







1 Department of Neurology, The First Hospital of Qiqihar (Southern Hospital Campus), 161005 Qiqihar, Heilongjiang, China
2 Department of Neurosurgery, The First Hospital of Qiqihar (Northern Hospital Campus), 161000 Qiqihar, Heilongjiang, China
†These authors contributed equally.
Abstract
Vascular dementia (VaD) is a prevalent cognitive disorder associated with cerebrovascular pathologies, in which hippocampal dysfunction plays a critical role. Impaired zinc homeostasis, mediated by the zinc transporters 3 (ZnT3) and transmembrane protein 163 (TMEM163), has been implicated in neuronal damage. However, the underlying mechanisms remain unclear. The purpose of this study is to elucidate the molecular mechanisms by which these zinc transporters may contribute to VaD pathogenesis, and to determine whether these mechanisms are involved in the development of VaD.
Rat primary hippocampal neurons were subjected to oxygen-glucose deprivation (OGD) to simulate ischemic conditions. To investigate the roles of ZnT3 and TMEM163, we employed siRNA-mediated silencing and plasmid-based overexpression. Neuronal viability was assessed using the methyl thiazolyl tetrazolium (MTT) assay, while apoptosis was quantified via TdT-mediated dUTP nick-end labeling (TUNEL) staining. Intracellular and extracellular zinc levels were measured using FluoZin-3 fluorescence and ELISA, respectively. Protein and mRNA expression levels were analyzed by western blot and real-time quantitative polymerase chain reaction (RT-qPCR). Protein-protein interactions were examined through co-immunoprecipitation, and subcellular localization was determined via cell surface biotinylation.
OGD induced significant extracellular zinc overload, neuronal apoptosis, and reduced cell viability, concomitant with upregulated expression of ZnT3 and TMEM163 (all p < 0.001). Overexpression of these transporters exacerbated zinc efflux and neuronal damage under OGD, whereas their silencing attenuated zinc overload and neuronal degeneration (p < 0.001). Co-immunoprecipitation confirmed a physical interaction between ZnT3 and TMEM163. Furthermore, OGD triggered the translocation of both proteins from the cell membrane to the cytoplasm (p < 0.001), suggesting ischemia-induced dysregulation of zinc transport dynamics. These findings demonstrate that ZnT3 and TMEM163 cooperatively modulate zinc homeostasis and that their dysregulation during OGD contributes to neuronal injury.
ZnT3 and TMEM163 are critical regulators of zinc homeostasis in hippocampal neurons. Their dysregulation under ischemic conditions promotes extracellular zinc overload and exacerbates neuronal damage, highlighting their potential therapeutic relevance in VaD.
Keywords
- zinc transporter 3
- TMEM163
- zinc
- hippocampus
- vascular dementia
Vascular dementia (VaD) represents a prevalent central nervous system disorder that arises from cerebrovascular pathologies and manifests as progressive cognitive impairment. Epidemiological studies have consistently demonstrated that VaD ranks as the second most common form of dementia after Alzheimer’s disease (AD), accounting for approximately 15–30% of all dementia cases worldwide [1]. Clinically, VaD patients exhibit heterogeneous cognitive symptoms that vary depending on the specific brain regions affected by vascular lesions. Notably, deficits in short-term memory, working memory, attention, and executive functions have been strongly associated with ischemic damage and neuronal loss in the hippocampal formation [2].
