






 , Yuemei Zhao 2,†, Shouhui Zhong 2, Lina Chen 2, Xing-Yue Wang 3, Anqi Zheng 2, Jiwei Zuo 2, Mingjun Wu 4, Qiao Cheng 5,*
, Yuemei Zhao 2,†, Shouhui Zhong 2, Lina Chen 2, Xing-Yue Wang 3, Anqi Zheng 2, Jiwei Zuo 2, Mingjun Wu 4, Qiao Cheng 5,* , Kai-Fu Tang 1,*
, Kai-Fu Tang 1,*1 Key Laboratory of Molecular Biology on Infectious Disease, Ministry of Education, Chongqing Medical University, 400016 Chongqing, China
2 Key Laboratory of Diagnosis and Treatment of Severe Hepato-Pancreatic Diseases of Zhejiang Province, The First Affiliated Hospital of Wenzhou Medical University, 325015 Wenzhou, Zhejiang, China
3 Department of Nuclear Medicine, The Second Affiliated Hospital of Nanjing Medical University, 210003 Nanjing, Jiangsu, China
4 Institute of Life Science, Chongqing Medical University, 400016 Chongqing, China
5 Department of Breast and Thyroid Surgery, The First Affiliated Hospital of Chongqing Medical University, 400016 Chongqing, China
†These authors contributed equally.
Abstract
Transcription of the hepatitis B virus (HBV) is regulated by the acetylation status of H3/H4 histones bound to the covalently closed circular DNA (cccDNA) minichromosome. Thus, this study aimed to investigate whether sirtuin 7 (SIRT7), an NAD⁺-dependent acetylated lysine 18 of histone H3 (H3K18Ac) deacetylase, represses HBV transcription by catalyzing H3K18 deacetylation on the cccDNA minichromosome.
We investigated the effects of SIRT7 overexpression/knockdown on HBV transcription and histone acetylation in HepG2.2.15 cells, which constitutively express HBV, and HBV-infected HepG2-NTCP cells. Viral RNA levels and hepatitis B surface and e antigen levels were quantified using reverse transcription quantitative polymerase chain reaction and enzyme-linked immunosorbent assays, respectively. Meanwhile, Western blotting was used to determine protein expression levels. The association between DNA damage-binding protein 1 (DDB1) and cccDNA and the acetylation status of cccDNA minichromosome-associated H3K18 were analyzed using chromatin immunoprecipitation. Co-immunoprecipitation was used to detect protein–protein interactions. Data were statistically analyzed using Student’s t-test.
This study demonstrated that SIRT7 enhanced rather than repressed HBV transcription. Mechanistically, SIRT7 promoted HBV transcription by upregulating DDB1, a protein essential for HBV transcription, and by facilitating the recruitment of DDB1 to the cccDNA minichromosome. SIRT7 knockdown inhibited DDB1 expression and reduced DDB1 enrichment on the cccDNA, thereby suppressing HBV transcription. Conversely, SIRT7 overexpression upregulated DDB1 expression and enhanced DDB1–cccDNA binding, eventually promoting HBV transcription.
SIRT7 enhances HBV transcription by upregulating DDB1 expression and promoting an association between DDB1 and cccDNA.
Keywords
- hepatitis B virus
- gene expression regulation
- sirtuin 7
- DDB1
- histone acetylation
Hepatitis B virus (HBV) infection, which frequently progresses to severe clinical outcomes, including liver cirrhosis, hepatic failure, and hepatocellular carcinoma, continues to pose a substantial challenge to global public health [1, 2, 3, 4]. Alarmingly, hepatitis B- and C-related mortality increased by approximately 200,000 cases between 2019 and 2022, underscoring the urgent need for improved therapeutic interventions [5]. HBV is a hepatotropic DNA virus that maintains persistent infection through its unique genomic architecture, that is, a 3.2-kb, partially double-stranded, relaxed circular DNA that converts into covalently closed circular DNA (cccDNA) post-infection [6, 7]. This nuclear-resident cccDNA forms minichromosomes by associating with host histones and chromatin remodelers, serving as the transcriptional template for viral RNAs [8, 9, 10].
The epigenetic plasticity of cccDNA minichromosomes constitutes a central mechanism of HBV persistence [11]. HBV transcription is regulated by the acetylation status of H3/H4 histones bound to cccDNA. Class I histone deacetylase inhibitors induce hyperacetylation of cccDNA-associated H4 and enhance HBV transcription [12]. Dynamic histone post-translational modifications, including activating marks (H3K4me3 and H3K27ac) and repressive marks (H3K9me3 and H3K27me3), contribute to viral transcription and the outcome of chronic HBV infection [11, 12, 13, 14]. The viral HBx protein hijacks the epigenetic networks by recruiting host acetyltransferases to enhance histone acetylation and promote viral transcription [11, 13].
Sirtuins (SIRTs), a conserved family of NAD+-dependent histone deacetylases [15], play pivotal roles in regulating diverse cellular processes in various diseases, including HBV infection [16, 17, 18]. SIRTs can regulate HBV transcription via multiple molecular mechanisms [19]. For example, during HBV infection, SIRT3, SIRT4, and SIRT6 suppress viral replication [20, 21, 22]. Upregulation of SIRT1, SIRT2, SIRT5, and SIRT7 expression in HBV-infected hepatocytes results in widespread reduction of histone acetylation [23, 24]. SIRT7 is an NAD+-dependent acetylated lysine 18 of histone H3 (H3K18Ac) deacetylase [25]. Genome-wide binding analyses have demonstrated that SIRT7 binds to specific target gene promoters to mediate transcriptional repression through H3K18Ac deacetylation [26, 27]. Despite evidence suggesting that SIRT7 suppresses HBV transcription by desuccinylating histone H3 on cccDNA minichromosomes [28], it is not clear whether SIRT7 also inhibits HBV transcription through H3K18Ac deacetylation. Therefore, in this study, we aimed to investigate whether SIRT7 represses HBV transcription by catalyzing H3K18 deacetylation on the cccDNA minichromosome.
