








1 The Research Center for Bone and Stem Cells, Department of Anatomy, Histology and Embryology, Nanjing Medical University, 211166 Nanjing, Jiangsu, China
2 Calcium Research Laboratory, McGill University Health Centre and Department of Medicine, McGill University, Montreal, QC H4A 3J1, Canada
3 Department of Plastic Surgery, Affiliated Friendship Plastic Surgery Hospital of Nanjing Medical University, 210029 Nanjing, Jiangsu, China
Abstract
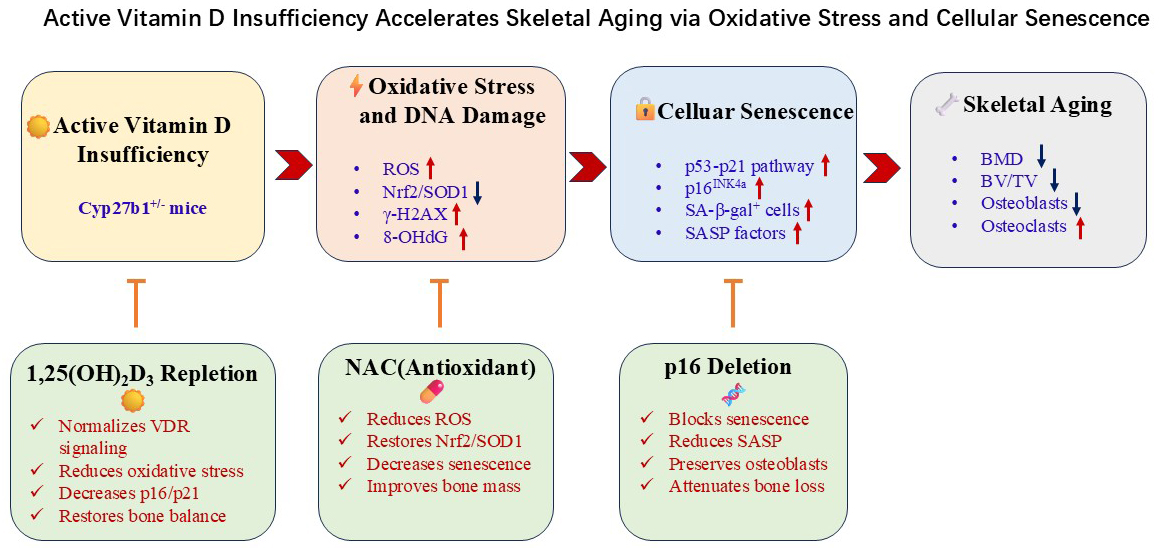
Vitamin D is essential for skeletal health, but its role in redox homeostasis and cellular senescence during aging in vivo is unclear. We therefore investigated whether active vitamin D insufficiency accelerates bone loss via oxidative stress and senescence pathways.
Male wild-type (WT) and Cyp27b1 haploinsufficient mice (modeling vitamin D insufficiency) were treated with N-acetylcysteine (NAC) or 1,25-dihydroxyvitamin D3 [1,25(OH)2D3]. Double-mutant p16-/-Cyp27b1+/- mice were used to assess the role of the tumor suppressor protein p16. Mice were maintained until 8 months of age in a specific pathogen-free facility. Outcomes included lifespan (n = variable per group, monitored daily); generation of oxidative stress (determined by serum malondialdehyde [MDA] levels via assay kit); generation of bone reactive oxygen species [ROS] (determined via flow cytometry), development of DNA damage (indicated by 8-hydroxy-2′-deoxyguanosine [8-OHdG] and γ-H2A.X generation and determined via immunohistochemistry and Western blot); and senescence (assessed by generation of β-galactosidase [β-gal], p16, and senescence-associated secretory phenotype [SASP] cytokines as determined via staining, blot, and real-time reverse transcription polymerase chain reaction). Additionally, bone microarchitecture was examined via micro-computed tomography and histomorphometry. Data from at least 5 mice per group were analyzed using unpaired Student’s t-test for two-group comparisons and two-way analysis of variance for multi-group comparisons, with significance at p < 0.05.
Compared with wild-type controls, Cyp27b1+/- mice showed a significantly shorter lifespan, higher oxidative stress, greater DNA damage, increased senescence markers, and lower trabecular bone volume (all p < 0.05). In Cyp27b1+/- mice, treatment with either N-acetylcysteine or 1,25(OH)2D3 significantly improved survival, reduced oxidative stress and DNA damage, attenuated senescence markers, and increased bone volume relative to untreated Cyp27b1+/- mice (p < 0.05 for all relevant comparisons; n = 5 per group). Genetic deletion of p16 in Cyp27b1+/-mice similarly increased bone volume and reduced senescence-associated readouts compared with Cyp27b1+/- controls (p < 0.05; n = 5).
Active vitamin D insufficiency accelerates skeletal aging in vivo through a pathway involving reactive oxygen species-DNA damage-p16/senescence-associated secretory phenotype. Antioxidants, vitamin D repletion, or p16 inhibition rescued bone loss, highlighting redox-senescence axes as potential therapeutic targets for osteoporosis.
Graphical Abstract
Keywords
- vitamin D
- oxidative stress
- p16 tumor suppressor protein
- cellular senescence
- osteoporosis
- N-acetylcysteine
Osteoporosis and age-related skeletal fragility remain major public health challenges driven by an imbalance between bone formation and resorption that emerges with aging [1, 2]. Accumulating evidence implicates oxidative stress and cellular senescence as central, convergent mechanisms that impair osteoblast function, enhance osteoclastogenesis, and damage bone microarchitecture [3, 4, 5, 6]. In parallel, vitamin D signaling via the vitamin D receptor (VDR) contributes to skeletal health not only through mineral homeostasis but also by modulating redox balance, DNA damage responses, and inflammatory programs in bone cells [7, 8, 9, 10]. However, how insufficiency of active vitamin D (1,25(OH)2D3) integrates with oxidative stress to trigger p53–p21 and p16INK4a checkpoints, establish a stable senescence-associated secretory phenotype (SASP), and accelerate skeletal aging in vivo remains incompletely defined.
In the aging skeleton, reactive oxygen species (ROS) activate the DNA damage response and checkpoint pathways that can culminate in senescence, with p21 often marking an initial arrest and p16INK4a enforcing a more durable state linked to SASP-mediated paracrine effects on bone remodeling [3, 4, 5, 6, 11, 12]. Independent studies demonstrate that enhancing antioxidant defenses or targeting senescent cells improves bone mass and strength in preclinical models, underscoring the therapeutic potential of redox–senescence modulation [4, 13, 14]. Vitamin D/VDR signaling intersects with chromatin remodeling and cell-cycle regulators and has been reported to influence oxidative stress handling in multiple cell lineages, suggesting a plausible route by which active vitamin D sufficiency might restrain senescence initiation and maintenance in bone [9, 10, 15, 16, 17, 18, 19]. Yet, direct in vivo evidence connecting active vitamin D insufficiency to increased skeletal ROS, DDR activation, and a transition from p21- to p16-dominant senescence programs is limited.
Here, using genetic and pharmacological perturbations, we delineate a vitamin D–redox–senescence axis that controls skeletal remodeling in vivo. We show that active vitamin D insufficiency elevates ROS, suppresses nuclear factor erythroid 2-related factor 2 (Nrf2)/superoxide dismutase 1 (SOD1) defenses, and increases DNA damage in bone, leading to augmented p16-associated senescence and SASP, impaired osteoblastogenesis, and increased osteoclast activity. Antioxidant supplementation or 1,25(OH)2D3 repletion reverses these changes, and genetic reduction of p16 attenuates bone loss by rebalancing formation and resorption. These findings integrate and extend prior observations on redox and senescence in skeletal aging and position active vitamin D sufficiency as a modifiable upstream determinant of bone health in aging.
