






 , Ning Chen 1, Huanhuan Feng 1, Xueli Du 1
, Ning Chen 1, Huanhuan Feng 1, Xueli Du 11 Depatment of Medicine, Luoyang Polytechnic, 471000 Luoyang, Henan, China
Abstract
The phycobilisomes (PBS) of cyanobacteria and red algae are unique light-harvesting protein-pigment complexes that utilize bilin derivatives for light absorption and energy transfer. These extramembranous mega-Dalton complexes are specifically organized and anchored to photosystem II (PSII) via the multi-domain core-membrane linker (LCM). While Arm2 is the longest segment in LCM domain, its specific functions remain uncharacterized.
A series of Synechocystis sp. PCC 6803 mutants with complete or partial deletions of Arm2 and its adjacent Rep domains within LCM were constructed. The assembled PBSs were isolatedand characterized using sucrose gradient ultracentrifugation, absorption and fluorescence spectroscopy, and SDS-PAGE. Physiological functions were further assessed by analyzing growth, photosynthetic performance, state transitions, and non-photochemical quenching (NPQ).
Our results reveal that the super-secondary element of helix-turn-helix of Arm2 is critical for assembling the two longitudinal halves of PBS. The truncation of either or both helices of Arm2 results in the specific degradation of the longitudinal half harboring the terminal emitter, ApcD. Consequently, these mutants were deficient in state transitions and exhibited accelerated recovery from orange carotenoid protein (OCP)-mediated NPQ. We also identified the Arm2(37–67) motif likely involved in attaching the rods to the core, whereas the Arm2(68–129) region had no significant impact on PBS assembly.
The helix-loop-helix element of Arm2 is essential for the longitudinal integrity of the PBS core and is a prerequisite for state transitions. These results suggest that state transitions may involve longitudinal rearrangements within the PBS structure, rather than lateral movements of the two halves, implicating that state transitions result from the longitudinal instead of the lateral moves of the two halves of the PBSs.
Keywords
- Synechocystis
- allophycocyanin
- phycocyanin
- linker
- energy transfer
- photoprotection
Phycobilisomes (PBSs) are mega-Dalton light-harvesting complexes in cyanobacteria and red algae, composed of dozens of phycobiliproteins that organize hundreds of bilin chromophores [1]. These complexes absorb light across a broad spectrum (480–660 nm) and efficiently channel the energy to the photosynthetic reaction centers. This transfer occurs either directly or assisted by the chlorophylls (Chls) in the photosystems’ core complexes [2, 3]. Final energy delivery is facilitated by two terminal emitters containing phycocyanobilin (PCB): allophycocyanin (APC) B (AP-B) and the core-membrane linker (LCM) [4, 5].
LCM is encoded by apcE gene and comprises an N-terminal
PCB-binding domain (ApcE
The connecting loops of Synechocystis’ LCM include Arm1(241–249)
that connects Rep1(250–400) to the PCB-binding domain (ApcE
In this study, we investigated the role of Arm2 in PBS assembly. We constructed
a series of Synechocystis mutants with complete or partial truncation of
Arm2 and/or deletions of its adjacent Rep2/Rep3 domains. Analysis of isolated
PBSs, we found that the truncation of either one or both helices of Arm2 in
LCM split the PBSs longitudinally into a simple PBS consisting of the APC
core and cyanobacterial phycocyanin (CPC) rod. Mutants with the damaged
helix-loop-helix element of Arm2 were deficient in state transitions and
exhibited accelerated recovery from non-photochemical quenching (NPQ) compared to
wild-type (WT) and Rep3-truncated Synechocystis
(
Synechocystis and its mutants were grown in BG-11 medium on a shaker (130 rpm) at 30 °C under continuous white light (20 µmol photons m-2s-1 for LL) [14]. BG-11 medium with 1.2% (w/v) agar containing 0.3% (w/v) sodium thiosulfate was solidified for plate cultures. The photomixotrophic growth was achieved by diluting an aliquot from a photoautotrophic culture in the log phase (OD730 ~1.0) to an OD730 ~0.05 with BG-11 containing 10 mM glucose. The growth and cell densities were monitored at OD730 on a Beckman DU800 spectrophotometer. The growth was evaluated based on the average of three parallel experiments. Chlorophyll a (Chl a) [15] or phycocyanin (PC) [16] concentrations were determined by absorption spectroscopy and calculated per cell [17].
All genetic manipulations were carried out according to standard protocols [18]. The apcE gene locus (slr0335) in Synechocystis encodes the LCM protein, which consists the following domains: the N-terminal loop(80–150), Arm1(241–249), Rep1(250–400), Arm2(401–534), Rep2(535–685), Arm3(686–714), and Rep3(715–860). To delete these domains and/or motifs, mutation plasmids were constructed as follows.