HepG2 human hepatocellular carcinoma cells and HBV-expressing HepG2.2.15 cells were obtained from Shanghai Xinyu Biotechnology Co., Ltd. (Shanghai, China). HepAD38 and HepG2 NTCP cell lines were provided by Prof. Deqiang Wang (Chongqing Medical University). All cells were cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco) at 37 °C in a humidified 5% CO2 atmosphere. All cell lines were authenticated using short tandem repeat profiling, and they tested negative for Mycoplasma contamination.
The SIRT7-overexpression plasmid Flag-SIRT7 has been described previously [25]. DNA damage-binding protein 1 (DDB1)-overexpression plasmid was provided by Prof. Bing Ni (Third Military Medical University, Chongqing, China). The pcDNA3.1 vector served as an empty control. The sequences of siRNAs used in this study are detailed in Supplementary Table 1. Plasmids and siRNAs were transfected into cells with Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions.
HBV particles were prepared as previously described [29]. Briefly, the supernatant from HepAD38 cells was concentrated by adding 4% PEG8000 and incubating at 4 °C overnight. The precipitate was collected using centrifugation and resuspended in Opti-MEM to approximately 1% of the original volume. For infection, HepG2-NTCP cells were incubated with HBV at a multiplicity of infection of 1000 genome equivalents per cell in the presence of 4% PEG8000 for 24 h at 37 °C. Subsequently, the cells were washed three times with PBS and maintained in fresh medium for another 24 h.
Cells were lysed using RIPA buffer containing protease inhibitor cocktails
(Roche; Basel, Switzerland). After centrifugation (12,000
Total RNA was extracted from cells with TRIzol reagent (Life Technologies, Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, cells were lysed with TRIzol, and then subjected to chloroform and phase separation via centrifugation. The RNA in the aqueous phase was precipitated with isopropanol and collected using centrifugation. The resulting RNA pellet was washed with 75% ethanol, briefly air-dried, and then dissolved in RNase-free water. The purified RNA was stored at –80 °C for subsequent analysis. RNA concentration and quality were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific), with an A260/A280 ratio of approximately 2.0 indicating high-purity RNA.
cDNA was generated from total RNA using the PrimeScript RT reagent kit (R323-01; Vazyme Biotech Co., Ltd., Nanjing, Jiangsu, China) according to the manufacturer’s instructions. The resulting cDNA was diluted 1:5 with nuclease-free water and stored at –20 °C until use. Quantitative polymerase chain reaction (qPCR) was performed using SYBR qPCR Master Mix (Vazyme Biotech) on an ABI 7500 FAST system (Life Technologies). Primer sequences utilized in this study are provided in Supplementary Table 2.
Cells were cross-linked with 1% formaldehyde for 10 min at room temperature,
and the reaction was quenched by adding 0.125 M glycine for 5 min. After two
washes with PBS, the cells were resuspended in SDS lysis buffer supplemented with
protease inhibitors (100 µL per 1
Secreted levels of HBsAg and HBeAg in the culture supernatant were quantified using commercial enzyme-linked immunosorbent assay (ELISA) kits (KeHua Biotech, Shanghai, China) according to the manufacturer’s instructions. Briefly, samples and standards were added to 96-well plates pre‑coated with capture antibodies specific to HBsAg or HBeAg and incubated for 1 h at 37 °C. After washing, HRP-conjugated detection antibodies were added, and the samples were incubated for 30 min at 37 °C. The color reaction was developed with 3,3′,5,5′-tetramethylbenzidine substrate and stopped by adding 2 M H₂SO₄. Absorbance at 450 nm was measured using a multifunctional microplate reader (Thermo Fisher Scientific). The concentrations of HBsAg and HBeAg were calculated using a standard curve.
The remaining supernatant was divided into 400-µL aliquots and
precleared with Protein A/G magnetic beads (Selleck Chemicals) at 4 °C
for 1 h. The beads were removed using a magnetic rack. The precleared supernatant
was incubated overnight at 4 °C with anti-SIRT7, anti-DDB1, or control
IgG. Protein A/G magnetic beads, pre-washed with IP buffer, were added to the
antibody-treated supernatants, which were then rotated at 4 °C for 2–3
h. After three washes with IP buffer, the bead-bound proteins were eluted by
heating in 1
Protein–protein interaction networks were constructed using data from the BioGRID (https://thebiogrid.org/) and STRING (https://www.string-db.org/) databases. Gene expression correlation analysis was performed with the GEPIA2 web tool (http://gepia2.cancer-pku.cn/).
All experiments were independently repeated at least three times. Data are
presented as mean
To determine whether SIRT7 suppresses HBV transcription by catalyzing H3K18 deacetylation on the cccDNA minichromosome, we first knocked down SIRT7 in HepG2.2.15 cells and HBV-infected HepG2-NTCP cells (Fig. 1A). Western blotting showed that SIRT7 knockdown elevated global H3K18 acetylation levels (Fig. 1A). Similarly, the ChIP assay indicated that SIRT7 knockdown enhanced H3K18 acetylation, specifically on the cccDNA minichromosome (Fig. 1B). Conversely, SIRT7 overexpression reduced the acetylation of both global and cccDNA-bound H3K18 (Fig. 1C,D).