Cyp27b1+/- mice were generated by intercrossing heterozygous parents
and genotyped using established protocols [20]. The p16-/- knockout mice
were kindly provided by Baojie Li (Shanghai Jiao Tong University, Shanghai,
China). The Cyp27b1+/-p16-/- double mutant mice were produced by sequential
breeding of Cyp27b1+/- and p16-/- mouse lines. After weaning at
postnatal day 21, Cyp27b1+/- mice were randomly assigned to one of three
intervention groups, with wild-type (WT) mice serving as controls. Group 1
received a standard rodent chow diet and sterile drinking water. To prevent
oxidation, group 2 was administered the antioxidant N-acetylcysteine (NAC; 1
mg/mL) in drinking water, which was refreshed every 48 hours. Group 3 received
subcutaneous injections of 1,25(OH)2D3 (0.1 µg/kg body weight) every other
day, prepared in ethanol and diluted in sterile phosphate-buffered saline (PBS;
final ethanol concentration
Serum malondialdehyde (MDA) levels, a marker of lipid peroxidation, were quantified using a commercial thiobarbituric acid reactive substances (TBARS) assay kit (#A003-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer’s instructions. Briefly, serum samples were mixed with reagents and incubated at 95 °C for 40 minutes. Absorbance was measured at 532 nm using a microplate spectrophotometer (Multiskan FC model, version 1.0 firmware; Thermo Fisher Scientific, Waltham, MA, USA). MDA concentrations were calculated based on a standard curve and normalized to total protein content.
ROS levels were measured in unfractionated total bone marrow cells prepared as a single-cell suspension that were flushed out from femurs of 8-momth-old WT, Cyp27b1+/-, Cyp27b1+/-+NAC, Cyp27b1+/-+1,25(OH)2D3 mice, by flow cytometry (Becton Dickinson, Heidelberg, Germany) based on fluorescence intensity measurements, as described previously [21].
Lumbar spine specimens from 8-month-old WT and Cyp27b1+/- mice were harvested and fixed in PLP fixative for 24 hours. Fixed samples were rinsed in PBS and scanned using X-ray radiography (Faxitron Bioptics, Tucson, AZ, USA) and micro-computed tomography (SkyScan 1276 model, Scan software version 1.18; Bruker MicroCT, Kontich, Antwerp, Belgium). Scanning parameters were set at 50 kV, 200 µA, and 9-µm voxel resolution. Three-dimensional reconstruction and quantitative analysis—including bone volume fraction (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp)—were performed using CTAn software (Bruker). Analyses followed standardized guidelines for bone microarchitecture assessment [22, 23].
Vertebral bone specimens were decalcified in 10% EDTA (pH 7.4) for 2–3 weeks,
processed through graded alcohols, embedded in paraffin, and sectioned at
5-µm thickness. Tissue sections were stained with hematoxylin and eosin
(H&E) for general histology, histochemically for total collagen, naphthol AS-MX
phosphate with fast Red TR for alkaline phosphatase (ALP) activity to identify
osteoblasts, followed by counterstaining with Vector methyl green as a nuclear
counterstain [24], and naphthol AS-BI phosphate with fast red violet LB for
tartrate-resistant acid phosphatase (TRAP) to identify osteoclasts. For
immunohistochemistry, antigen retrieval was performed using citrate buffer (pH
6.0) under heat-induced conditions. Sections were blocked with 5% normal goat
serum and incubated overnight at 4 °C with primary antibodies against
superoxide dismutase 1 (SOD1; catalog no. ab16831, lot #GR123456, dilution
1:200; Abcam, Cambridge, UK), 8-hydroxy-2′-deoxyguanosine (8-OHdG; catalog
no. ab48508, lot #GR234567, dilution 1:100; Abcam, Cambridge, UK), and
Proteins were extracted from vertebral bone tissues using RIPA lysis buffer
supplemented with protease and phosphatase inhibitors (Roche). Lysates were
homogenized, centrifuged (12,000
Total RNA was extracted from vertebral bone tissue using Trizol Reagent
(Invitrogen) according to the manufacturer’s protocol. RNA concentration and
purity were assessed by spectrophotometry (A260/A280 ratio
| Name | S/AS | Sequence (5′ to 3′) | Tm (°C) | bp |
| SOD1 | S | ATTACAGGATTAACTGAAGG | 50 | 238 |
| AS | CAATGATGGAATGCTCTC | |||
| Nrf2 | S | ACCAAGGGGCACCATATAAAAG | 60 | 114 |