A 0.8-kbp DNA fragment including arm2-rep2 and a 0.6-kbp DNA
fragment including arm3-rep3 of apcE was amplified
from Synechocystis genomic DNA by PCR using primers P1–P4
(Supplementary Table 1), as upstream and downstream targeting arms,
respectively. The restriction sites XhoI plus EcoRV and
EcoRV plus XbaI were introduced in the targeting arms,
respectively, for cloning the fragment into pBluescript. A DNA fragment
containing streptomycin resistance cassette was excised from plasmid pHB45 via
the restriction site SmaI and then inserted in the plasmid containing
the targeting arms via the restriction site EcoRV, thereby yielding
pBlue-
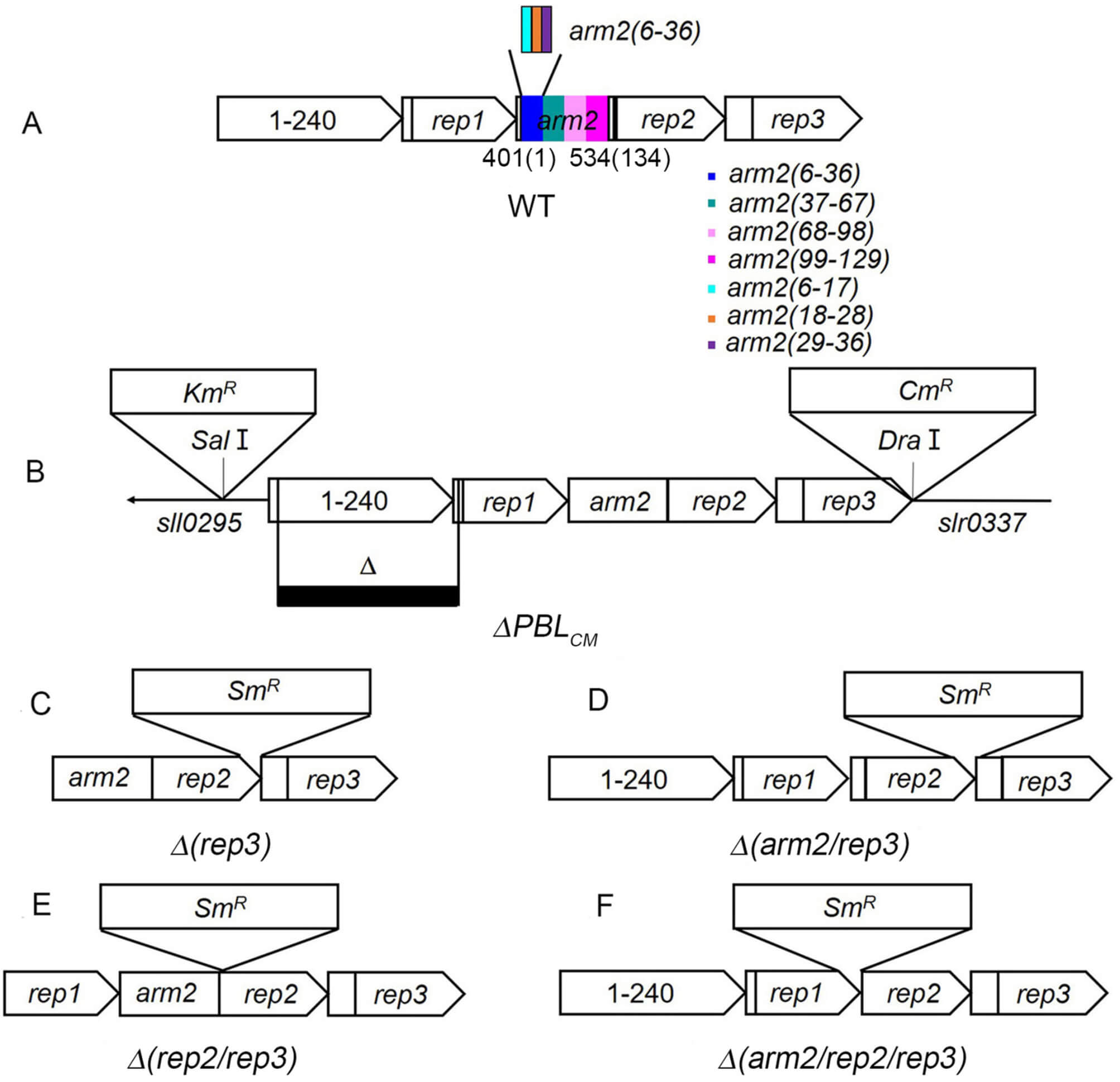 Fig. 1.
Fig. 1.
Construction and characterization of ApcE-mutants in
Synechocystis PCC 6803. (A) is the domain location in the ApcE protein
and (B) is the
A 1.7-kbp DNA fragment including
apcE
A 5.3-kbp DNA fragment, including
pBlue-apcE
The mutant strains were generated by transforming WT Synechocytis cells with the respective plasmids via homologous recombination [20]. Transformants were selected on BG-11 plates containing 12.5 µg/mL spectinomycin, with segregation ensured throughsuccessive rounds of selection at 25 µg/mL spectinomycin. The genotypes of all final mutant strains were confirmed by PCR using respective primer sets (Supplementary Fig. 2, Supplementary Table 1).
