged due to I/R. The uncoupled NO signalling likely plays a role under such acute I/R protocol, evidenced by increased iNOS expression (Fig. 4). Importantly, RIP3 inhibition, which has exhibited the capability to prevent I/R-induced mitochondrial swelling and plasma membrane disruption [10], attenuated these mitochondrial alterations, thereby highlighting an interlink between RIP3, mitochondrial events and associated necrosis-like cell death (Fig. 4).
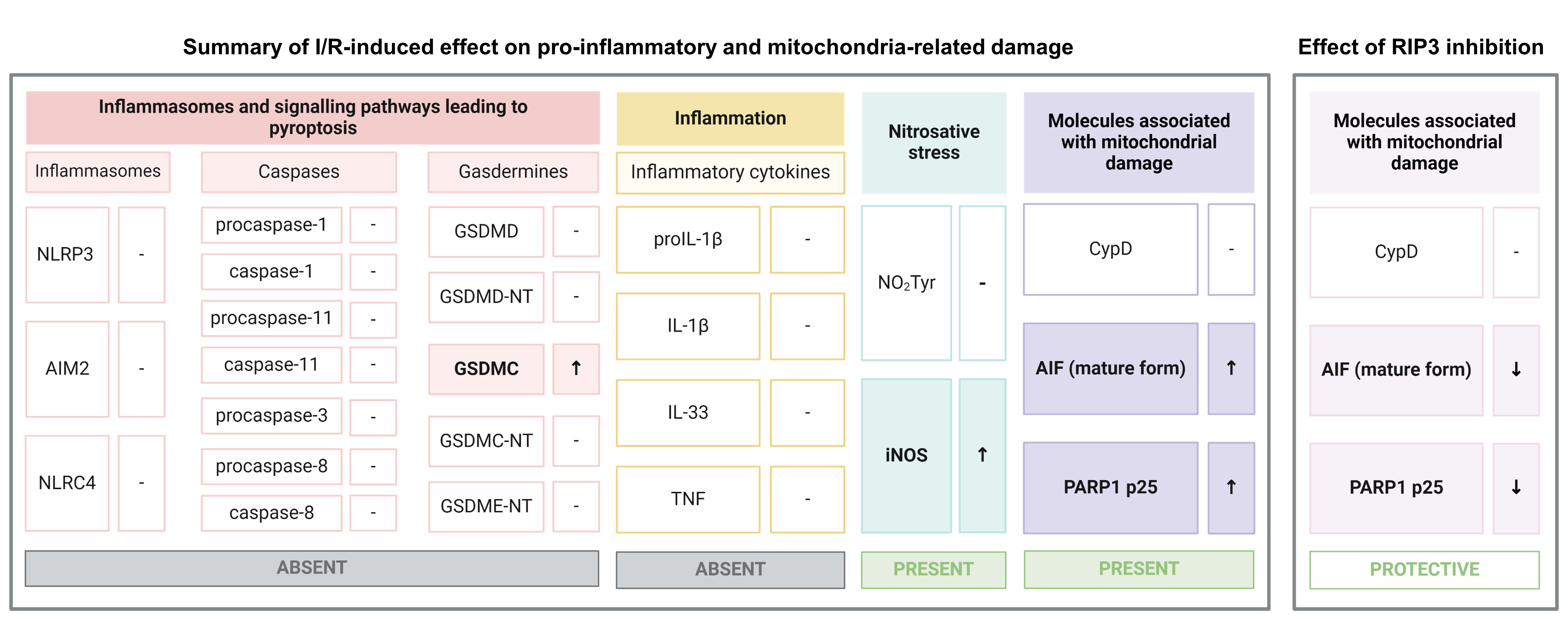 Fig. 4.
Fig. 4.