| AS | CTTCGCCGAGTTGCACTCA | |||
| Nqo1 | S | AGGATGGGAGGTACTCGAATC | 59 | 144 |
| AS | AGGCGTCCTTCCTTATATGCTA | |||
| GSR | S | GACACCTCTTCCTTCGACTACC | 60 | 116 |
| AS | CCCAGCTTGTGACTCTCCAC | |||
| Hmox1 | S | AAGCCGAGAATGCTGAGTTCA | 60 | 100 |
| AS | GCCGTGTAGATATGGTACAAGGA | |||
| Txnrd1 | S | CCCACTTGCCCCAACTGTT | 60 | 134 |
| AS | GGGAGTGTCTTGGAGGGAC | |||
| CAT | S | GCAGATACCTGTGAACTGTCCCT | 60 | 472 |
| AS | TTACAGGTTAGCTTTTCCCTTCG | |||
| p16 | S | AACTCTTTCGGTCGTACCCC | 62 | 364 |
| AS | GCGTGCTTGAGCTGAAGCTA | |||
| p21 | S | CAATCCTGGTGATGTCCGACCTGTT | 70 | 369 |
| AS | GAATCTTCAGGCCGCTCAGACACCA | |||
| Bmi1 | S | ATCCCCACTTAATGTGTGTCCT | 61 | 116 |
| AS | CTTGCTGGTCTCCAAGTAACG | |||
| TNF |
S | CCTGTAGCCCACGTCGTAG | 62 | 148 |
| AS | GGGAGTAGACAAGGTACAACCC | |||
| IL6 | S | TAGTCCTTCCTACCCCAATTTCC | 61 | 76 |
| AS | TTGGTCCTTAGCCACTCCTTC | |||
| ALP | S | CTTGCTGGTGGAAGGAGGCAGG | 55 | 393 |
| AS | GGAGCACAGGAAGTTGGGAC | |||
| Col-I | S | TCTCCACTCTTCTAGTTCCT | 55 | 269 |
| AS | TTGGGTCATTTCCACATGC | |||
| OPG | S | TGGAGATCGAATTCTGCTTG | 57 | 719 |
| AS | TCAAGTGCTTGAGGGCATAC | |||
| Runx2 | S | GTGACACCGTGTCAGCAAAG | 55 | 356 |
| AS | GGAGCACAGGAAGTTGGGAC | |||
| Osterix | S | ATGGCGTCCTCTCTGCTTG | 62 | 156 |
| AS | TGAAAGGTCAGCGTATGGCTT | |||
| OCN | S | CAAGTCCCACACAGCAGCTT | 55 | 370 |
| AS | AAAGCCGAGCTGCCAGAGTT | |||
| BSP | S | AGCAAGAAACTCTTCCAAGCAA | 60 | 134 |
| AS | GTGAGATTCGTCAGATTCATCCG | |||
| M-csf | S | GGCCTTGGAAGCATGTAGAGG | 62 | 104 |
| AS | GGAGAACTCGTTAGAGACGACTT | |||
| RANKL | S | GGTCGGGCAATTCTGAATT | 57 | 813 |
| AS | GGGGAATTACAAAGTGCACCAG | |||
| Trap | S | ACACAGTGATGCTGTGTGGCAACTC | 57 | 466 |
| AS | CCAGAGGCTTCCACATATATGATGG | |||
| CTSK | S | GAAGAAGACTCACCAGAAGCAG | 60 | 102 |
| AS | TCCAGGTTATGGGCAGAGATT | |||
| c-FMS | S | GGCTTGGCTTGGGATGATTCT | 62 | 126 |
| AS | GAGGGTCTGGCAGGTACTC | |||
| CTR | S | GCAACGCTTTCACTTCTGAGA | 62 | 297 |
| AS | GTTCCCACTGCATTGTCCACA | |||
| Nfatc-1 | S | GGAGCGGAGAAACTTTGCG | 61 | 94 |
| AS | GTGACACTAGGGGACACATAACT | |||
| Mmp13 | S | CTTCTTCTTGTTGAGCTGGACTC | 62 | 173 |
| AS | CTGTGGAGGTCACTGTAGACT | |||
| IL-1 |
S | CGAAGACTACAGTTCTGCCATT | 60 | 126 |
| AS | GACGTTTCAGAGGTTCTCAGAG | |||
| IL-1 |
S | GCAACTGTTCCTGAACTCAACT | 61 | 89 |
| AS | ATCTTTTGGGGTCCGTCAACT | |||
| GAPDH | S | TGGATTTGGACGCATTGGTC | 55 | 211 |
| AS | TTTGCACTGGTACGTGTTGAT |
bp, base pairs; Tm, Melting temperature; S, Sense; AS, Anti-sense; SOD1,
superoxide dismutase 1; Nrf2, nuclear factor erythroid 2-related factor 2; Nqo1,
NAD(P)H quinone dehydrogenase 1; GSR, glutathione reductase; Hmox1, heme
oxygenase 1; Txnrd1, thioredoxin reductase 1; CAT, catalase; Bmi1, B lymphoma
Mo-MLV insertion region 1 homolog; TNF
All data are presented as mean
To determine whether active vitamin D delays aging by suppressing oxidative stress and DNA damage, male Cyp27b1+/- mice were given either the antioxidant N-acetylcysteine (NAC) in drinking water from 3 weeks of age after weaning, or 1,25(OH)2D3 by subcutaneous injection. Age-matched WT and Cyp27b1+/- males on a standard diet served as controls. Survival was monitored across groups. In 8-month-old mice, serum MDA was measured biochemically; ROS levels in bone marrow cells were quantified by 2′,7′-Dichlorofluorescin diacetate (DCF-DA) staining and flow cytometry; and DNA damage and antioxidant markers in bone were assessed by immunohistochemistry, Western blots, and real-time RT–PCR.
Cyp27b1+/- mice on a standard diet had a mean lifespan of 241
days (range 80–534), which was extended to 398 days (182–636) by NAC and to 583
days (382–700) by 1,25(OH)2D3 (Fig. 1A). Relative to WT controls,
8-month-old Cyp27b1+/- mice exhibited significantly elevated serum
MDA and bone ROS, both of which were reduced by NAC or 1,25(OH)2D3
supplementation (Fig. 1B,C). In vertebrae from Cyp27b1+/- mice, the
proportion of SOD1-positive cells (Fig. 1D,E), SOD1 and Nrf2 protein levels (Fig. 1H,I), and mRNA levels of Nrf2 target genes (Nqo1, Gsr, Hmox1, Txnrd1,
and Cat; Fig. 1J) were all decreased, while DNA damage
markers—8OHdG-positive cells (Fig. 1F,G) and
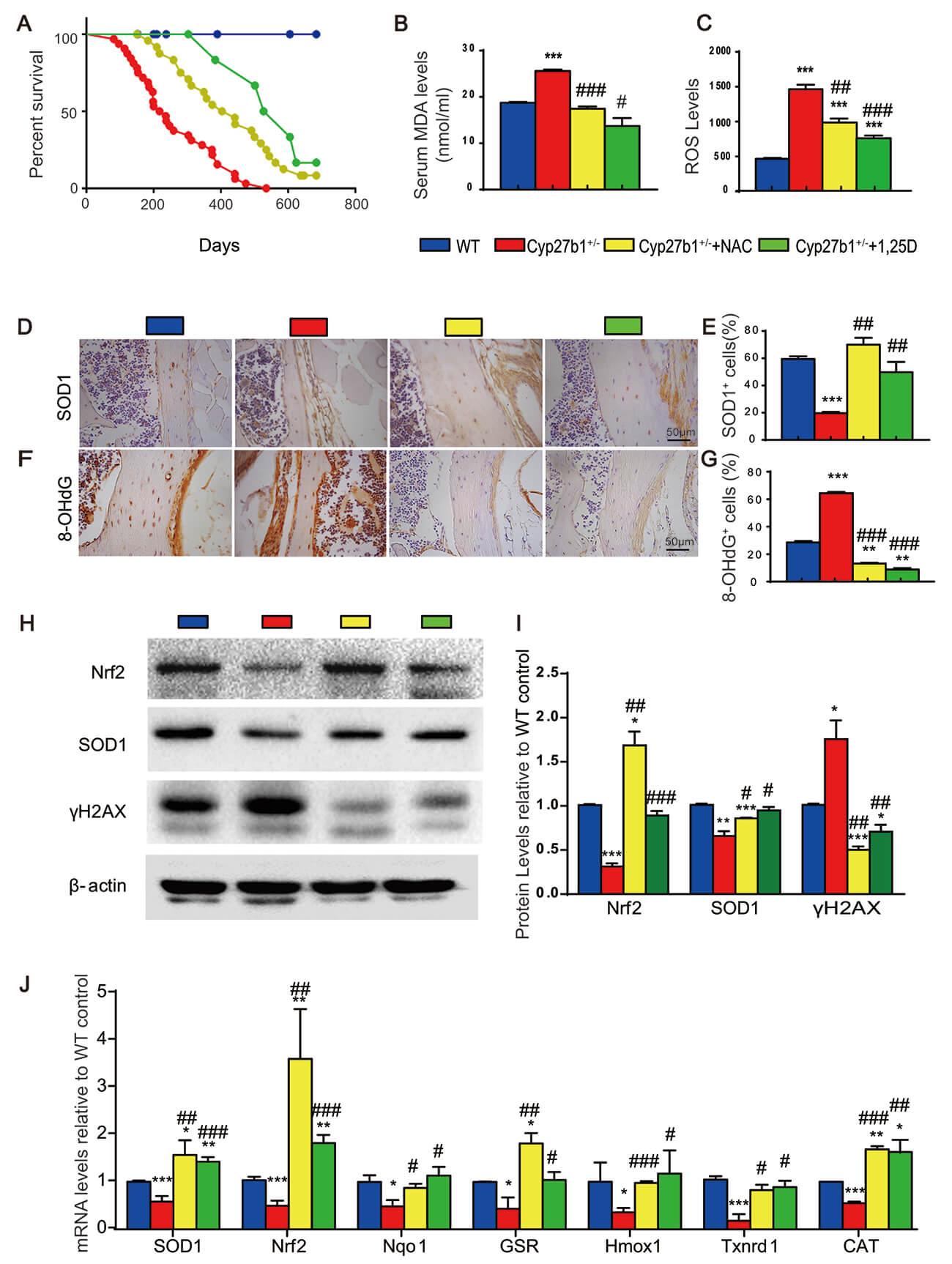 Fig. 1.
