1. Introduction
The best choice of therapeutic agent(s) to improve outcomes of cardiopulmonary
resuscitation (CPR) during cardiac arrest is still a matter of debate.
Circulating hormone epinephrine (Epi, adrenaline), produced and secreted by the
chromaffin cells of the adrenal medulla, with contributions from the sympathetic
nervous system neurotransmitter norepinephrine (NE, noradrenaline), is
responsible for generating the “fight-or-flight” response of our bodies to
environmental stressful stimuli or traumatic insults [1, 2, 3]. Epi and NE exert
their effects in cells via adrenergic receptors (ARs), all nine subtypes of which
are class A (rhodopsin-like) G protein-coupled receptors (GPCRs) [4]: three
1-ARs (1A, 1B, 1D),
primarily exerting vasopressor actions [5]; three 2-ARs
(2A, 2B, 2C), primarily functioning
as sympatho-inhibitory autoreceptors in central and peripheral nervous systems
[6]; and three ARs (1, 2, 3),
primarily coupling to adenylyl cyclase (AC) stimulation and cyclic adenosine
monophosphate (cAMP) signaling [7]. The AR subtypes share some actions,
e.g., in adipocytes [7], but have very distinct functions, as well: for instance,
while both 1- and 2ARs exert positive inotropy,
chronotropy, dromotropy, and lusitropy in the myocardium, 3AR
exerts negative inotropy [8]. In addition, 1- and
3ARs are almost exclusively located at sites receiving direct
sympathetic innervation, and thus, are ideally placed to respond to neuronally
released NE [4]. Conversely, 2AR is located at various peripheral
organs and tissues that usually lack sympathetic innervation, and thus, is
ideally suited to respond to the circulating hormone Epi.
NE and Epi activate all 1ARs, all 2ARs, and the
1AR equipotently. Whereas Epi is more potent at 1AR
& 2AR activation than at AR activation [9], NE is more
potent at 3AR activation than Epi is and, in fact,
3AR is the only AR subtype for which Epi has low affinity [10]. The
exact opposite is true for the 2AR: NE has by far the lowest
affinity for this AR subtype, much lower than that of Epi. In this opinion
article, we argue that this is exactly what makes Epi much more efficacious in
the treatment of cardiac arrest.
2. Why Epi Is the Sole Endogenous 2AR Agonist Hormone:
The Answer is Tyr308
NE has ~10–15-fold lower affinity for human 2AR
than Epi [11]; yet, both NE and Epi make contacts with the exact same amino acid
residues in the agonist binding (orthosteric) pockets of both 2AR
and 1AR [12, 13]. Yet, Epi displays far superior potency and
efficacy at the human 2AR over NE, as measured in a cAMP
accumulation assay in human lymphocytes [14]. In human atrial and ventricular
tissue biopsies from patients, NE displayed 20-fold lower affinity for
2AR over 1AR [15]. This, coupled with the fact that
1AR, which both NE and Epi activate equipotently, is much
(~10-fold) less efficacious at producing cAMP in human heart than
2AR is [15], translates into a significantly (5-fold) higher
potency for Epi over NE at stimulating cardiac contractility: 1 µM of NE
produces the same degree of positive inotropy as 200 nM of Epi in human atrial
myocardium [15]. It is thus clear that NE, at normal physiological
concentrations, does not really activate the 2AR subtype, similarly
to dopamine [14], and thus, Epi is essentially the only endogenous catecholamine
that activates the 2AR, at least at physiological concentrations.
The only structural difference between NE and Epi is the presence of one methyl
group on the positively charged (protonated)-NH2 group, absent in NE (Fig. 1A, Ref. [16]). This methyl substitution increases the basicity (protonation) of
Epi’s nitrogen atom (secondary amines like Epi are generally more basic than
primary amines like NE). It also increases ligand affinity for the human
2AR dramatically: when this N-methyl group is added to NE, affinity
for the 2AR increases ~60-fold [17]. How exactly
this N-methyl group confers this dramatic difference in 2AR
affinity is still under investigation. However, the orientation of the
catecholamine in the orthosteric pocket, a “groove” formed by Asp113 of TM3 &
Asn312 of TM7 on one end, and Ser203/Ser204/Ser207 of TM5 on the other (Fig. 1A),
gives some crucial hints [16]. The N-methyl group is well positioned to interact
with the extracellular top of TM7 at the extracellular “lid” of the pocket.