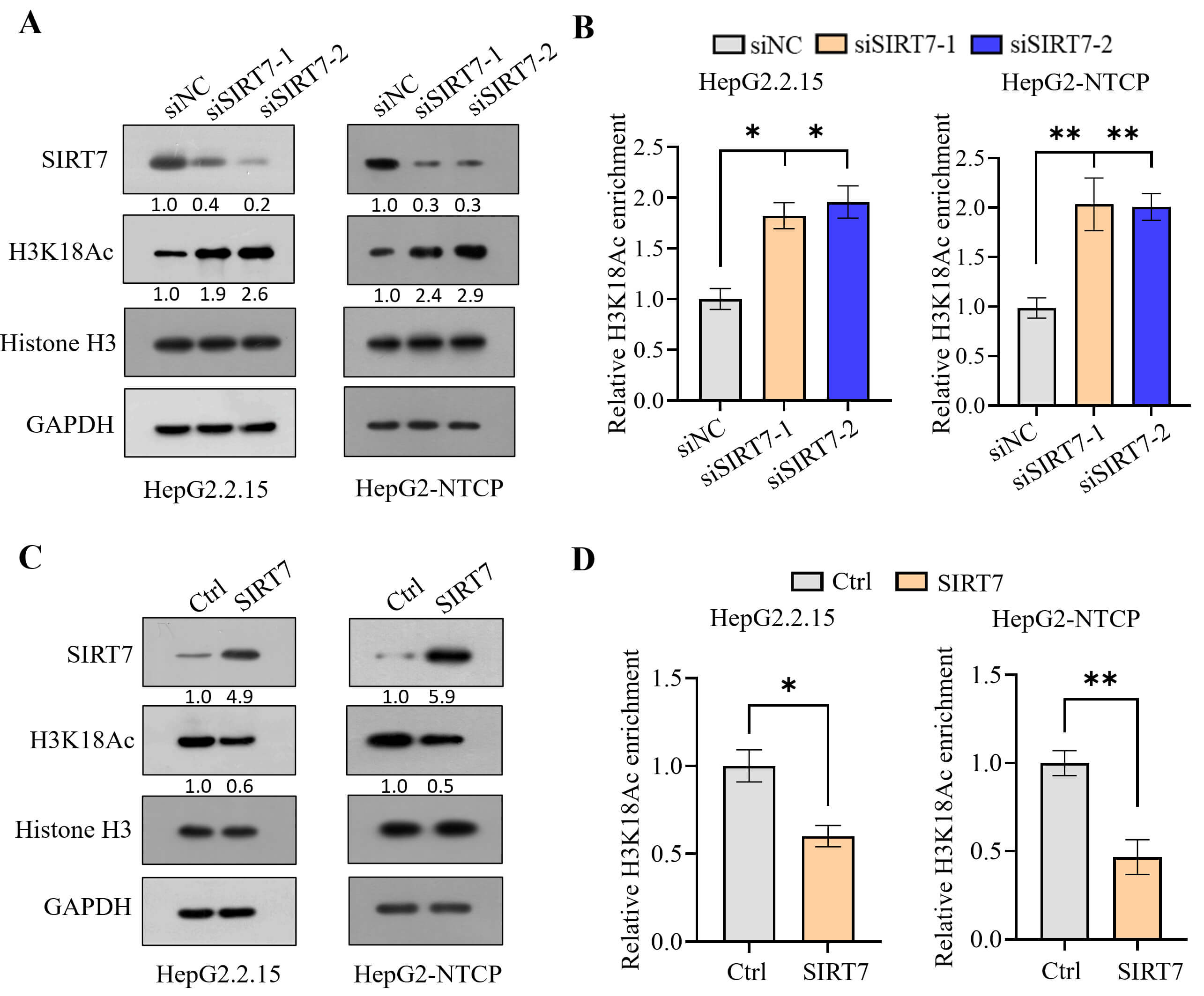 Fig. 1.
Fig. 1.
Sirtuin 7 (SIRT7) suppresses histone acetylation. (A,B) Changes
in H3K18 acetylation levels in HepG2.2.15 cells and Hepatitis B virus
(HBV)-infected HepG2-NTCP cells following SIRT7 knockdown, as detected
using western blotting (A) and Chromatin Immunoprecipitation (ChIP) assay (B).
(C,D) SIRT7 overexpression decreased H3K18 acetylation, as detected
using western blotting (C) and ChIP assay (D). *p
Contrary to our initial hypothesis, ELISA showed that SIRT7 knockdown significantly reduced the secretion of both HBeAg and HBsAg into the culture supernatant (Fig. 2A). Correspondingly, the qPCR analysis indicated decreased expression of total HBV RNA and the 3.5-kb pre-core/pre-genome (pC/pg) RNA (Fig. 2B). Consistent with the loss-of-function results, SIRT7 overexpression increased the levels of viral antigens and RNAs (Fig. 2C,D). The ChIP assay revealed that SIRT7 knockdown and overexpression decreased and increased RNA polymerase II occupancy on cccDNA, respectively (Fig. 2E,F). Moreover, we found that SIRT7 overexpression increased the level of histone H3 lysine 36 trimethylation (H3K36me3)—a marker of transcriptional elongation—on cccDNA, whereas its knockdown decreased this modification (Fig. 2G,H). These findings suggest that SIRT7 facilitates HBV transcription.