Fig. 1.
NAC or exogenous 1,25(OH)2D3 extends lifespan and
suppresses oxidative stress and DNA damage in Cyp27b1+/- mice.
(A) Kaplan–Meier survival curves for WT, Cyp27b1+/-, and
Cyp27b1+/- mice supplemented with NAC or 1,25(OH)2D3 on
a standard diet; groups compared by log-rank test. (B) Serum malondialdehyde
(MDA) concentrations reflecting systemic lipid peroxidation in 8month-old mice.
(C) Reactive oxygen species (ROS) levels in bone marrow cells. (D)
Immunohistochemistry for SOD1-positive cells in vertebral sections (Scale bar =
50 µm) and (E) quantification of positive area. (F) Immunohistochemistry
(Scale bar = 50 µm) and (G) quantification for 8-OHdG-positive cells
indicating oxidative DNA damage. (H,I) Representative Western blots and
quantification of Nrf2, SOD1, and
To test whether NAC or 1,25(OH)2D3 can rescue bone loss induced by active vitamin D haploinsufficiency, we analyzed vertebral phenotypes in the four groups of 8-month-old mice by imaging and histology. Compared with WT, Cyp27b1+/- mice showed significant reductions in vertebral bone mineral density (BMD), bone volume, trabecular number and thickness, total collagen-stained area, and osteoblasts with reduced ALP-positive area; in addition, there was increased trabecular separation and greater TRAP-positive osteoclast surface (Fig. 2A–M). NAC or 1,25(OH)2D3 supplementation markedly improved these parameters, reversing bone loss, low BMD, trabecular architectural deterioration, collagen reduction, osteoblast depletion, and osteoclast expansion (Fig. 2A–M). These results suggest that active vitamin D counters osteoporosis by limiting oxidative stress, thereby promoting osteoblast-mediated bone formation and restraining osteoclast-mediated bone resorption.
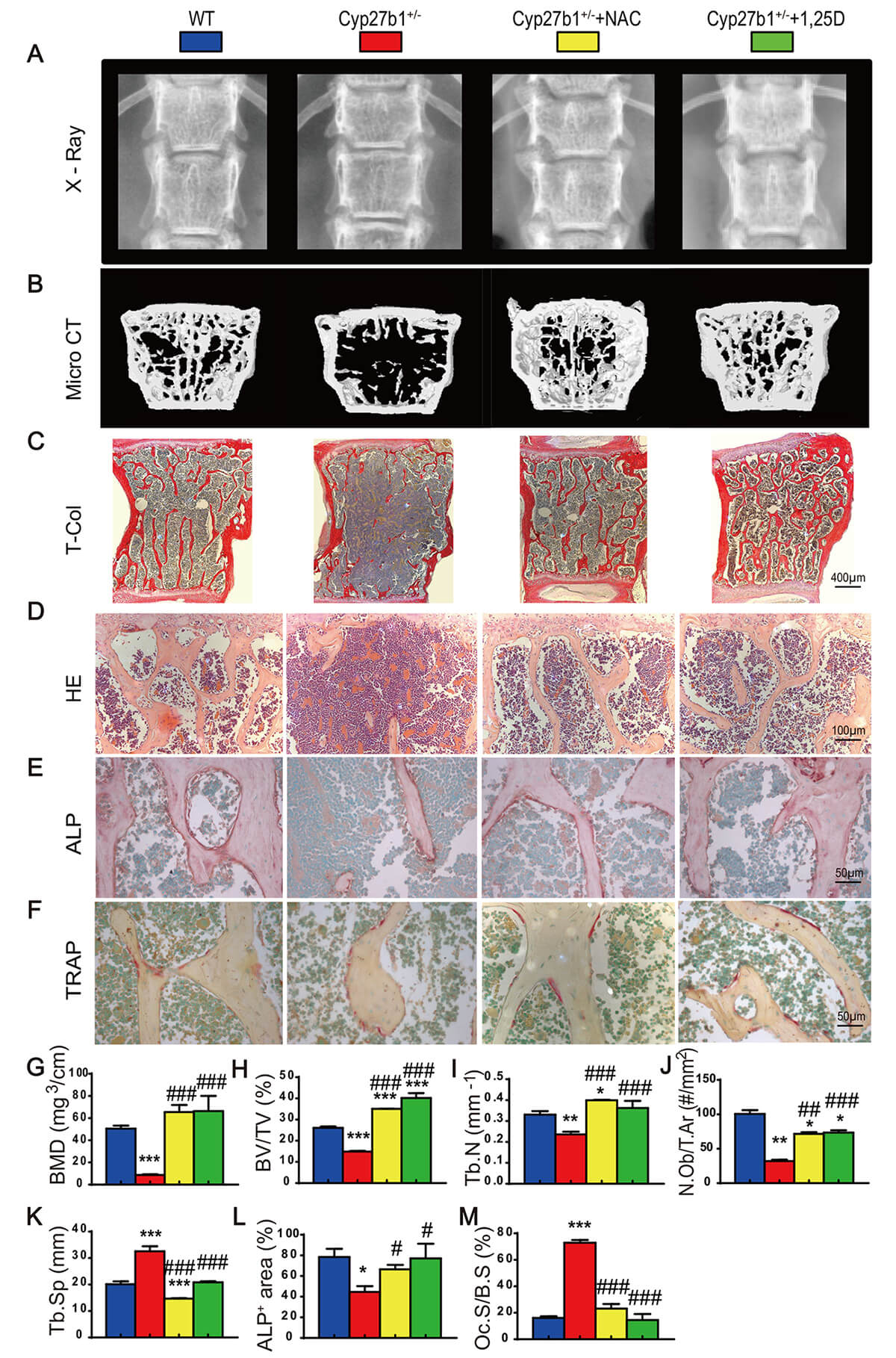 Fig. 2.
Fig. 2.