Zinc, an essential trace element crucial for numerous biological processes
including DNA function, protein catalysis, and neural signaling, has emerged as a
key player in post-ischemic hippocampal dysfunction when its homeostasis becomes
disrupted [3]. Under pathological conditions, excessive zinc accumulation within
neurons triggers cell death pathways through activation of endoplasmic reticulum
stress, particularly in vulnerable regions such as the ischemic penumbra and
hippocampal CA1/CA3 subfields [4, 5]. In the extracellular compartment, zinc
overload contributes to blood-brain barrier disruption by promoting ferroptosis
and accelerating amyloid-beta (A
The SLC30A gene family encodes zinc transporter (ZnT) proteins, which serve as primary regulators of cellular zinc homeostasis by controlling cytosolic zinc storage and efflux [9]. Among these transporters, ZnT3 stands out as the most abundantly expressed isoform in the hippocampus, where it plays a specialized role in zinc translocation into synaptic vesicles [10]. Clinical investigations have revealed significant correlations between cerebrospinal fluid ZnT3 levels and cognitive performance scores in AD patients [11]. This association may stem from hypoxia-induced ZnT3 dysregulation in the hippocampal CA1 region, where excessive zinc efflux leads to extracellular zinc accumulation and subsequent neuronal injury [12]. However, it is important to note that the role of ZnT3 in neurodegeneration appears to be complex and context-dependent. For instance, some studies have reported that ZnT3 knockout can exacerbate cognitive deficits and synaptic dysfunction, suggesting a protective role for synaptic zinc under certain conditions [13, 14, 15].
Beyond ZnT3, recent research has identified transmembrane protein 163 (TMEM163) as a novel zinc efflux transporter localized to both cell membranes and lysosomes, where it appears to function synergistically with other zinc regulatory proteins in the human brain [16]. Genetic studies have linked specific TMEM163 variants to early-onset Parkinson’s disease in Chinese populations, while experimental knockdown of TMEM163 in zebrafish models results in significant locomotor deficits and myelin abnormalities [17]. Intriguingly, elevated TMEM163 expression has been observed in specific microglial subpopulations even within non-lesional brain tissue from multiple sclerosis patients [18]. Structural analyses have further demonstrated that TMEM163 can form both homodimers and heterodimers with various ZnT family members, suggesting potential functional interactions [19]. These findings strongly imply that ZnT3 and TMEM163 may cooperate structurally and functionally to maintain normal neuronal physiology. However, whether dysregulation of these transporters and consequent zinc homeostasis disruption represents a critical mechanism underlying hippocampal neuronal damage and cognitive impairment in VaD remains an important unanswered question. In the present investigation, we systematically examined the roles of ZnT3 and TMEM163 in zinc regulation under both physiological conditions and oxygen-glucose deprivation (OGD) stress. Our study specifically focused on their effects on hippocampal neuronal function, subcellular distribution patterns, and potential protein-protein interactions. Through comprehensive experimental approaches, we aimed to elucidate the molecular mechanisms by which these zinc transporters may contribute to VaD pathogenesis.
Primary hippocampal neurons (PHNs; CP-R107) were obtained from Pricella Biotechnology Co., Ltd. (Wuhan, Hubei, China) and cultured in complete medium at 37 °C under 5% CO2 and saturated humidity. For subculturing, cells were dissociated with 1 mL of 0.25% trypsin for 1 min, neutralized with 5 mL of complete medium, gently pipetted to achieve a single-cell suspension, and returned to the incubator for further culture. A cellular model of oxygen-glucose deprivation (OGD) was constructed. PHNs were washed twice with phosphate-buffered saline (PBS), incubated in a glucose-free medium, and placed in a hypoxic chamber (1% O2, 5% CO2 and 94% N2) at 37 °C for 2–6 hours to simulate ischemia. The cell line was validated by STR profiling and tested negative for mycoplasma.
siRNA sequences targeting the coding regions of ZnT3 (NM_001013243.1, Sangon, Shanghai, China) and TMEM163 (NM_001110763.2, Sangon, Shanghai, China) were designed using the NCBI database (Supplementary Table 1). A scrambled siRNA (si-NC) served as the negative control. For transfection, 2 µL of siRNA duplexes and 10 µL of RNA Fit transfection reagent (HB-RF-1000, Hanbio, Shanghai, China) were separately diluted in 200 µL of Opti-MEM medium (31985088, Gibco, New York, USA), incubated at room temperature for 10 min to form complexes, and added to cell cultures. Cells were then incubated for 48 h to achieve gene silencing (si-ZnT3 and si-TMEM163).