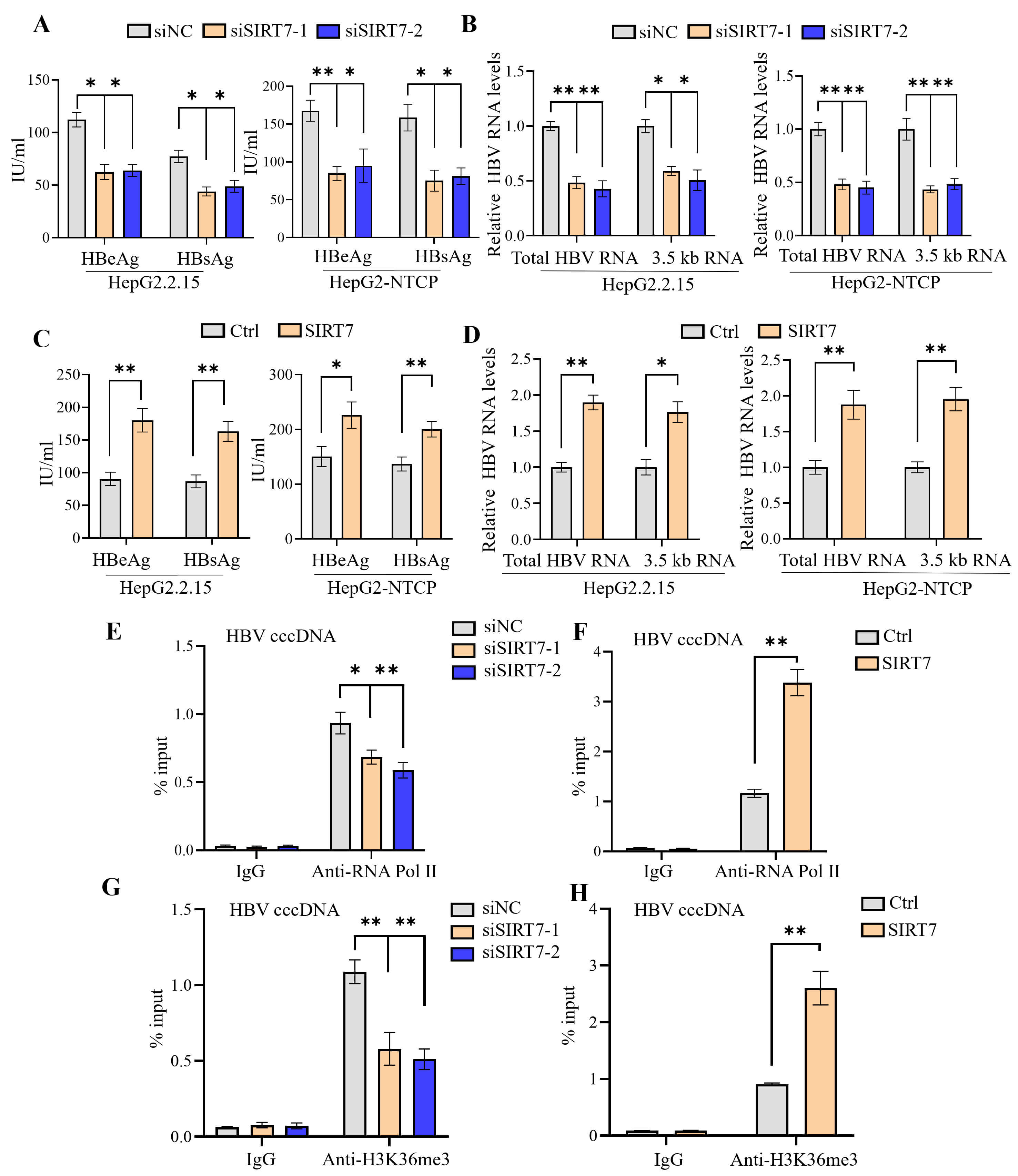 Fig. 2.
Fig. 2.
SIRT7 promotes HBV transcription. (A,B) Following
siRNA-mediated SIRT7 depletion, HBeAg and HBsAg secretion in culture supernatants
was quantified using ELISA (A). qPCR analysis of total HBV RNA and 3.5-kb pC/pg
RNA (B). (C,D) ELISA-based analysis of secreted HBeAg/HBsAg (C) and qPCR analysis
of total HBV RNA and 3.5-kb pC/pg RNA levels (D), which revealed the functional
consequences of SIRT7 overexpression. (E,F) ChIP assay of RNA polymerase
II occupancy on cccDNA in SIRT7-knockdown (E) or
SIRT7-overexpressing HBV-infected HepG2-NTCP cells (F). (G,H) ChIP assay
of the levels of H3K36me3 on cccDNA in SIRT7-knockdown (G) or
SIRT7-overexpressing HBV-infected HepG2-NTCP cells (H). *p
To elucidate the molecular mechanism by which SIRT7 promotes HBV transcription, we first identified potential SIRT7-interacting proteins by screening the BioGRID and STRING databases. Preliminary data suggested that SIRT7 physically interacts with DDB1, an adaptor protein of the CUL4 E3 ubiquitin ligase complex (Fig. 3A). The co-immunoprecipitation analysis revealed an interaction between SIRT7 and DDB1 (Fig. 3B,C). The bioinformatic analysis using the GEPIA2 tool demonstrated that the mRNA levels of SIRT7 positively correlated with those of DDB1 in various human tissues, including the liver tissue (Fig. 3D and Supplementary Fig. 1). SIRT7 knockdown led to reduced DDB1 expression at both the protein (Fig. 3E) and mRNA levels (Supplementary Fig. 2A), whereas SIRT7 overexpression had the opposite effect (Fig. 3F and Supplementary Fig. 2B). The ChIP assay revealed the binding of DDB1 to cccDNA, as well as attenuation of this interaction upon SIRT7 knockdown (Fig. 3G).