NAC or exogenous 1,25(OH)2D3 ameliorates bone loss due
to Cyp27b1 haploinsufficiency. (A) Lumbar spine Xrays. (B) MicroCT scans and 3D
reconstructions of lumbar vertebrae. (C) Total collagen staining of vertebral
sections (Scale bar = 400 µm). (D) Hematoxylin and eosin (H&E) staining
(Scale bar = 100 µm). (E) Alkaline phosphatase (ALP) staining (Scale bar =
50 µm). (F) Tartrate-resistant acid phosphatase (TRAP) staining (Scale bar
= 50 µm). (G) Bone mineral density (BMD, mg/cm3). (H) Trabecular bone
volume fraction (BV/TV, %). (I) Trabecular number (Tb.N, #/mm). (J) Osteoblast
number (N.Ob/T.Ar, #/mm2). (K) Trabecular separation (Tb.Sp, µm). (L)
ALP-positive area (%). (M) Osteoclast surface (Oc.S/B.S, %). Data are mean
To evaluate skeletal aging, senescence-associated markers were examined in
vertebrae from the four groups of 8-month-old mice using immunohistochemistry,
Western blots, and real-time RT–PCR. Relative to WT, Cyp27b1+/-
mice showed increased
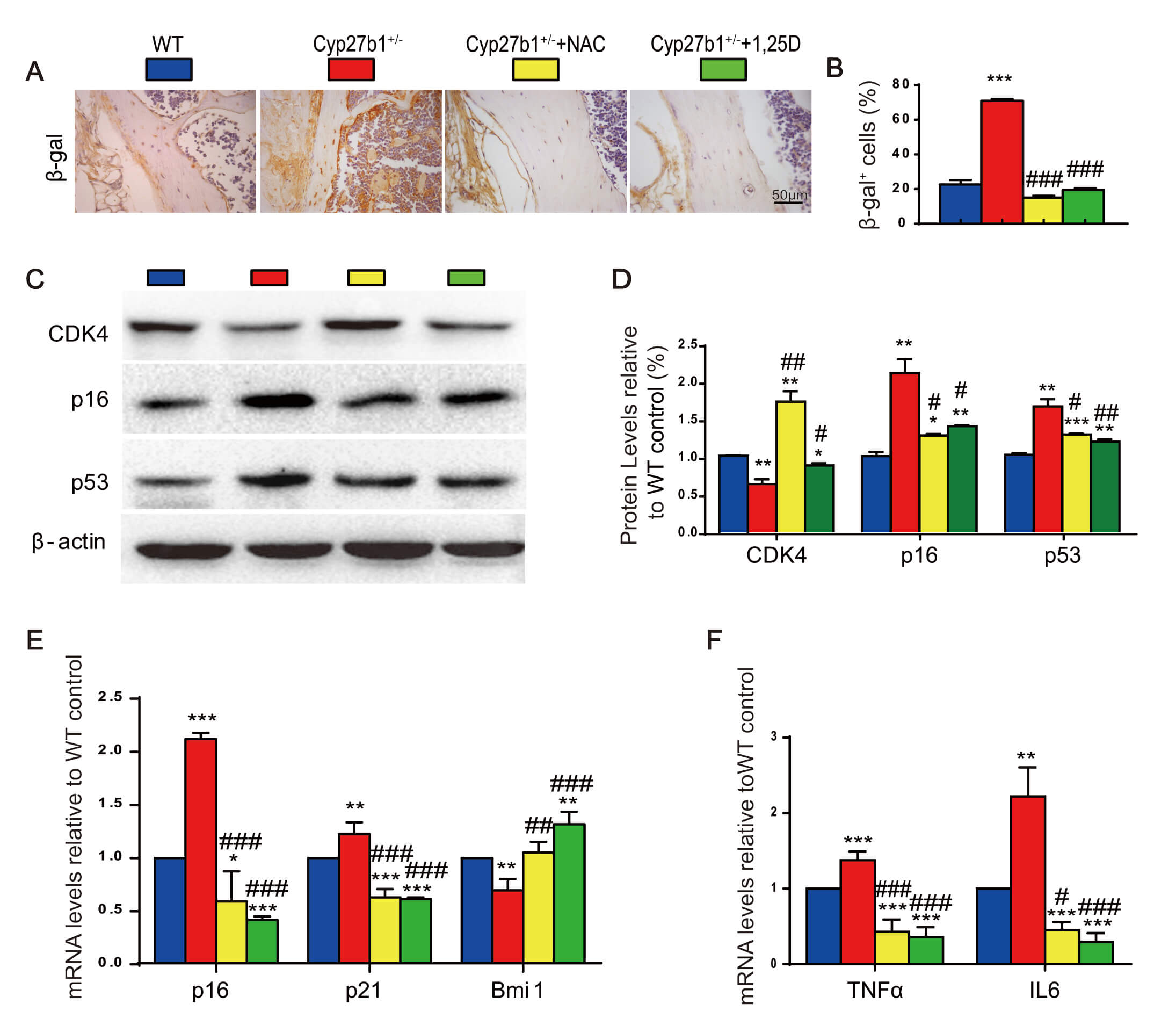 Fig. 3.
Fig. 3.
NAC or exogenous 1,25(OH)2D3 delays skeletal aging
caused by Cyp27b1 haploinsufficiency. (A)
Given the robust upregulation of p16 in bone, we generated double-mutant mice lacking p16 on a Cyp27b1+/- background (p16-/-Cyp27b1+/-), and compared their skeletal phenotypes at 8 months on a standard diet, with WT, p16-/-, and Cyp27b1+/- littermates. Imaging and histology showed that, relative to WT, Cyp27b1+/- mice had significantly reduced BMD, bone volume, and trabecular number and thickness, along with increased trabecular separation (Fig. 4A–G). Strikingly, p16 deletion in Cyp27b1+/- mice restored bone mass, BMD, and trabecular number and thickness, and reduced trabecular separation (Fig. 4A–G). These data demonstrate that p16 loss effectively corrects bone loss resulting from active vitamin D haploinsufficiency.
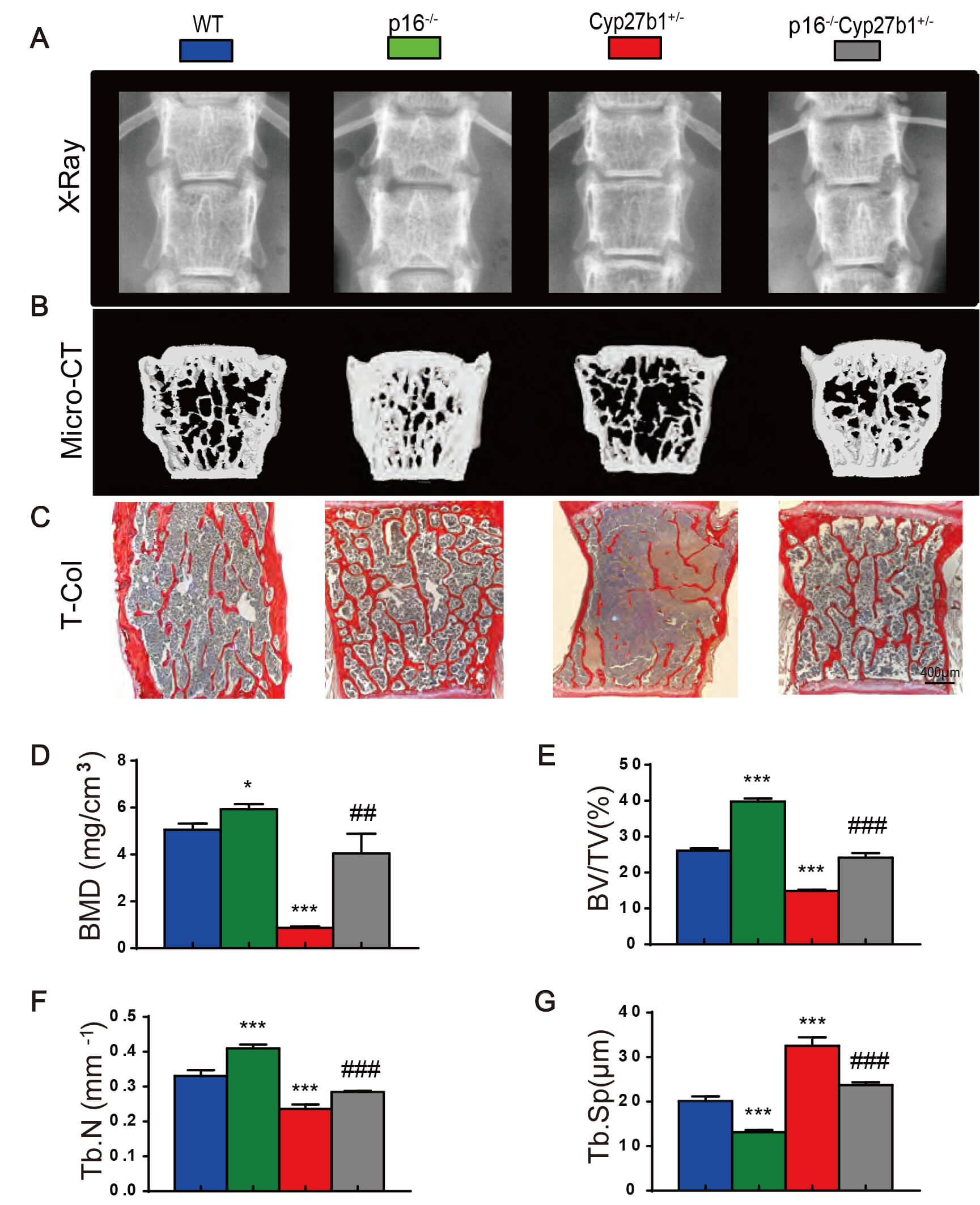 Fig. 4.