Although it does not contact the agonist in the pocket directly, Tyr308 of TM7 is
in close proximity (within 8 Å) to amino acids that do contact the ligand,
specifically Asn312 (Fig. 1A) [18]. Notably, all other AR subtypes, the
1AR included, have phenylalanine (F7.35) at this TM7 position
(position 7.35 based on the Ballesteros-Weinstein numbering) [13]. Given that
tyrosine forms both polar (hydrogen bond) interactions via its hydroxyl group and
hydrophobic (Van der Waals) interactions via its phenyl group (Fig. 1A), Tyr308
is ideally placed to coordinate the entry of the N-methyl group of Epi into the
pocket and to stabilize binding (prevent dissociation) to the 2AR
(Fig. 1A). This is because the N-methyl group also makes polar, via the
protonated amino group (-+NH2), and hydrophobic, via the methyl group
(-CH3), interactions. Indeed, the “on” (receptor association) rate of Epi
for the 2AR has been estimated to be ~14-fold
faster than that of NE [11], and Tyr308 of the 2AR has been
documented to be the main determinant of 2AR-selective affinity for
AR agonists via both hydrophobic (with the phenyl) and polar (with the
-OH group) interactions [19]. Importantly, Tyr308 both increases the association
and decreases the dissociation rates of Epi on the 2AR [19]. In
contrast, by lacking this N-methyl substitution, NE is probably incapable of
interacting with Tyr308 (the NE molecule is not long enough to sterically reach
Tyr308 for strong interaction) and thus, quickly dissociates from the
2AR’s orthosteric pocket. Hence, only very high NE concentrations
can activate the 2AR [14]. Interestingly, Tyr308 has been reported
to be essential for the Gs protein-“biased” agonism of the 2AR,
as well [20]. 2AR’s Tyr7.35 appears necessary for efficient
activation of the Gs/AC/cAMP signaling pathway by the receptor. Since
1AR has Phe instead of Tyr at this position, this could underlie,
in part, the reduced efficacy, versus 2AR, of 1AR at