The room temperature absorption spectra of the cell samples and the isolated PBS fractions were recorded on a UV-9000S spectrophotometer with a slit width of 1 nm in a 2-mm cuvette with high scattering property. At the corresponding peak wavelengths, molar extinction coefficients of 1185 mM-1cm-1 and 770 mM-1cm-1 were used for a trimer of PC (620 nm) and APC (650 nm), respectively. According to the absorption spectra of CPC [21], the molar extinction coefficient of 266 mM-1cm-1 was used for a trimer of PC (650 nm), while the molar extinction coefficient of 295 mM-1cm-1 was used for a trimer of APC in 620 nm according to the absorption spectra of APC [22]. The concentrations of CPC and APC were calculated according to established methods [16, 23]. The equations used were:
Spectra were acquired using an F-320 Fluorolog spectrofluorimeter, with slit widths set at either 5 or 10 nm. Synechocystis WT and mutant strains were cultivated under normal light until reaching an optical density of A730~0.8. For measurement, the chlorophyll a (Chl a) concentration of the cell suspensions was adjusted to either 3 or 5 µg/mL, corresponding to the excitation wavelengths of 580 nm or 430 nm, respectively. Subsequently, 800 µL aliquots of the adjusted cells were dark-adapted for 5 minutes in fresh BG11 medium prior to spectral recording. All experiments were performed with at least three independent biological replicates.
The 77-K fluorescence emission spectra were monitored using a Horiba Fluorolog spectrofluorimeter with a slit width of 8 nm. The cells at CChl = 3 µg/mL (580 nm excitation) were collected by centrifugation and suspended in an equivalent volume of fresh BG11 containing 40% (v/v) glycerol and 25 mM HEPES-NaOH (pH 7.5). In all cases, whole cells were dark-adapted for 15 min before the measurements. Then, the spectra were recorded corresponding to State II. For State I spectra, cells were illuminated with 55 µmol photons m-2s-1 of blue light for 5 min. Then, 800 µL suspensions were quickly frozen in quartz tubes by immersion in liquid nitrogen for 10 s. The excitation was measured at 580 nm, and emission was scanned at 600–800 nm [24, 25]. Date are from at least three biological replicates.
The light response curves, NPQ, and state transitions were monitored on a PAM fluorometer (PAM 2500; Walz, Effelrich, Germany) [26, 27]. Mutant and WT cells were grown under similar conditions (A730 = 0.6) and estimated on dark-adapted (15 min) whole cells at a chlorophyll concentration of 3 mg/L. F0 is a minimal fluorescence level determined by illuminating dark-adapted cells with a low intensity of red-modulated light (pulses of 1 s, 1.6 kHz, 0.024 µmol photons m2s-1). For the measurements of state transitions [28], cells were dark-adapted, irradiated with blue (55 µmol photons m2s-1) or orange light (20 µmol photons m2s-1), and subjected to saturating pulses (2000 µmol photons m2s-1, 30 s) to measure the Fm′ levels (maximum fluorescence under illumination).
All NPQ induction and recovery experiments were carried out in the presence of
chloramphenicol (30 g/mL), a protein synthesis inhibitor, to inhibit protein
synthesis [26]. For the measurements of the light response curve, cells (CChl =
20–30 mg/L) were irradiated with various intensities (0–1400 µmol
photons m2s-1) after dark adaptation (15 min) and then subjected to
saturating pulses (2000 µmol photons m2s-1, 30 s) to
measure the Fm′ levels. The initial slope of the rapid light response curve
(alpha), the maximal rate of electron transfer (ETRmax), and the minimal
saturated light intensity at semi-saturated light intensity (Ik) were calculated
from the PAM data using the analysis software [29, 30]. Data are presented as
mean
Protein concentration was quantified using the Bradford method [31] with bovine serum albumin (BSA) as a standard. The proteins from sucrose-containing fractions were precipitated by 50% (NH4)2SO4 and collected by centrifugation to remove the sucrose. Then, the samples were diluted with distilled water and precipitated by 10% trichloroacetic acid (TCA) to remove the ammonia. The remaining TCA was removed by acetone extraction. Subsequently, the obtained samples were dried, solubilized in the sample buffer [32], and analyzed by SDS-PAGE using Laemmli buffer system [33]. Finally, the proteins were stained with Coomassie brilliant blue [34].
PBSs were isolated from Synechocystis cells by an
ultracentrifugation-based protocol [35]. Briefly, cells were lysed with 2% (v/v)
Triton X-100, and the lysate was clarified by centrifugation to remove unbroken
cells, debris, and chlorophyll. The resulting supernatant (2 mL) was then layered
onto sucrose step-gradients for ultracentrifugation. The gradients, prepared in
12-mL tubes with 0.8 M KPB (pH 7.2), consisted of 1 mL of 2.0 M, 2.5 mL of 1.0 M,
2 mL of 0.75 M, 2 mL of 0.5 M, and 1.5 mL of 0.25 M sucrose solutions.
Centrifugation was performed at 230,000
Electron microscopy was performed on a HITACHI H-7650 transmission electron
microscope operated at 100 kV. The images were recorded with a 1024
Arm2 in apo-protein of LCM (ApcE) from Synechocystis constitutes
401–534 aa. Thus, we re-numbered Arm2 as 1–134 aa (Fig. 1A). Strikingly, the
amino acid sequence of Arm2 of the red alga Griffithsia pacifica was
highly similar to that of Synechocystis (Supplementary Fig.