Summary of main findings on pyroptotic,
pro-inflammatory and mitochondria-related events due to early reperfusion and the
effect of RIP3 inhibition. Symbols: “-”, no statistically significant change
in protein expression compared to the control group; “
Pyroptosis has been studied in myocardial I/R injury in models of in
vivo [21, 22, 23], as well as under conditions outside of the living organism,
in vitro isolated myocardial cells [24, 25], and ex vivo hearts
[26, 27]. Here, we used a model of short reperfusion of previously ischemic
Langendorff-perfused hearts in order to examine whether the very first minutes
are critical for pyroptosis signalling because necroptosis was unlikely activated
under such conditions [10]. The expression of the main pro-pyroptotic
inflammasomes such as NLRP3, NLRC4 and AIM2 was unchanged which is in contrast
with the studies employing a longer reperfusion phase (20 minutes–24 hours)
[26, 27, 28, 29, 30, 31, 32]. Likewise, the downstream markers of both canonical and non-canonical
pyroptosis, including caspase-1, caspase-11, GSDMD, IL-1
From the aforementioned discussion, the observed necrosis-like injury of the heart during the first minutes of reperfusion is unlikely to be due to intracardiac signalling of pyroptosis and potentially originates from other mechanisms. Thus, to address this issue we looked at mitochondria-associated pro-death events. We evaluated mitochondrial swelling, nitrosative stress, CypD, PARP1 p25 and AIF expression. Tyrosine nitration and CypD levels were not changed due to I/R, whereas we showed upregulation of iNOS, PARP1 p25 and AIF after 10-minute reperfusion. In line with our data, the increase in iNOS and AIF was also found in the hearts subjected to the prolonged periods of reperfusion, (90 minutes–2 hours) [37, 38]. In these studies employing a model of ex vivo perfused hearts, the nuclear fraction of AIF increased, while the mitochondrial fraction decreased. Similar changes in AIF fractions were observed during 3-hour reperfusion under in vivo conditions [39]. PARP-1, which seems to exert profound mitochondria-related effects on cellular energetics potentially culminating in cellular demise [40, 41], was also reported to be increased after 1–4 hours of in vivo reperfusion [41].
Because RIP3 can act as an upstream protein capable of activating proteins including PARP1 p25 and AIF as well as calcium-sensitive regulation of mPTP opening and thereby producing subsequent mitochondrial swelling and damage [10], we investigated the effect of RIP3 inhibition. We report for the first time that inhibition of RIP3 significantly reduced the expression of PARP1 p25 and mature AIF. As it was accompanied by the mitigation of mitochondrial swelling, it can be suggested that the changes in these proteins might underlie, at least in part, the cardioprotection of a pharmacological approach targeting RIP3. In agreement with our work is a study employing different tissue and a different model of I/R, it has been shown that RIP3 inhibition also significantly limited neuronal death in a potential AIF-dependent manner in global cerebral I/R injury [42].
While this study provides novel insights into I/R mediated cardiac damage and likely RIP3 involvement in the underlying mechanisms, several limitations should be acknowledged. First, as an ex vivo investigation, it captures an ultra-acute state, which may not fully mimic the complexity of in vivo conditions or encompass the broader spectrum of clinical scenarios. Additionally, the findings are predominantly derived from SDS-PAGE/Western Blot analyses, which, while informative, do not enable precise localisation or identification of the specific cardiac cell types affected.
Another limitation is the use of a single dose of the RIP3 inhibitor (GSK′872). Moreover, the study would have benefited from complementary approaches, such as employing genetic knockout models, to provide more comprehensive mechanistic insights. These limitations underscore the need for further research to validate and expand upon the current findings.
Collectively, the data from this and our previous study [10] suggest that early minutes of reperfusion of the isolated hearts lead to the plasma membrane rupture that is not due to necroptotic nor pyroptotic cell death and that non-inflammatory, mitochondrial RIP3-mediated mechanisms might underlie the myocardial damage. Further studies are needed to examine the participation of RIP3 in such PARP1 p25 and AIF changes and the relevance of RIP3 inhibition as a novel cardioprotective strategy for acute I/R-associated events.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conceptualisation—AA; methodology—AM, CH, IJ, PM, DO, MSS; writing- original draft preparation—AM, CH, AA; writing- review and editing—AM, AA, CH, IJ, MSS; supervision—AA; interpretation of data for the work—AM, CH, MSS, AA. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Approval for the study was granted by the Animal Welfare Ethical Review Board at the University of Bristol (ethical approval number UB/15/017). Procedures adhered to the Guide for the Care and Use of Laboratory Animals issued by the US National Institutes of Health (Guide, NRC 2011) and complied with the UK’s Animals Act of 1986.
The figures in this manuscript were created with BioRender.com. URL: https://www.biorender.com/.
This research was funded by grants VEGA 1/0078/25, VEGA 1/0016/20, APVV-20-0242, UK/1181/2024 and the EU Next Generation EU through the Recovery and Resilience Plan for Slovakia under the project No. 09I03-03-V04-00231.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.