producing cAMP [15]. The significantly larger (by 27 amino acids) third
intracellular loop of 1AR, compared to that of 2AR,
is another reason postulated for this reduced efficacy [15]. Taken together,
Tyr308 controls both orthosteric agonist affinity (potency) and, partly, cAMP
signaling efficiency (agonist efficacy) at the human 2AR.
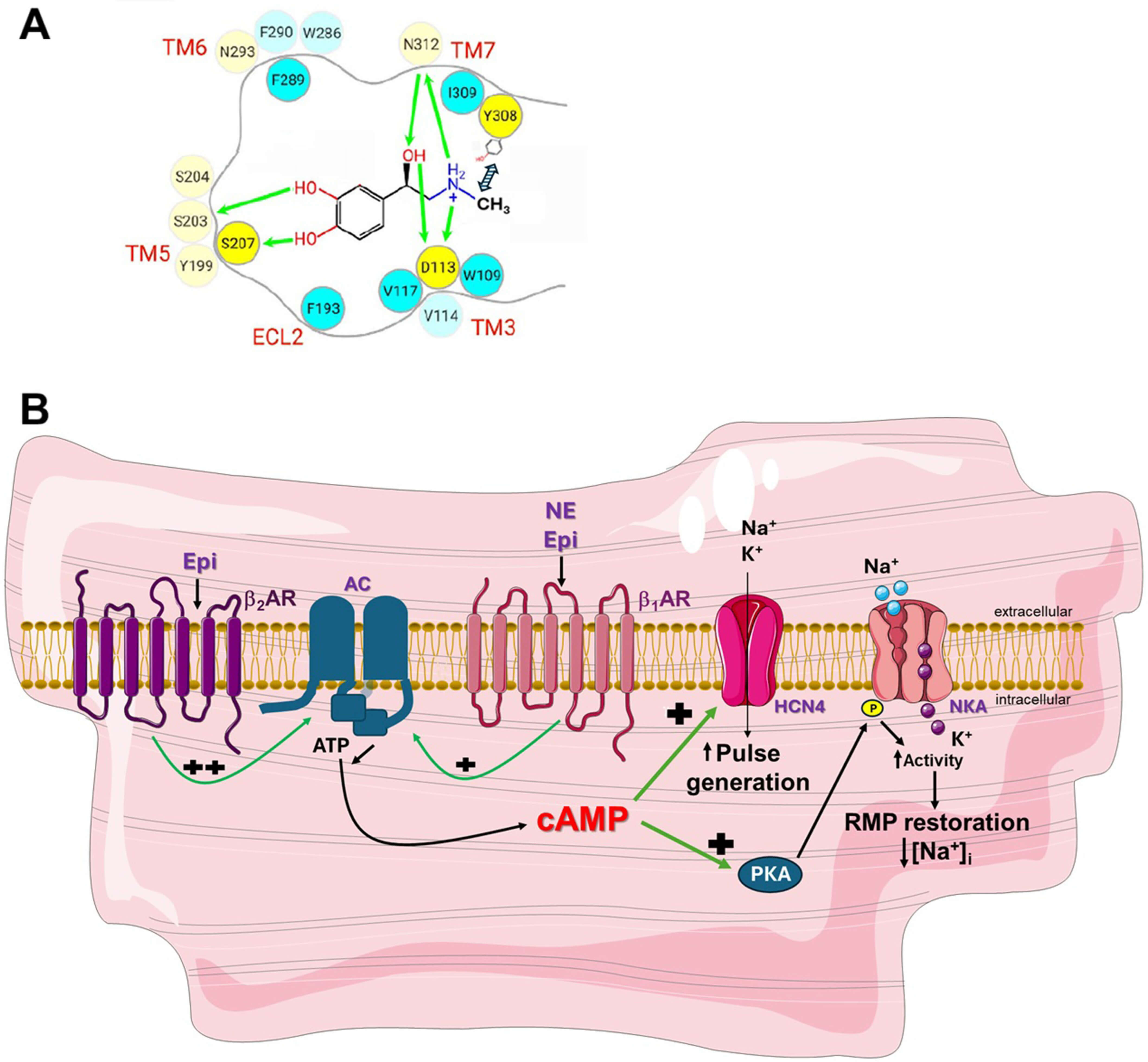 Fig. 1.
Fig. 1.
2AR activation boosts the efficacy of Epi in
cardiac arrest. (A) The amino acids of the orthosteric pocket of the human
2AR occupied by the Epi molecule, including Tyr7.35 (Y308). Y308
does not contact Epi directly in the pocket but nevertheless facilitates its
docking and binding to the pocket thanks to proper coordination of the N-methyl
group. In contrast, this methyl group is absent in NE and thus, Y308 cannot do
the same for NE. The green arrows indicate polar (hydrogen bond or ionic)
interactions. Fig. 1A is modified with permission from Ref. [16]. (B) Beneficial
actions in cardiac arrest of cardiomyocyte-residing 2ARs via cAMP
towards pulse generation and contractile function restoration. Because Epi
activates the 2AR, in addition to the 1AR, and
2AR is more potent at generating cAMP (i.e., at activating AC) than
1AR is, Epi activates HCN4 channels in SA nodal cardiomyocytes to
increase pulse generation, and NKA in working (contracting) cardiomyocytes to
restore resting membrane potential & excitability, more robustly than NE does.
“++” denotes higher potency than “+”. AC, Adenylyl cyclase; AR, Adrenergic
receptor; ATP, Adenosine triphosphate; cAMP, Cyclic adenosine monophosphate; ECL,
Extracellular loop; Epi, Epinephrine; HCN4, Hyperpolarization-activated cyclic
nucleotide-gated channel-4; [Na+]i, Intracellular Na+ concentration; NE, Norepinephrine; NKA, Na+/K+-ATPase; P,
Phosphorylation (of phospholemman); PKA, Protein kinase A; RMP, Resting membrane
potential; TM, Transmembrane helix. Fig. 1B was created using images from Servier
Medical Art Commons Attribution 3.0 Unported License.