1A). Based on the 3.5 Å PBS structure from the red alga Griffithsia
pacifica [8], the conservative motif of Arm2(6–36) in Griffithsia
pacifica formed the super-secondary structure of
The assembly of PBSs in the absences of Rep3 remains a subject of debate. The
classic model of PBSs of Synechocystis suggests that Pep3 is essential
for Cylinder 3 attachment, with its lost resulting in a simple, two-cylinder core
(Cylinder1 and 2) [7]. In contrast, the structural study on PBS on the red alga
Griffithsia pacifica [8], Cylinder3 is lost in the absence of Rep3, and
then Cylinder1 and 2 are disconnected. Given the symmetry of PBSs, Cylinder1 and
2 are same, and we would only isolate a simple PBS of Cylinder1 or 2 in the core.
Next, we isolated the PBSs from
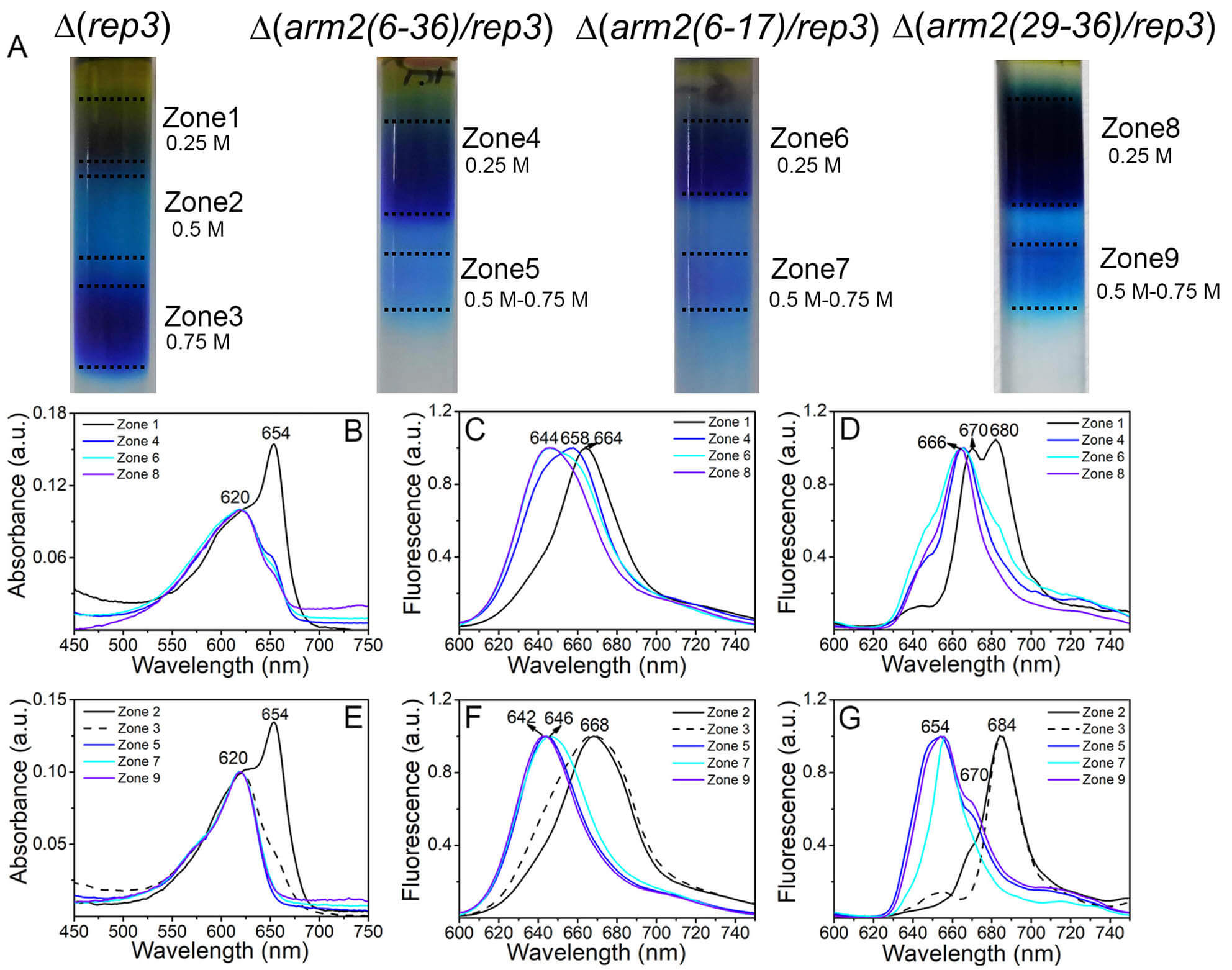 Fig. 2.
Fig. 2.