Fig. 4.
p16 deletion mitigates bone loss due to Cyp27b1
haploinsufficiency. (A) Lumbar spine Xrays. (B) MicroCT and 3D reconstructions.
(C) Total collagen staining of vertebral sections (Scale bar = 400 µm). (D)
Bone mineral density (BMD, mg/cm3). (E) Trabecular bone volume fraction
(BV/TV, %). (F) Trabecular number (Tb.N, #/mm). (G) Trabecular separation
(Tb.Sp, µm). Data are mean
To determine whether p16 deletion ameliorates impaired bone formation and excessive resorption, we assessed osteogenic and osteoclastic indices in vertebrae from the four genotypes at 8 months. Compared with WT, p16-/- mice exhibited increased osteoblast numbers and ALP-positive area, with higher mRNA levels of osteogenic genes (ALP, ColI, osteoprotegerin (OPG), Runt-related transcription factor 2 (Runx2), Osterix, osteocalcin (OCN), and bone sialoprotein (BSP)). In contrast, Cyp27b1+/- mice had reduced osteoblast numbers and ALP-positive area, and downregulated osteogenic gene expression. Notably, p16 deletion in Cyp27b1+/- mice increased osteoblast numbers and ALP-positive area and upregulated osteogenic genes (Fig. 5A,B,D,E,H). On the resorptive side, Cyp27b1+/- mice displayed increased TRAP-positive osteoclast surface, an elevated receptor activator of nuclear factor kappa-B ligand (RANKL)/OPG mRNA ratio, and higher mRNA levels of resorption-associated genes (Mcsf, RANKL, TRAP, CTSK, cFMS, CTR, and Nfatc1). p16 deletion in Cyp27b1+/- mice reduced osteoclast surface and downregulated these resorptive genes (Fig. 5C,F,G,I). Thus, p16 loss rescues vitamin D haploinsufficiency–induced bone loss by promoting formation and inhibiting resorption.
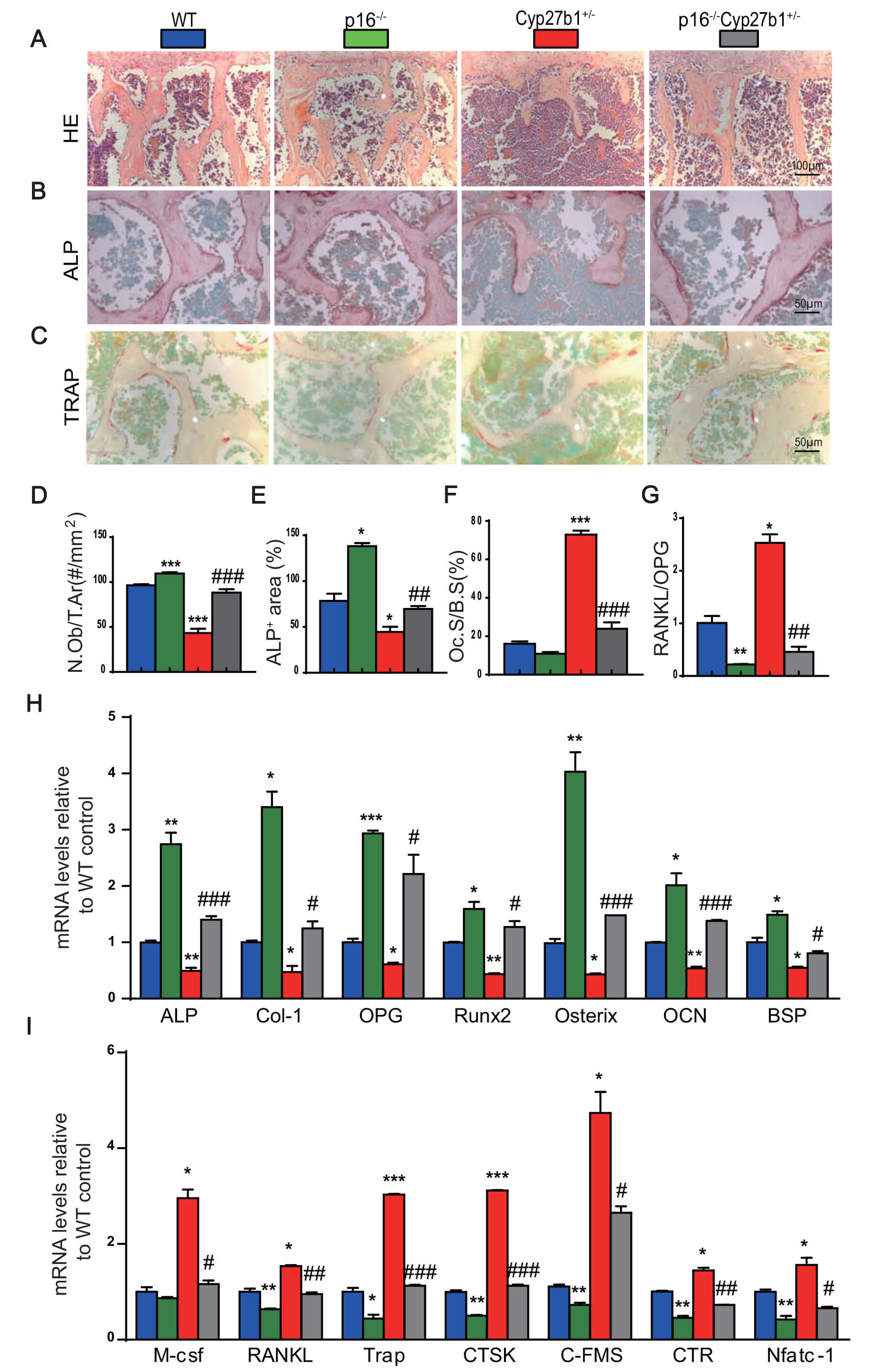 Fig. 5.