The coding sequences (CDS) of ZnT3 and TMEM163 were cloned into the pcDNA3.1 (SYNBIO, Sunzhou, China) vector to generate overexpression plasmids (pcDNA3.1-ZnT3 and pcDNA3.1-TMEM163) (Supplementary Table 2 and Supplementary Fig. 1). PHNs (80% confluent in 6-well plates) were transfected with 5 µg plasmid DNA mixed with 10 µL P3000 reagent (Thermo Fisher Scientific, MA, USA) and 3.75 µL Lipofectamine 3000 (ThermoFisher, MA, USA) in Opti-MEM. After 15 min of incubation, the mixture was added to cells for 24–48 h to induce overexpression (OE-ZnT3 and OE-TMEM163).
PHNs (2000 cells/well in 96-well plates) were treated as per experimental groups (n = 6 replicates/group). After 6 h, 10 µL MTT (C0009S, Beyotime, Shanghai, China) (5 mg/mL) was added to each well, followed by 4 h of incubation. Formazan crystals were dissolved in DMSO, and absorbance at 570 nm was measured (Thermo Fisher Scientific, MA, USA).
PHNs were fixed with 4% paraformaldehyde (30 min), permeabilized with 0.3%
Triton X-100, and labeled using a TUNEL kit (Roche, Basel, Switzerland).
Apoptotic nuclei (TUNEL-positive) and total nuclei (DAPI-stained, C1006,
Beyotime, Shanghai, China) were counted under a fluorescence microscope (BZ-H4XD,
Keyence, Osaka, Japan). The apoptosis rate was calculated as (TUNEL⁺ cells/total
cells)
Supernatants were centrifuged, and zinc levels were measured using a commercial ELISA kit (ab277419, Abcam, Cambridge, UK). A standard curve (0–5 nmol/well) was generated, and absorbance at 560 nm was recorded.
PHNs were seeded in 6-well plates (2
PHNs were loaded with FluoZin™-3 AM (F24195, Thermo Fisher Scientific, MA, USA) (5 µM, 30 min at 37 °C), washed, and incubated in PBS for 30 min. Fluorescence (ex/em: 494/525 nm) was quantified using a fluorescence microscope (BZ-H4XD, Keyence, Japan).
PHNs were washed with PBS, lysed with RIPA buffer, and centrifuged to collect supernatants for total protein extracts. Protein quantification was used the BCA assay with a standard curve generated by diluting standards. Samples were mixed with Loading Buffer and stored at –20 °C. SDS-PAGE was performed using 12% resolving and 5% stacking gels. Proteins were electrophoresed, transferred to PVDF membranes, and blocked. Then the membranes were incubated with primary antibodies ZnT3 (1:1000, 67262-1-Ig, Proteintech), TMEM163 (1:1000, PA5-114329, ThermoFisher), GAPDH (1:10000, ab70700, Abcam) at 4 °C overnight and incubated with Goat Anti-Rabbit IgG H&L/HRP (1:20000, ab205718, Abcam) or Goat Anti-Mouse IgG H&L/HRP secondary antibodies (1:10000, ab205719, Abcam) for 2 h. Chemiluminescent detection and image capture followed, with protein expression analyzed using ImageJ (v1.8.0, National Institutes of Health, Bethesda, MD,USA).
Total RNA was extracted from PHNs of logarithmic phase using the FastPure RNA kit (R0017M, Beyotime, Shanghai, China), following cell lysis and column purification with ethanol treatment. RNA concentration and purity were assessed using Nano 6000 (Nano-600, JIAPENG, Shanghai, China). cDNA was synthesized from 500 ng RNA using HiScript III RT SuperMix with gDNA wiper. RT-qPCR was performed in a 20 µL reaction containing diluted cDNA, Nuclease-free water, Taq Pro Universal SYBR qPCR Master Mix, and gene-specific primers on the CFX96 Touch instrument (CFX96 Touch, Bio-rad, Shanghai,China). The results were calculated by the 2-ΔΔCt method. The sequences accessions of the primers are: mouse ZnT3 (5′-AGACCACCCGCCTAGTGAG-3′ forward, 5′-CTGAGACACGCGGTTCTTGT-3′ reverse), mouse TMEM163 (5′-CCCAGTCGTCCACCATCAAA-3′ forward, 5′-AAGCACCATAGGTGGCTTCC-3′ reverse), and GAPDH (5′-CAATCCTGGGCGGTACAACT′ forward, 5′-TACGGCCAAATCCGTTCACA-3′ reverse). Gene mRNA levels were normalized to those of GAPDH. All samples were run in duplicate.