Characterization of the PBSs of
We next analyzed PBSs from the double mutant
According to the PBS structure from the red alga Griffithsia pacifica [8], the Arm2 element of helix-loop-helix is inserted between one normal APC disk
and two APC disks, one of which has a LCM terminal emitter and the other has
AP-B terminal emitter in one basal cylinder (i.e., Cylinder1 or 2). When one
helix or both helices of Arm2 were truncated, a simple PBS containing only
APC/CPC was generated such that the other longitudinal half PBS with the APC
disks harboring the terminal emitters (LCM and ApcD) was lost. The cell
spectra showed a high fluorescence of these mutants at 660 nm
(Supplementary Fig. 5A–C) that disappeared completely at 683 nm
(Supplementary Fig. 5D–F). Upon excitation at 430 nm and at 580 nm, the
corresponding fluorescence confirmed that the energy transfer from APC to the
terminal emitters of PBSs was disrupted [19]. Moreover, due to the absence of
terminal emitters, the energy transfer from the PBSs to PSs was disrupted
(Supplementary Fig. 5). The photosynthetic capacities
(Supplementary Table 3) were much lower than those of
Therefore, the Arm2 element is responsible for the longitudinal assembly of PBSs. The loss of the element results in the dissociation of half of the PBS harboring the terminal emitters, while the other half, i.e., the simple PBS with only APC and CPC is retained in the cells. The cell spectra showed the absence of 77 K fluorescence at 684–685 nm corresponding to the terminal emitters (Supplementary Fig. 6, Supplementary Table 5). Hence, the dissociated parts of the PBSs could be degraded via specific phycobiliprotein