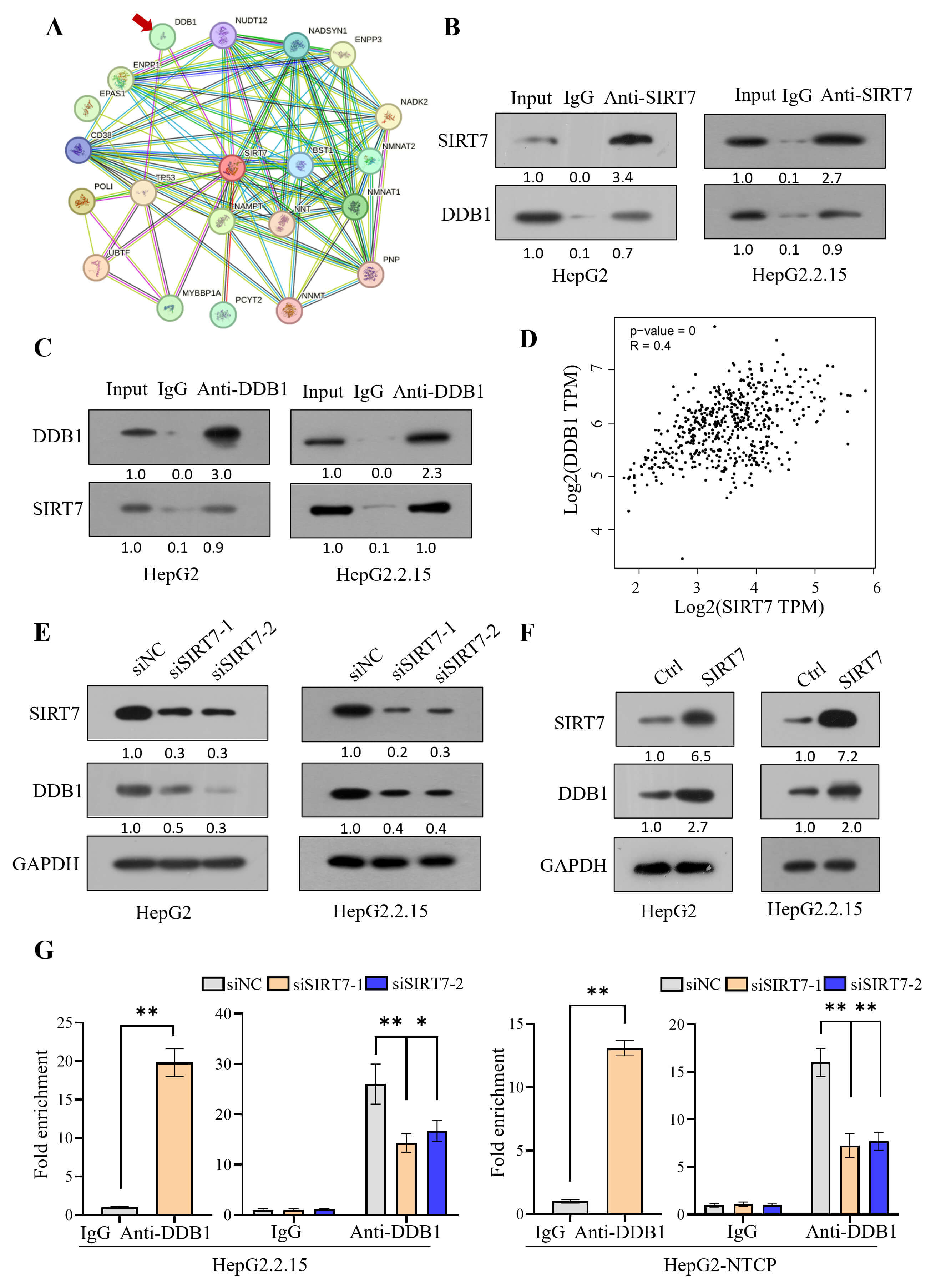 Fig. 3.
Fig. 3.
SIRT7 interacts with DDB1 to regulate HBV transcription. (A)
Bioinformatic analysis (BioGRID/STRING databases) identified DNA damage-binding
protein 1 (DDB1) (indicated by red arrow) as a potential SIRT7-interacting
protein. (B,C) Reciprocal co-immunoprecipitation assays using anti-SIRT7 (B) and
anti-DDB1 (C) antibodies confirmed the interaction between SIRT7 and DDB1 in
HepG2 and HepG2.2.15 cells. (D) GEPIA2 revealed a significant positive
correlation between SIRT7 and DDB1 expression in liver tissues. (E,F) Modulation
of SIRT7 expression regulates DDB1 protein levels in HepG2 (left) and
HepG2.2.15 (right) cells. (G) ChIP demonstrated DDB1 binding to HBV cccDNA, which
was attenuated upon SIRT7 knockdown. *p
Consistent with our previous study results [30], DDB1 knockdown led to repressed HBV transcription (Fig. 4A,B), whereas DDB1 overexpression had the opposite effect (Fig. 4C,D). To investigate whether SIRT7 facilitates HBV transcription by promoting the interaction between DDB1 and cccDNA, we overexpressed DDB1 in SIRT7-knockdown cells (Fig. 5A). The ELISA showed that DDB1 overexpression partially reversed the inhibitory effect of SIRT7 knockdown on HBeAg and HBsAg secretion (Fig. 5B), whereas the qPCR analysis indicated that DDB1 overexpression alleviated the inhibitory effect of SIRT7 knockdown on total HBV RNA and 3.5-kb pC/pg RNA expression (Fig. 5C).