3. Epi in Cardiac Arrest: 2AR Activation is Key
3.1 Clinical Evidence for the Use of Epi in Cardiac Arrest
Their 1AR-dependent vasopressor actions form the basis for the
use of catecholamines in cardiac arrest (or asystole) and during CPR [21]. In
cardiac arrest, coronary artery perfusion pressure drops precipitously because
the heart has stopped beating, and, since coronary perfusion pressure is defined
as the difference between aortic and right atrial blood pressures, vasopressors
that acutely raise arterial blood pressure are indicated to enhance coronary
perfusion and restart the heart, i.e., to induce the so-called “return of
spontaneous circulation” (ROSC) [22]. Thus, alongside NE and Epi, potent
vasopressors, such as vasopressin and phenylephrine, are sometimes used during
CPR. Epi has historically been the agent of choice for cardiac arrest/CPR with
several studies/meta-analyses supporting its utility in this medical emergency
setting [23, 24, 25, 26, 27], showing consistent increases in ROSC and return of pulse in
CPR-receiving subjects, as well as in survival to hospital admission and even
hospital discharge. Indeed, the latest American Heart Association’s guidelines
recommend administering 1 mg of epinephrine intravenously (IV) or intraosseously
(IO) every 3–5 minutes for adult cardiac arrest [27]. Epi can also augment the
effect of vasopressin and other vasopressors on ROSC and survival rate in cardiac
arrest [26]. However, a few trials have cast doubt on the actual benefit of Epi
for long term cardiac arrest outcomes, particularly with respect to neurological
recovery [21, 23]. Indeed, 2AR activation by Epi can substantially
increase myocardial oxygen consumption to meet the demands of significantly
enhanced contractility, which may lead to non-favorable long term survival
outcomes, especially in ischemic heart disease patients [21, 28]. In addition,
2AR activation leads to vasodilation, which may affect local
microcirculation negatively, particularly in the brain, due to perturbations in
small vessel (arterioles/capillaries)-mediated tissue perfusion [28].
Nevertheless, other studies have failed to demonstrate any inferiority of Epi in
cardiac arrest compared to NE [29] and the overall picture regarding Epi’s
utility in cardiac arrest is still fuzzy. This is probably because: (a) each
study’s findings may not be generalizable to all patients; (b) there are several
factors, unrelated to Epi’s effects, affecting neurological outcomes in cardiac
arrest survivors, as well as methodological problems in some of the studies [24];
and, perhaps most importantly, (c) since Epi increases the chances of
post-cardiac arrest survival, the total number of survivors having received Epi
during CPR is higher, which can skew the number of survivors that do not recover
neurologically towards Epi in an unfavorable manner. In other words, the main
goal of Epi administration in cardiac arrest is ROSC and return of pulse, so the
patient can escape instant death and hopefully survive either to hospital
admission or, if already hospitalized, to discharge.
3.2 Physiological Mechanisms of Epi in Cardiac Arrest
Apart from Epi-activated 1AR-mediated vasoconstriction that
enhances coronary perfusion, Epi-activated 2ARs in the airways
dilate the bronchi and lungs, an action crucial not only in treatment of
anaphylactic shock but also during CPR to improve blood oxygenation [30]. In
addition, cAMP elevation inside cardiomyocytes by Epi-activated
1ARs and 2ARs affords two additional important
benefits in cardiac arrest. One of these benefits is stimulation of the
sinoatrial (SA) node, the natural pacemaker of the myocardium, to increase the
If current via enhanced opening of hyperpolarization-activated cyclic
nucleotide-gated (HCN)-4 channels [31, 32]. These channels are directly bound and
operated by cAMP and their opening results in increased beating frequency, i.e.,
elevated heart rate (Fig. 1B). It is well established that 2ARs are
abundant in the SA node (in fact, more abundant than in the rest of the atria)
and mediate HCN4 channel activation and positive chronotropy alongside
1ARs [33, 34, 35]. Given that cardiac 2ARs are 10 times
more potent at cAMP synthesis than their 1AR counterparts [15] and
that Epi activates both subtypes equipotently, unlike NE that activates the
1AR but not the 2AR subtype at normal concentrations,
it follows that Epi can stimulate the pacemaker activity of the SA nodal
cardiomyocytes much more robustly than NE (Fig. 1B). This would be consistent
with the long-reported greater potency of Epi over NE at stimulating
contractility, as well (higher heart rate normally leads to increased
contractility) [15]. Being able to stimulate pacemaker activity more robustly via
greater cAMP production driven by both 1- and 2ARs
gives Epi a unique ability to generate heartbeats (pulse), and thus, a
significant advantage over NE (and other vasopressors) in treatment of cardiac
arrest.