PHNs were lysed in RIPA buffer with PMSF, and protein concentrations were determined using BCA assay. For co-immunoprecipitation (Co-IP), 500 µL cell lysates (200–1000 µg protein) were pre-cleared with Protein A/G Agarose at 4 °C for 1 h. Samples were incubated overnight with 2 µg primary antibodies (same antibodies as Western blot) or control IgG at 4 °C. Immune complexes were captured by adding Protein A/G Agarose, washed with lysis buffer and boiled in SDS-PAGE loading buffer. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and detected by Western blot using specific antibodies. Protein bands were visualized using chemiluminescence reagents and quantified with Image J.
PHNs (1.5
Statistical analysis was conducted using GraphPad Prism (v9.4.0, San Diego, CA, USA). Data are
presented as mean
We began by establishing an in vitro model of cerebral ischemia using
oxygen-glucose deprivation (OGD) in primary hippocampal neurons (PHNs).
Hypoxic-hypoglycemic conditions markedly reduced the fluorescence intensity of
NeuN, a specific neuronal nuclear marker (p
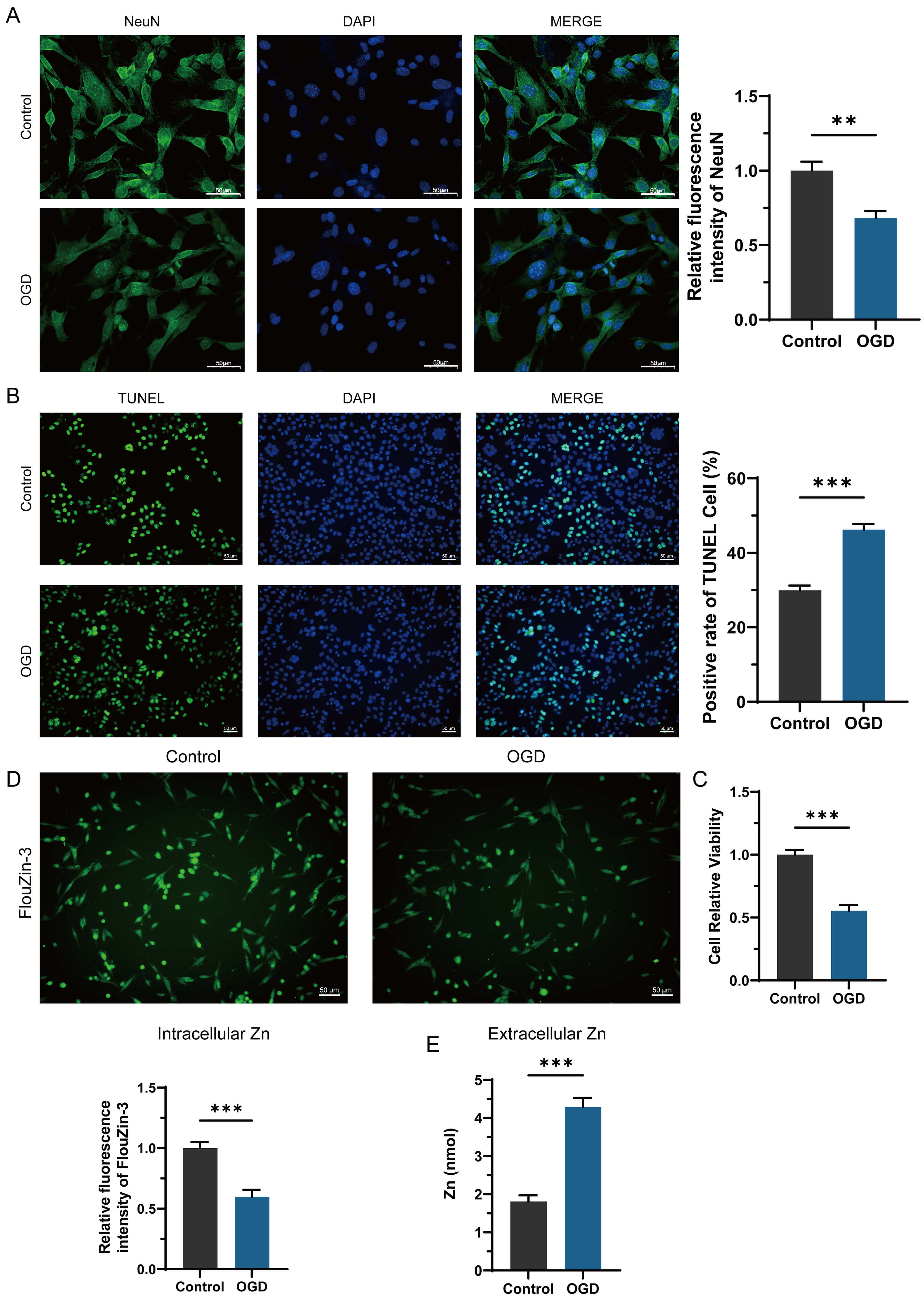 Fig. 1.