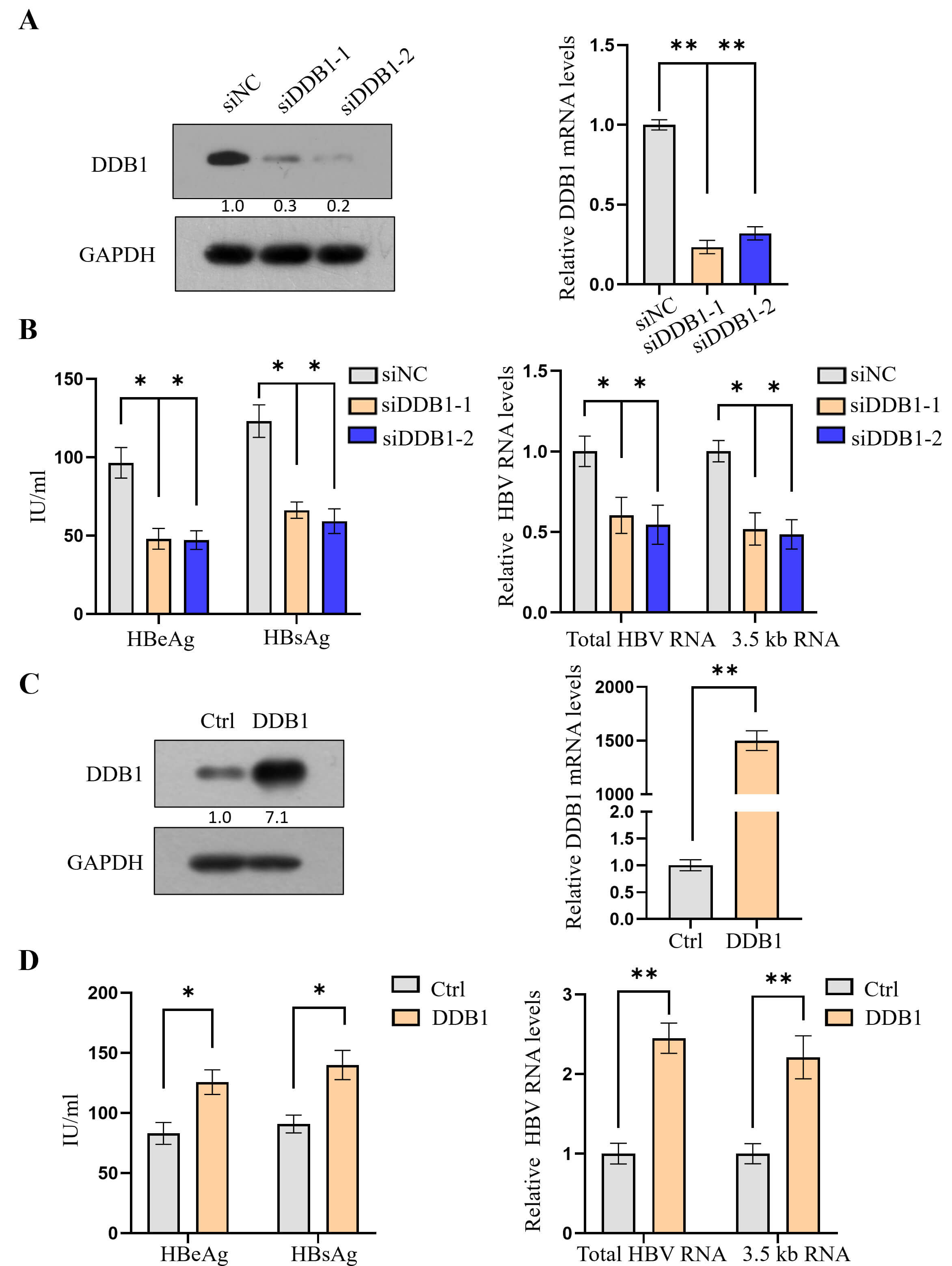 Fig. 4.
Fig. 4.
DDB1 mediates the SIRT7-dependent regulation of HBV
transcription. (A) Effective knockdown of DDB1 in HepG2.2.15 cells was
confirmed using western blotting and qPCR analysis. (B) DDB1 depletion
significantly reduced HBV transcription, as evidenced by decreased HBeAg/HBsAg
secretion and viral RNA levels. (C) Successful ectopic expression of
DDB1 was verified using western blotting. (D) DDB1
overexpression enhanced HBV transcription, increasing both viral antigen and RNA
levels. *p
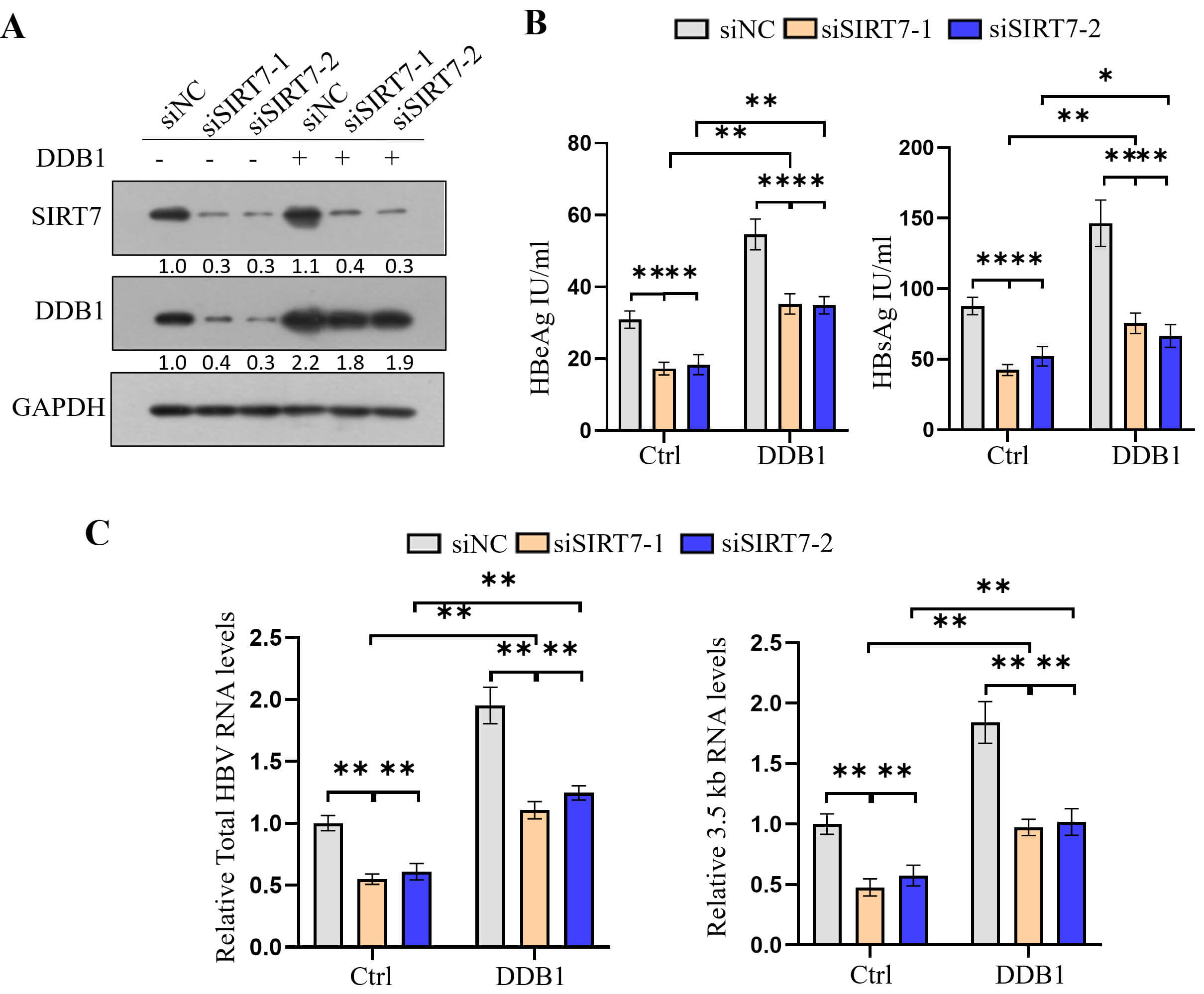 Fig. 5.