degradation routes [38, 39]. One terminal emitter, ApcD, could undergo de-chromophorylation for its degradation [22]. The other terminal emitter, LCM, degraded slowly, and hence, a little LCM (the small peak at ~670 nm in Fig. 2 and Supplementary Fig. 6) was detected in the isolated PBSs from some mutants of the damaged Arm2 element.
To further investigate the roles of the motif of Arm2(6–36), we isolated the
PBSs of
The ultracentrifugation of
Conversely,
Cyanobacterial NPQ and state transitions affect the balance between energy transfer and dissipation within the PBSs and the distribution of excitation energy from the PBSs to the PSs [41]. Experimentally, states I and II are induced by blue LL and orange LL irradiation, respectively, such that the energy is preferentially transferred to photosystem I (PSI) and from PBS to PSII [42]. When state transitions occur at a low irradiation intensity, the high irradiation intensity quenches the non-photochemical PBSs, triggered by OCP [19, 43].
Pulse amplitude-modulated fluorometer (PAM) fluorescence technique is often used
to study the energy transfer between PBSs and PSs. In PAM measurements,
dark-adapted cells have a low dark maximal fluorescence (Fmd), but when they
are continuously irradiated by blue LL (55 µmol photons m2/s),
the Fm′ increases rapidly and arrives at a maximal Fm′ fluorescence
level (Fmb′), indicating that the cells have undergone the transition to
state I. Thus, the ratio of Fvb/Fvd (FV = Fm – F0)
indicated the ability of mutants to transform state II
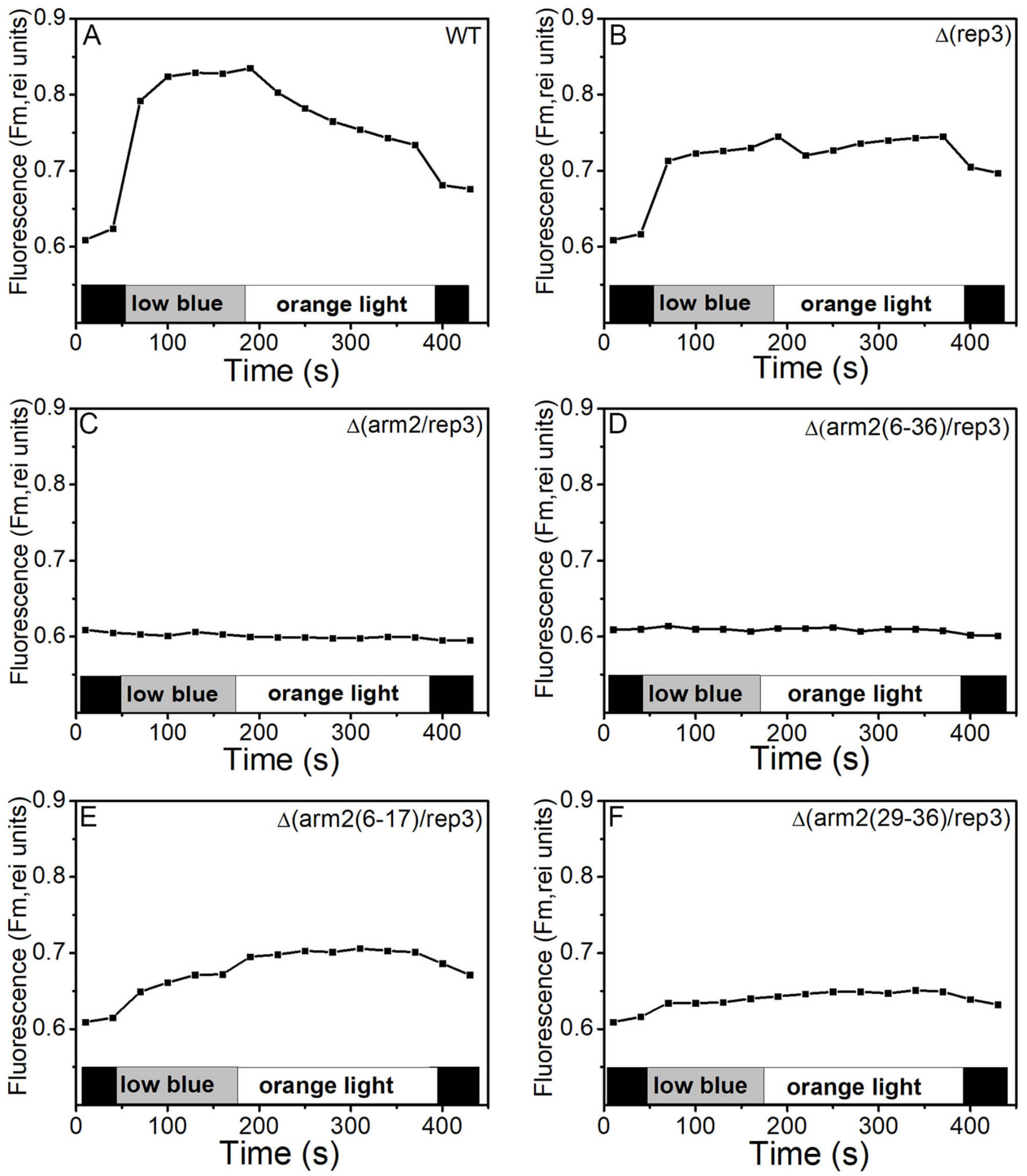 Fig. 3.
Fig. 3.
State transitions in WT,
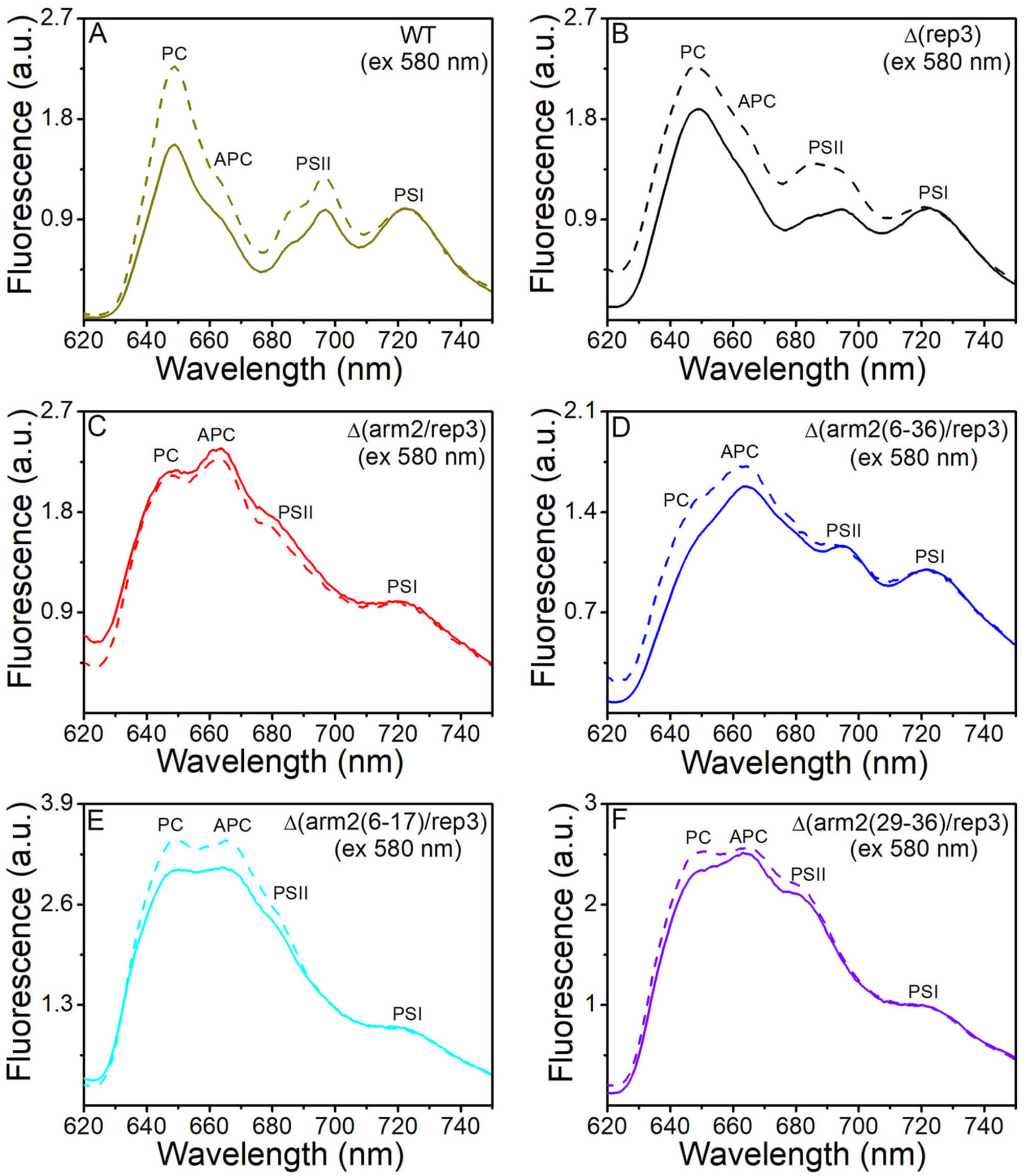 Fig. 4.