Fig. 5.
p16 deletion corrects the reduced bone formation and the
increased bone resorption in Cyp27b1 haploinsufficiency. (A) H&E staining
(Scale bar = 100 µm). (B) ALP staining (Scale bar = 50 µm). (C) TRAP
staining of vertebral trabeculae (Scale bar = 50 µm). (D) Osteoblast number
per trabecular area (N.Ob/T.Ar, #/mm2). (E) ALP-positive area (%). (F)
Osteoclast surface per bone surface (Oc.S/B.S, %). (G)
RANKL/OPG mRNA ratio in vertebral bone. (H) Relative mRNA
levels of osteogenic marker genes, including alkaline phosphatase (ALP),
collagen type I (Col I), osteoprotegerin (OPG), Runt-related
transcription factor 2 (Runx2), osterix (OSX), osteocalcin
(OCN), and bone sialoprotein (BSP). (I) Relative mRNA levels of
osteoclastic marker genes, including macrophage colony-stimulating factor
(M-csf), receptor activator of nuclear factor kappa-B ligand
(RANKL), tartrate-resistant acid phosphatase (TRAP), cathepsin
K (CTSK), colony-stimulating factor 1 receptor (c-FMS),
calcitonin receptor (CTR), and nuclear factor of activated T cells
cytoplasmic 1 (Nfatc1). Data are mean
We next examined whether p16 deletion mitigates skeletal cell senescence and
SASP. Relative to WT, Cyp27b1+/- mice exhibited increased
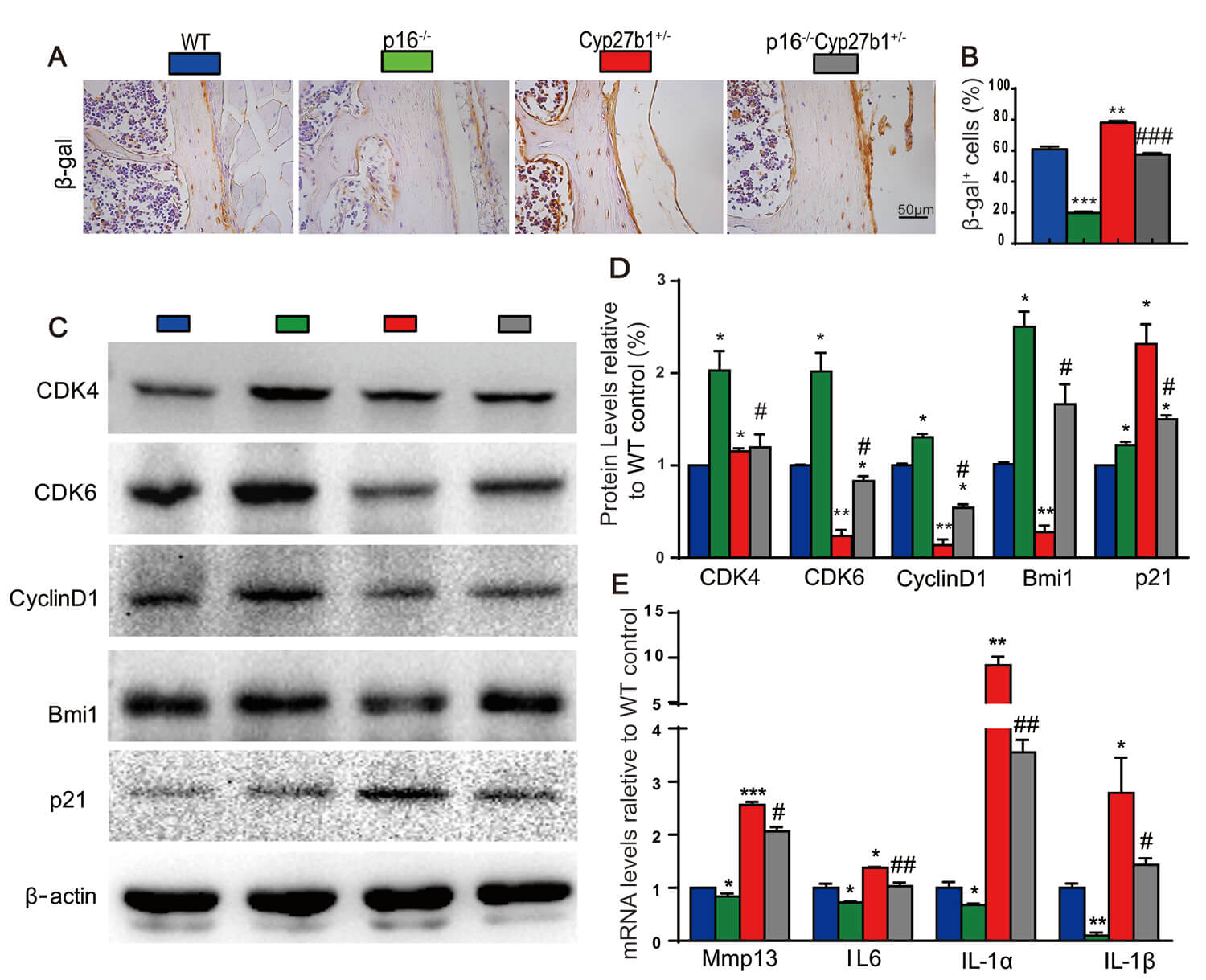 Fig. 6.
Fig. 6.
p16 deletion suppresses skeletal cell senescence induced by
Cyp27b1 haploinsufficiency. (A)
Our findings define a vitamin D–redox–senescence axis that governs skeletal remodeling during aging by linking active vitamin D insufficiency to heightened ROS, attenuated Nrf2/SOD1 defenses, and increased DNA damage, culminating in p53–p21 and p16 activation, SASP induction, suppressed osteoblastogenesis, elevated osteoclast activity, and trabecular deterioration [3, 5, 12, 25]. Both NAC and 1,25(OH)2D3 reversed these molecular and cellular abnormalities, and genetic attenuation of p16 restored bone mass by enhancing formation and reducing resorption while dampening SASP, underscoring senescence as a causal node in this pathway [4]. Mechanistically, our data support a model in which sufficient 1,25(OH)2D3 constrains oxidative and genotoxic stress to prevent sustained activation of senescence checkpoints and pro-osteoclastogenic SASP factors that impair osteoblast lineage commitment and promote osteoclastogenesis [3, 4, 5, 12, 25]. These results are consistent with literature linking ROS and DDR signaling to bone aging and demonstrating that bolstering antioxidant defenses curtails osteoclastogenesis and preserves osteoblast function [26, 27, 28, 29, 30]. They also converge with reports that VDR signaling interfaces with chromatin and cell-cycle regulators (for example, EZH2–p16) to restrain senescence programs in bone [31, 32]. At the tissue level, NAC and 1,25(OH)2D3 improved osteoblast indices, collagen deposition, and microarchitecture while reducing osteoclast surface, in line with studies showing that mitigating oxidative stress shifts the formation–resorption balance toward anabolism and that perturbation of redox sensors such as Nrf2 alters this balance [28, 29, 30, 33]. The reduction in osteoclast surface, together with vitamin D/VDR modulation of osteoclastogenic mediators, provides a plausible mechanism for antiresorptive benefit in aging bone [34, 35]. Collectively, these data highlight redox–senescence signaling as a tractable therapeutic target and suggest that maintaining sufficient active vitamin D or pharmacologically buffering ROS may mitigate skeletal aging by limiting p16-driven chronic senescence.
In parallel, in WT cohorts, long-term NAC (1 mg/mL in drinking water) and 1,25(OH)2D3 (0.1 µg/kg, s.c.) did not alter survival compared with untreated WT controls up to at least 600 days of age, indicating no lifespan extension under these regimens. Consistent with prior literature, neither intervention produced adverse skeletal phenotypes; NAC modestly improved redox status with trends toward preservation of age-sensitive trabecular microarchitecture [36], while 1,25(OH)2D3 maintained calcium homeostasis and attenuated age-related declines in bone formation indices, accompanied by activation of Nrf2- and VDR-dependent signaling [18]. These observations align with established evidence that vitamin D signaling mitigates skeletal aging and that antioxidant modulation of ROS can temper senescence-associated bone deterioration [37], providing contextual support for our mechanistic findings while explaining the omission of detailed WT control data from the main text.