Fig. 5.
DDB1 overexpression rescues HBV transcription
suppression caused by SIRT7 knockdown. (A) Western blotting verified
simultaneous SIRT7 knockdown and DDB1 overexpression in
HepG2.2.15 cells. (B) ELISA showed that DDB1 overexpression rescued
SIRT7 knockdown-mediated reduction in HBeAg and HBsAg secretion. (C)
qPCR analysis demonstrated that DDB1 overexpression reversed
SIRT7 knockdown-induced decrease in total HBV RNA and 3.5-kb pC/pg RNA
levels. *p
To confirm that SIRT7 facilitated HBV transcription by promoting DDB1–cccDNA interaction, we knocked down DDB1 in SIRT7-overexpressing cells (Fig. 6A). The ELISA revealed that DDB1 knockdown repressed the SIRT7 overexpression-induced increase in HBeAg and HBsAg secretion (Fig. 6B), whereas qPCR analysis indicated that DDB1 knockdown blocked the SIRT7 overexpression-induced increase in total HBV RNA and 3.5-kb pC/pg RNA expression (Fig. 6C).
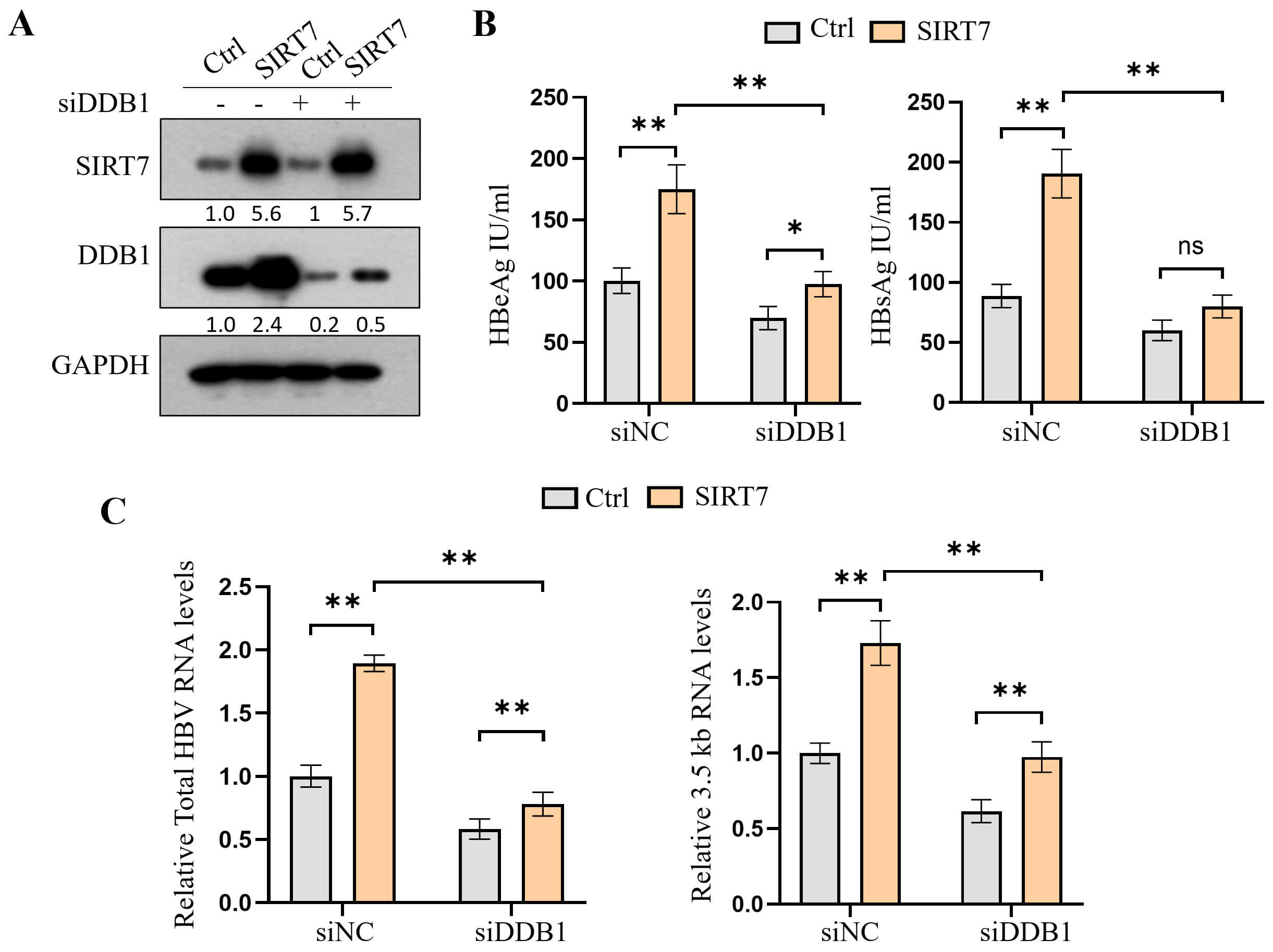 Fig. 6.