Fig. 4.
State transitions in WT,
Next, the NPQ performance of WT and mutants was compared (Fig. 5). The results
showed strong blue light-induced PBS fluorescence quenching (about 14%) in WT,
while that in
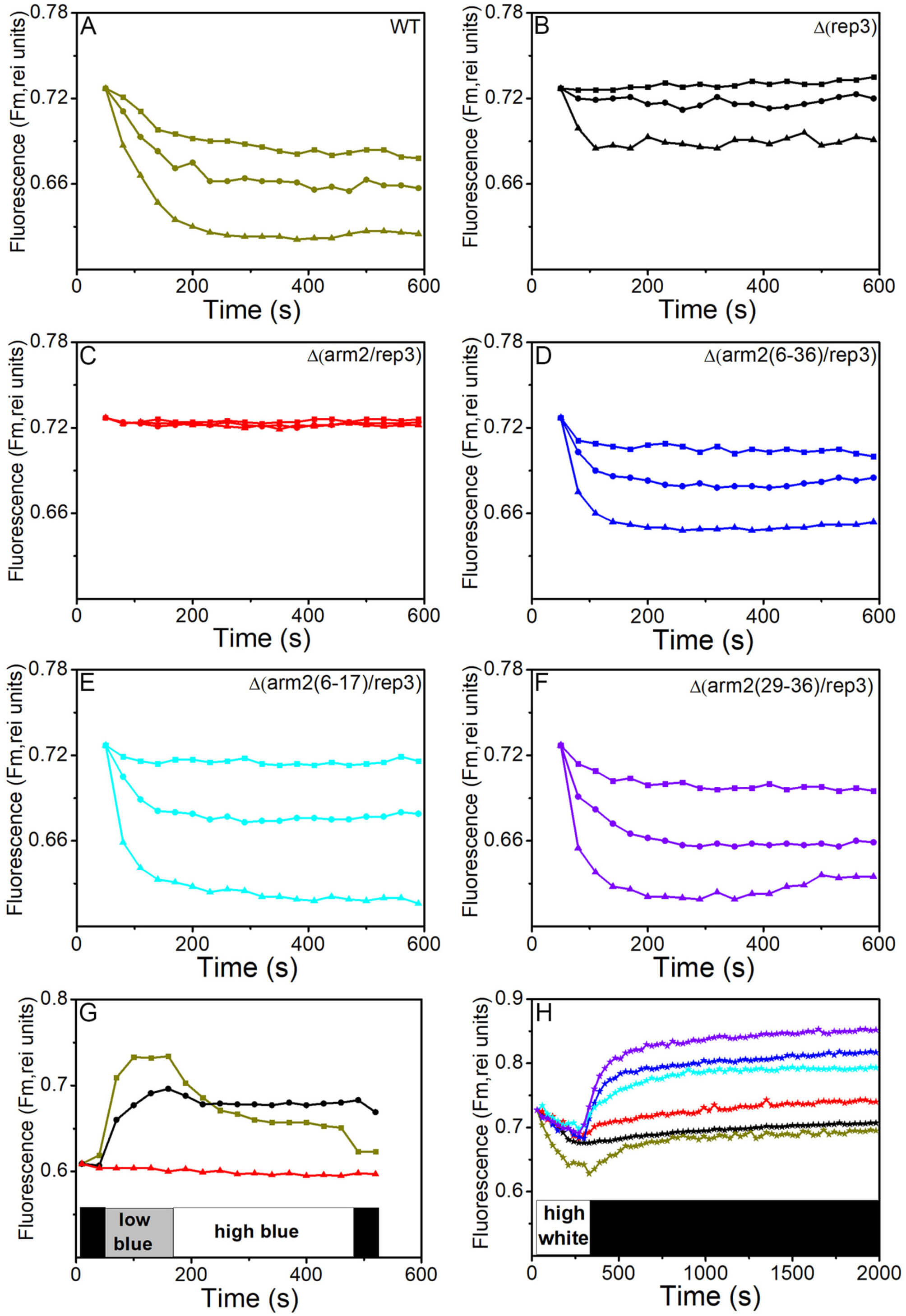 Fig. 5.
Fig. 5.
Changes in fluorescence levels induced by different intensities
of blue light and dark recovery of quenched fluorescence measured in a PAM
fluorometer. Dark-adapted Synechocystis WT cells (dark yellow, A),
Our study establishes the critical role of the Arm2 helix-loop-helix element in the longitudinal assembly and functional integrity of the PBS in Synechocystis sp. PCC 6803. The structural and functional defects observed in our Arm2 truncation mutants provide new insights into PBS architecture, state transitions, and photoprotection.