Finally, the other cAMP-dependent benefit that is therapeutically exploited by
using Epi in cardiac arrest is stimulation of the sodium-potassium pump
(Na+/K+-ATPase, NKA) (Fig. 1B). Protein kinase A (PKA), the main
effector of cAMP, activates NKA via phosphorylation of FXYD1 (also known as
phospholemman, PLM), a protein that physically interacts with NKA reducing its
affinity for intracellular Na+ and thus, its activity [36, 37]. PKA-dependent
phosphorylation releases PLM inhibition of NKA, robustly increasing NKA activity
[37, 38]. Since NKA-dependent Na+ efflux and K+ influx are essential for
restoration of the resting membrane potential and gradients of these two cations,
NKA activation is an integral part of the adrenergic “fight-or-flight” response
[37] (Fig. 1B). It is also essential for reduction of intracellular [Ca2+]
(via the Na+/Ca2+ exchanger) [38] (Fig. 1B). High intracellular
[Na+] suppresses excitability of working cardiomyocytes due to perturbation
(suppression) of normal Na+ currents, on which fast depolarization (Phase 0
of the action potential) depends [39]. Therefore, NKA stimulation by cAMP is
crucial for maintaining cardiac function, particularly in ischemic (hypoxic)
cardiomyocytes, where NKA activity is low due to energy (ATP) depletion.
2AR, probably again due to its greater potency at increasing cAMP,
stimulates NKA activity more robustly than 1AR does: the
2AR-selective agonist salbutamol is equipotent to Epi but 100-fold
more potent than a 1AR-selective agonist at stimulating NKA in rat
soleus muscle [40]. Together with activation of both 1- and
2ARs, this means that Epi stimulates cardiomyocyte NKA more
robustly than NE does (Fig. 1B), which is also consistent with the
well-documented, clinically, Epi-induced hypokalemia (due to high NKA activity)
[41]. In conclusion, Epi stimulates cardiac NKA more robustly than NE and other
vasopressors used in cardiac arrest, which is another crucial beneficial
mechanism by which Epi can restore cardiac pulse and contraction, i.e., increase
ROSC.
4. Conclusions & Future Perspectives
Epi is a particularly useful drug (the agent of choice) in cardiac arrest thanks
to its unique, among vasopressor hormones/agents, activation of cardiac
2ARs. 2AR activation results in profound increases in
potency and efficacy of Epi, compared to NE and other non-catecholamine
vasopressors, towards pulse generation in the SA node and contractility
restoration in the working myocardium via NKA activation. Of course, other
important mechanisms for cardiomyocyte homeostasis, such as NCX and
sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) activation [42], may also
mediate Epi’s superior potency & efficacy. Being able to exert this lifesaving
action in the extremely dire situation of cardiac arrest via 2AR
activation might be one of the reasons why only the 1AR (not the
2AR) is downregulated in human chronic heart failure; however,
2AR may also be desensitized and dysfunctional in this disease
[43]. It could also explain why circulating Epi, contrary to NE, is not elevated
in chronic human heart failure [44]: perhaps the body keeps Epi levels low in
this disease state, so it can increase them to activate the 2AR
only at moments of absolute life-or-death emergencies, such as cardiac arrest or
asphyxiation due to acute airway obstruction.
Abbreviations
AC, Adenylyl cyclase; AR, Adrenergic receptor; Epi, Epinephrine; NE, Norepinephrine; cAMP, Cyclic adenosine monophosphate; HCN4, Hyperpolarization-activated cyclic nucleotide-gated channel-4; NCX, Na+/Ca2+ exchanger; NKA, Na+/K+-adenosine triphosphatase (ATPase); PKA, Protein kinase A; ROSC, Return of spontaneous circulation; SA, Sinoatrial; SERCA, Sarco(endo)plasmic reticulum Ca2+-ATPase.
Author Contributions
AL conceived the article, performed literature research, and wrote the manuscript. AJM, RAS, and VLA assisted with literature research and contributed to the writing of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
We thank all the peer reviewers for their opinions and suggestions that helped us improve our manuscript.
Funding
A.L. is supported by a National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI) grant (R01 #HL155718-01). R.A.S. is supported by an American Foundation for Pharmaceutical Education (AFPE) Gateway to Research Scholarship (#333609-2025).
Conflict of Interest
The authors declare no conflict of interest.
Declaration of AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work the authors used Gemini for assistance with literature search. No AI tool was used for drafting of the manuscript. The authors take full responsibility for the content of the publication.



 , Alexis J. M’Sadoques 1, Renee A. Stoicovy 1, Victoria L. Altsman 1
, Alexis J. M’Sadoques 1, Renee A. Stoicovy 1, Victoria L. Altsman 1