Fig. 6.
DDB1 knockdown reverses SIRT7
overexpression-induced promotion of HBV transcription. (A) Western blot showing
the levels of the indicated protein in HepG2.2.15 cells co-transfected with
SIRT7-overexpression plasmid and DDB1-specific siRNA. (B)
ELISA-based analysis showed that DDB1 depletion abolished SIRT7-mediated
increase in HBeAg and HBsAg secretion. (C) qPCR results demonstrating that
DDB1 knockdown reversed SIRT7-induced increase in total HBV RNA and
3.5-kb pC/pg RNA levels. *p
HBV cccDNA accumulates in the nucleus as chromatin-like minichromosomes assemble with histones and non-histone proteins [14]. Enhanced acetylation of H3/H4 histones bound to the cccDNA minichromosomes has been shown to promote HBV transcription [12]. In this study, although SIRT7 deacetylated H3K18 on the cccDNA minichromosome, we found that SIRT7 enhances HBV transcription, rather than repressing it. Mechanistically, we demonstrated that SIRT7 promotes HBV transcription by upregulating DDB1 expression and facilitating the recruitment of DDB1 to cccDNA minichromosomes.
DDB1 plays a pivotal role in HBV transcription, primarily by serving as a critical cellular adaptor hijacked by the viral HBx protein. HBx recruits DDB1 to assemble a CUL4–DDB1 E3 ubiquitin ligase complex, which is exploited to target host restriction factors for proteasomal degradation [31, 32, 33, 34]. A major substrate of this complex is the structural maintenance of chromosomes 5/6 complex, which strongly suppresses viral transcription from cccDNA. Degradation of this complex effectively relieves transcriptional repression, enabling robust HBV gene expression and transcription [31, 32, 33, 35]. Similarly, HBx–DDB1 targets WDR77, a subunit of protein arginine methyltransferase 5, for proteasomal degradation to reduce the levels of repressive histone marks on cccDNA and further amplify viral transcription [36]. Structural studies have confirmed the essential role of HBx–DDB1 interaction in HBV transcription [32]. Moreover, DDB1 can promote HBV transcription via an HBx-independent mechanism [37]. In this study, we demonstrated that DDB1 is associated with cccDNA; DDB1 knockdown suppressed HBV transcription, whereas DDB1 overexpression promoted HBV transcription.
The role of SIRT7 in HBV transcription remains controversial. Sekiba K et al. [31] reported that SIRT7 suppresses HBV transcription, whereas Deng et al. [19] reported that several SIRT family members, including SIRT7, can promote HBV transcription. Our results are consistent with those of Deng et al. [19]. Existing evidence and our findings suggest that SIRT7 paradoxically plays a dual role in regulating HBV transcription—promoting HBV transcription by upregulating DDB1 expression and facilitating DDB1 recruitment to the cccDNA minichromosome, while concurrently repressing HBV transcription through the following mechanisms. First, SIRT7 may suppress HBV transcription by catalyzing histone H3 desuccinylation on cccDNA minichromosomes [28]. Second, SIRT7 deacetylates DDB1 and suppresses activity of the CUL4–DDB1 E3 ubiquitin ligase complex [38]; therefore, SIRT7 may inhibit HBV transcription by suppressing the proteasomal degradation of host restriction factors. Finally, we showed that SIRT7 deacetylates H3K18 on cccDNA minichromosomes; hence, SIRT7 may inhibit HBV transcription by decreasing the acetylation of cccDNA-bound histones. Further studies are required to determine which mechanism dominates under which conditions.
Our study revealed that SIRT7 paradoxically promotes viral transcription, despite deacetylating H3K18 on the cccDNA minichromosome. Mechanistically, SIRT7 promotes DDB1 expression and facilitates its recruitment to cccDNA, which in turn enhances HBV transcription.
All data supporting the findings of this study are available within the article and are also accessible from the lead contact on request.
Conceptualization: FC, YMZ, KFT; Methodology: FC, YMZ, SHZ, LNC, XYW, AQZ, JWZ, MJW, QC; Investigation: FC, YMZ, SHZ, LNC, XYW, AQZ, JWZ, MJW, QC, KFT; Visualization: FC, YMZ; Data curation: FC, YMZ; Resources: KFT; Funding acquisition: KFT, MJW; Project administration: KFT, QC; Supervision: KFT, QC; Writing—original draft: FC; Writing—review & editing: KFT. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Prof. Bing Ni (Third Military Medical University, Chongqing, China) for providing the DDB1 overexpression plasmid and Prof. Deqiang Wang (Chongqing Medical University) for providing HepAD38 and HepG2 NTCP cells. We also wish to thank Editage for editing the manuscript.
This research was funded by the Chongqing Medical University Program for Youth Innovation in Future Medicine, grant number W0084.
The authors declare no conflict of interest. Given his role as editorial board member, Kai-Fu Tang had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Ananda Ayyappan Jaguva Vasudevan.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL46445.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.