Structural Role of Arm2 and PBS Assembly. Our data demonstrate that the helix-loop-helix element of Arm2 (residues 6–36) is essential for connecting the two longitudinal halves of the PBS core. Truncation of this element leads to the specific loss of the half-core containing the terminal emitters LCM and ApcD, resulting in simplified APC/CPC complexes (Fig. 2, Supplementary Figs. 5,6). This phenotype aligns with the proposed location of Arm2 in the Griffithsia pacifica PBS structure [8], where it bridges APC hexamers within a basal cylinder. Our previous study, informed by homology modeling [12] and based on low-resolution molecular structure of cyanobacterial PBS [48], positions the Synechocystis Arm2 helix-loop-helix similarly, providing a structural basis for its essential role in core integrity. The CPC/APC molar ratio of ~1.66 in these simplified PBSs (Supplementary Table 2) is consistent with a structure where one APC hexamer associates with three CPC hexamers, a configuration potentially favored in the absence of the full core constraint. The subsequent degradation of the dissociated emitter-half suggests active quality control mechanisms for unassembled PBS components [38, 39]. Beyond the core, the Arm2(37–67) region, though unstructured, appears to facilitate the attachment of CPC rods to the core (Supplementary Fig. 10), highlighting the multi-functional nature of this linker domain. In contrast, the distal regions of Arm2(68–129) have minimal impact on assembly under our conditions.
Implications for State Transitions. A key finding is the abolition of state transitions in mutants lacking the Arm2 helix-loop-helix element (Figs. 3,4). The current understanding of state transitions involves redistribution of excitation energy between PSII and PSI, potentially through PBS movement [42, 49]. While some models propose lateral PBS displacement or detachment from the membrane [49, 50], our results suggest that longitudinal rearrangements within the PBS core itself, facilitated by Arm2, are crucial. The loss of state transitions in our mutants likely stems from a dual defect: (1) the physical absence of one terminal emitter-bearing half-core, which disrupts the energy delivery network to both photosystems, and (2) the weakened association of the simplified PBSs with the thylakoid membrane, potentially impeding the physical mobility or conformational changes required for state transitions. Our data do not rule out detachment/reattachment mechanisms [49, 50] but emphasize that the integrity of the core structure, maintained by Arm2, is a prerequisite for these processes.
OCP-mediated NPQ and PBS Integrity. The mutants with damaged Arm2 exhibited faster recovery from OCP-related NPQ (Fig. 5H), despite retaining significant quenching capacity. This accelerated recovery is consistent with a model where the simplified PBSs, due to their altered conformation and/or reduced size, bind the activated OCP with lower affinity, allowing for its more rapid release upon signal cessation. The core region, particularly the APC cylinders, is a proposed binding site for OCP [51, 52, 53]. Structural alterations in this region, as caused by Arm2 truncation, could directly affect OCP docking stability. While this interpretation is plausible, direct evidence from OCP binding affinity assays with purified WT and mutant PBSs would be needed to conclusively support this hypothesis, representing an important direction for future research.
In summary, the Arm2 linker in LCM acts as a critical structural organizer for the PBS core. Its helix-loop-helix element is indispensable for longitudinal assembly, state transitions, and fine-tuning OCP-related photoprotection, underscoring the importance of linker domains in the structure and function of massive photosynthetic antenna complexes.
This study demonstrates that the helix-loop-helix element of Arm2 is critical
for the longitudinal assembly of PBS. Deletion of the Rep3 domain resulted in an
intact PBS core comprising only Cylinder1 or 2. Additionally, when one or both
helices of Arm2 are truncated, simple PBS containing only APC/CPC could be
generated, while the other longitudinal half of PBSs containing the terminal
emitters (LCM and ApcD) may be degraded and lost. Consequently, in the
isolated
AP-B, allophycocyanin B; APC, allophycocyanin; ApcE, apo-protein of LCM; Chl, chlorophyll; CPC, cyanobacterial phycocyanin; F0, minimal fluorescence level; Fm, maximal fluorescence level in dark-adapted samples; Fm′, maximum fluorescence under illumination; Fs, steady-state fluorescence level; Fmd, maximum fluorescence in darkness; Fmb′, maximum fluorescence under blue-light illumination; FV, variable fluorescence = Fm – F0; Fvd, variable fluorescence in darkness; Fvb, variable fluorescence under blue-light illumination; HL, high light; KPB, potassium phosphate buffer; LCM, phycobilisome core-membrane linker; LL, low light; NPQ, non-photochemical quenching; OCP, orange carotenoid protein; PAM, a pulse amplitude-modulated fluorometer; PBS, phycobilisome; PCB, phycocyanobilin; PSI and PSII, photosystem I and II; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
The data supporting the results are included in this article and the supporting information files.
NNN, NC, HHF and XLD contributed to the study conception and design. NNN and NC carried out the studies, participated in collecting data, and drafted the manuscript. HHF and XLD performed the statistical analysis and participated in its design. NNN participated in acquisition, analysis, or interpretation of data and draft the manuscript. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the Key Research Projects of Higher Education Institutions in Henan Province (26B230010) and the Research Project of Luoyang Polytechnic (2024046).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL47370.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.