It is important to acknowledge the inherent complexities of skeletal aging and to interpret our findings within this broader biological context. While the antioxidant NAC effectively rescued the bone phenotype in our model, supporting a key role for oxidative stress, its broad mechanism of action means that its benefits may extend beyond the direct quenching of reactive oxygen species to influence other redox-sensitive signaling pathways [38]. Likewise, while genetic deletion of p16Ink4a provides strong evidence for its role as a key effector in this process, this intervention may have pleiotropic effects. Cellular senescence is a fundamentally bivalent process, with beneficial roles in tumor suppression and tissue repair that may be compromised by its systemic elimination [3, 4]. The long-term consequences of constitutive p16 deletion, including potential impacts on cancer incidence or wound healing, were not assessed in this study and represent an important area for future investigation [26]. Therefore, the vitamin D–redox–senescence axis identified here should be viewed as a critical contributor to skeletal aging, which likely interacts with other well-established regulators such as hormonal fluctuations, mechanical loading, and systemic inflammation to determine the final bone phenotype.
While both NAC and exogenous 1,25(OH)2D3 significantly rescued vitamin D
deficiency-induced phenotypes, our study was not designed for direct therapeutic
comparison. Notably, 1,25(OH)2D3 more completely normalized lifespan and
senescence markers, suggesting more targeted pathway correction. Beyond its role
as a glutathione precursor, NAC’s benefits likely involve pleiotropic mechanisms
including direct ROS scavenging, NF-
Genetic evidence from p16 deletion strengthens the causal link between
senescence and osteopenia in the presence of vitamin D insufficiency. Removing
p16 normalized bone mass, increased osteogenic programs, reduced osteoclast
surface and RANKL/OPG ratio, and suppressed SASP. This mirrors evidence that
targeting senescent cells prevents age-related bone loss and rejuvenates
remodeling [26, 27] and supports p16 as a key effector coupling redox/genotoxic
stress to skeletal aging [31, 32, 42]. It is important to contextualize the role
of the Senescence-Associated Secretory Phenotype (SASP) as both a consequence of
cellular stress and a driver of tissue pathology. The SASP is highly
heterogeneous, but the components we identified as elevated in the bone of
Cyp27b1+/- mice—including the pro-inflammatory cytokines
TNF
Cellular senescence in bone reflects convergent yet distinct checkpoint programs governed by the p53–p21CIP1 and p16INK4a–RB pathways. Acute oxidative stress commonly activates the DNA damage response (DDR), leading to p53 stabilization and transcriptional induction of p21, which enforces an initial, often reversible growth arrest to facilitate repair [5, 6, 12, 46]. In contrast, chronic or unresolvable stressors—including sustained ROS and mitochondrial dysfunction—favor engagement of p16INK4a, promoting a more durable RB-mediated arrest associated with a stable SASP [3, 5, 6, 12, 25]. In our model of active vitamin D insufficiency, we observed a rapid increase in p21 coincident with early ROS elevations, consistent with DDR activation, followed by a predominant and persistent upregulation of p16 that correlated with osteopenic remodeling and SASP enrichment. This temporal sequence supports a two-stage paradigm in osteolineage cells: p53–p21 predominates during the initiation phase, whereas p16 maintains long-term senescence and reinforces paracrine dysfunction affecting osteoclastogenesis and osteoblast differentiation [3, 5, 6, 12, 25]. Notably, antioxidant intervention attenuated p21 induction and blunted subsequent p16 accumulation, implicating oxidative stress as a shared upstream driver. These findings align with reports that p21-associated arrest can be reversible and less pro-inflammatory, whereas p16-driven senescence is more stable and SASP-potent, thereby exerting stronger deleterious effects on bone turnover [3, 4, 5, 6, 12, 25, 47]. Thus, while p16 appears to be the principal mediator of the sustained senescent phenotype underlying skeletal aging in our model, the p53–p21 axis likely acts as an early sentinel whose modulation may determine whether cells recover or transition to p16-dependent chronic senescence.
Beyond bone, lifespan extension with NAC and 1,25(OH)2D3 suggests organismal benefits from mitigating oxidative stress and senescence, echoing reports that senescence modulation extends healthspan and that systemic redox improvements benefit multiple tissues [14, 23, 26]. Translationally, maintaining vitamin D sufficiency may protect bone by preserving intracellular redox and genomic stability. Antioxidants and senescence-modulating approaches (senolytics/senomorphics) could complement vitamin D repletion in osteoporosis—particularly in vitamin D insufficiency—with careful attention to safety and dosing [14, 23, 26, 28]. Because pharmacologic 1,25(OH)2D3 can cause hypercalcemia, physiological vitamin D repletion and bone-targeted or intermittent regimens for active analogs warrant consideration [34].
Clinical implications emerge on several fronts. First, maintaining adequate vitamin D status may protect bone not only by optimizing calcium–phosphate balance but also by preserving intracellular redox and genomic stability in skeletal cells [4, 7, 9, 18, 19, 20, 48, 49, 50]. Second, antioxidants such as NAC and agents that activate Nrf2 could complement vitamin D repletion to dampen oxidative stress–driven bone aging, though dosing, duration, and offtarget effects require careful evaluation [37, 51]. Third, senescencetargeted strategies (senolytics/senomorphics) merit exploration in osteoporosis, particularly when vitamin D insufficiency is present, with attention to safety and tissue specificity [4, 14, 50]. Because pharmacologic 1,25(OH)2D3 can cause hypercalcemia, translational paths should prioritize physiological vitamin D repletion (cholecalciferol/25OHD) and consider bonetargeted or intermittent regimens for active analogs [7, 9, 18, 48, 49, 52].
The therapeutic potential of vitamin D in modulating senescence pathways suggests promising avenues for clinical intervention, particularly when combined with emerging senolytic agents. Preclinical evidence, including data presented in this study, indicates that vitamin D receptor activation can suppress senescence-associated secretory phenotype (SASP) and enhance clearance of senescent cells, potentially augmenting the efficacy of senolytics such as dasatinib and quercetin [14]. A combinatorial approach may allow for lower, safer doses of both agents, reducing the risk of adverse effects. However, the use of active vitamin D analogs, such as calcitriol, carries a well-documented risk of hypercalcemia, especially in older adults and individuals with impaired renal function [7]. To mitigate this, strategies including intermittent dosing regimens, close monitoring of serum calcium and parathyroid hormone levels, and patient selection based on vitamin D deficiency status may be employed [41]. Furthermore, the use of less calcemic vitamin D analogs or prodrug formulations currently under investigation could offer a safer profile for long-term use in aging populations [53]. Future clinical trials should evaluate the safety and efficacy of such combination therapies, including active vitamin D or analogs with anti-oxidants and/or senolytics to ameliorate age-related conditions such as osteoporosis.
Active vitamin D insufficiency orchestrates a ROS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
DM conceived and supervised the project, secured funding, provided research resources and facilities, validated findings, and led manuscript writing and revision. WQ performed primary experiments, analyzed data, and developed experimental methodologies. MH and LC conducted supporting experiments and assisted with data analysis and methodology development. DG made subbstantial contributions to the conception or design of the work and made critical revision to the article for important intellectual content. All authors approved the final version of this manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The use of animals in this study was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (Approval number: IACUC-1802007), ensuring compliance with China’s ethical guidelines for laboratory animal care and use.
Not applicable.
This work was supported by grants from the National Natural Science Foundation of China (81730066 to DM).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.