Fig. 1.
OGD induces neuronal loss and zinc efflux in PHNs. (A) NeuN fluorescence intensity was significantly reduced in PHNs after OGD, scale bar = 50 μm.
(B) OGD increased the apoptosis rate of PHNs, scale bar = 50 μm. (C) Neuronal
viability declined under OGD conditions. (D) Decreased intracellular zinc levels,
measured by FluoZin-3 fluorescence, were elevated post-OGD, scale bar = 50 μm. (E)
Extracellular zinc efflux, measured by ELISA. Data represent mean
Having observed aberrant zinc efflux, we sought to identify the transporters
responsible. We focused on ZnT3, given its predominant expression in hippocampal
neurons and its critical role in synaptic zinc transport and its documented
involvement in cerebral ischemia. We therefore hypothesized that ZnT3 and TMEM163
might act in concert to mediate the pathological zinc efflux observed in our OGD
model. siRNA and the overexpression plasmid were designed for the manipulation of
ZnT3 or TMEM163 expression in PHNs, respectively. The interference and
overexpression efficiency were measured by Western blot and the results suggest
that the protein expression of ZnT3 and TMEM163 were silenced by siRNA
interference while the overexpression plasmid exhibited upregulation of ZnT3 and
TMEM163 in PHNs (Fig. 2A). Since extracellular zinc level was significantly
elevated by OGD, and ZnT3 is a high-expressed zinc transporter protein in neurons
that already been confirmed to be colocalized with TMEM163, we hypothesis that
both ZnT3 and TMEM163 and their expression may be affected with the changes of
culture environment. Our experiment suggests that OGD significantly upregulated
ZnT3 and TMEM163 at both protein and mRNA levels (p
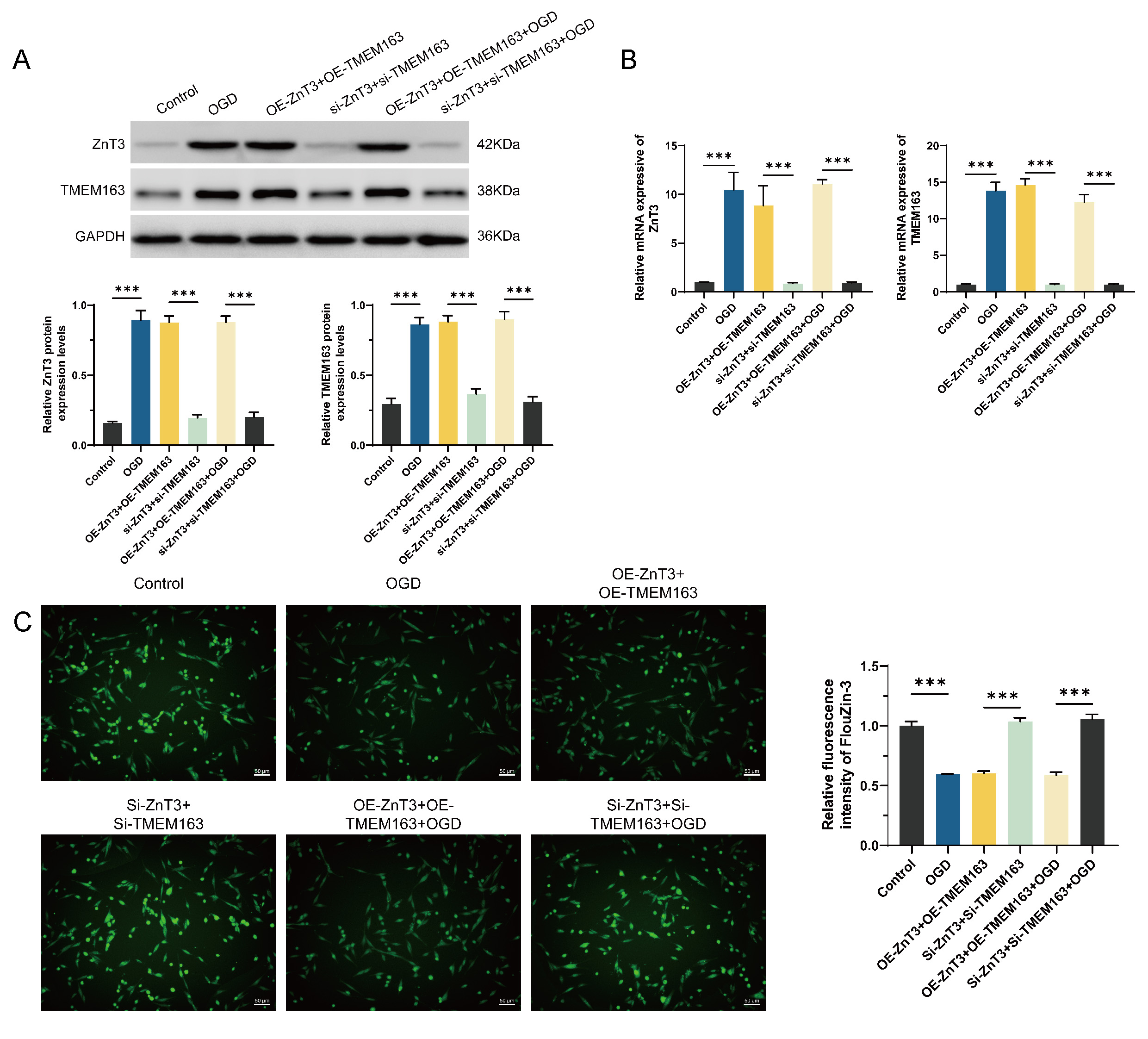 Fig. 2.
Fig. 2.
OGD alters ZnT3 and TMEM163 expression and zinc distribution in
PHNs. (A) Protein levels of ZnT3 and TMEM163 increased under OGD and
overexpression (OE) conditions but decreased after siRNA silencing. (B) Corresponding mRNA expression changes. (C) Intracellular zinc levels decreased
after OGD or OE but were restored by siRNA-mediated knockdown, scale bar = 50 μm. Data represent
mean
Since manipulating ZnT3/TMEM163 expression directly affected zinc flux, we next
asked whether this manipulation would correspondingly impact neuronal survival.
It was evident that OGD and OE-ZnT3/TMEM163 exacerbated PHNs damage, as
demonstrated by a reduction in NeuN-positive cells (indicating neuronal loss,
p
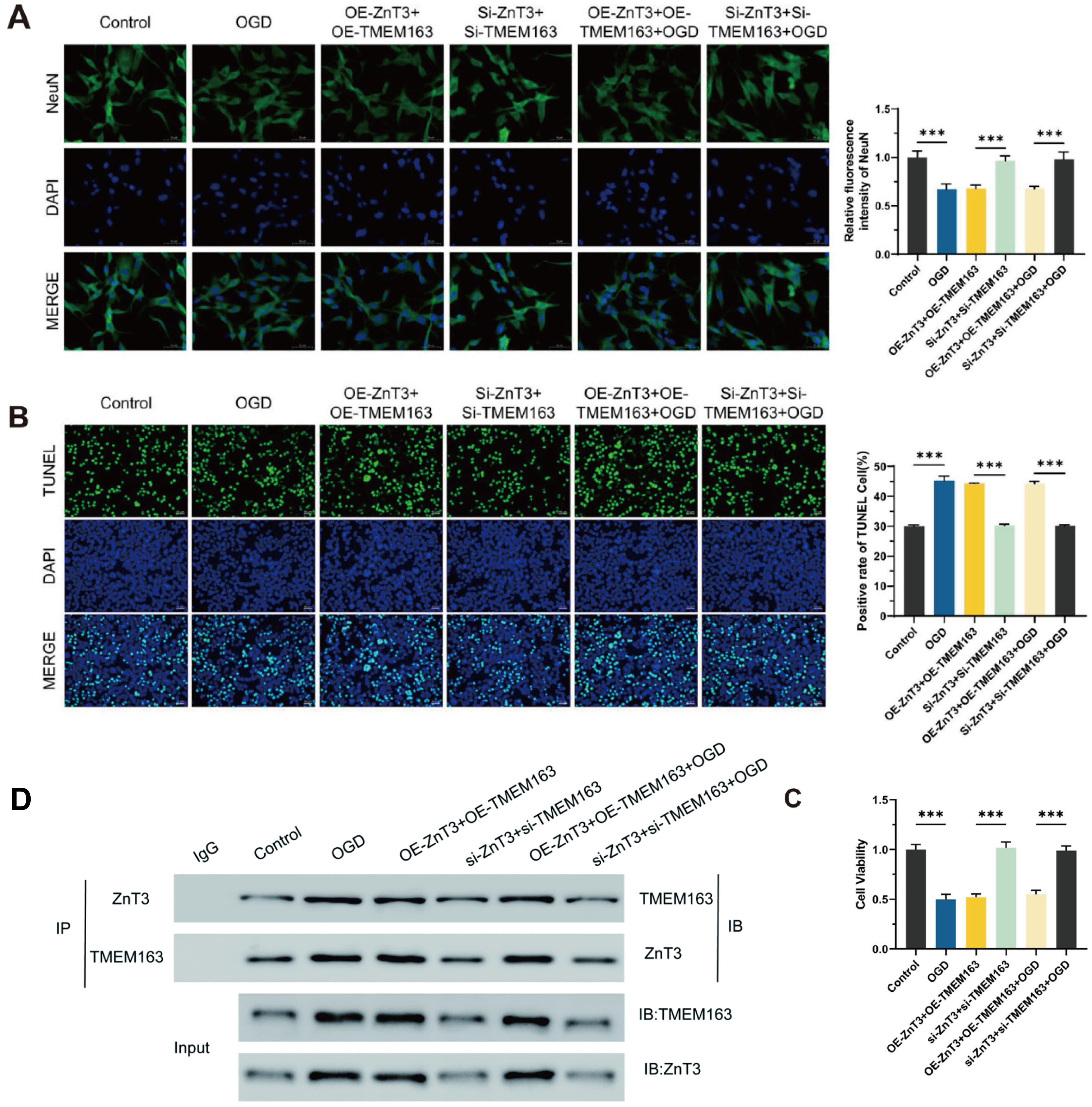 Fig. 3.
Fig. 3.
ZnT3 and TMEM163 contribute to OGD-induced PHNs degeneration.
(A) NeuN expression declined after OGD or OE but was preserved by
siRNA, scale bar = 50 μm. (B) Apoptosis increased under OGD/OE but decreased with
silencing, scale bar = 50 μm. (C) Neuronal viability followed similar trends. (D)
Co-immunoprecipitation confirms ZnT3-TMEM163 interaction. Immunoprecipitation assays detected ZnT3-TMEM163 complexes in all experimental
groups, regardless of expression levels. Data represent mean
The discovery of a ZnT3-TMEM163 complex prompted us to investigate how OGD regulates its activity. Since the subcellular localization of membrane transporters is a critical regulatory mechanism for their function, we assessed whether OGD affects the distribution of ZnT3 and TMEM163 between the cell membrane and cytoplasm. Cell surface biotinylation assays revealed that OGD induced a significant translocation of both transporters from the plasma membrane to the cytoplasm (Fig. 4). This redistribution was prevented by siRNA knockdown. This OGD-induced relocalization away from the membrane may represent a maladaptive response that disrupts precise zinc compartmentalization, ultimately contributing to the observed extracellular zinc overload and toxicity.
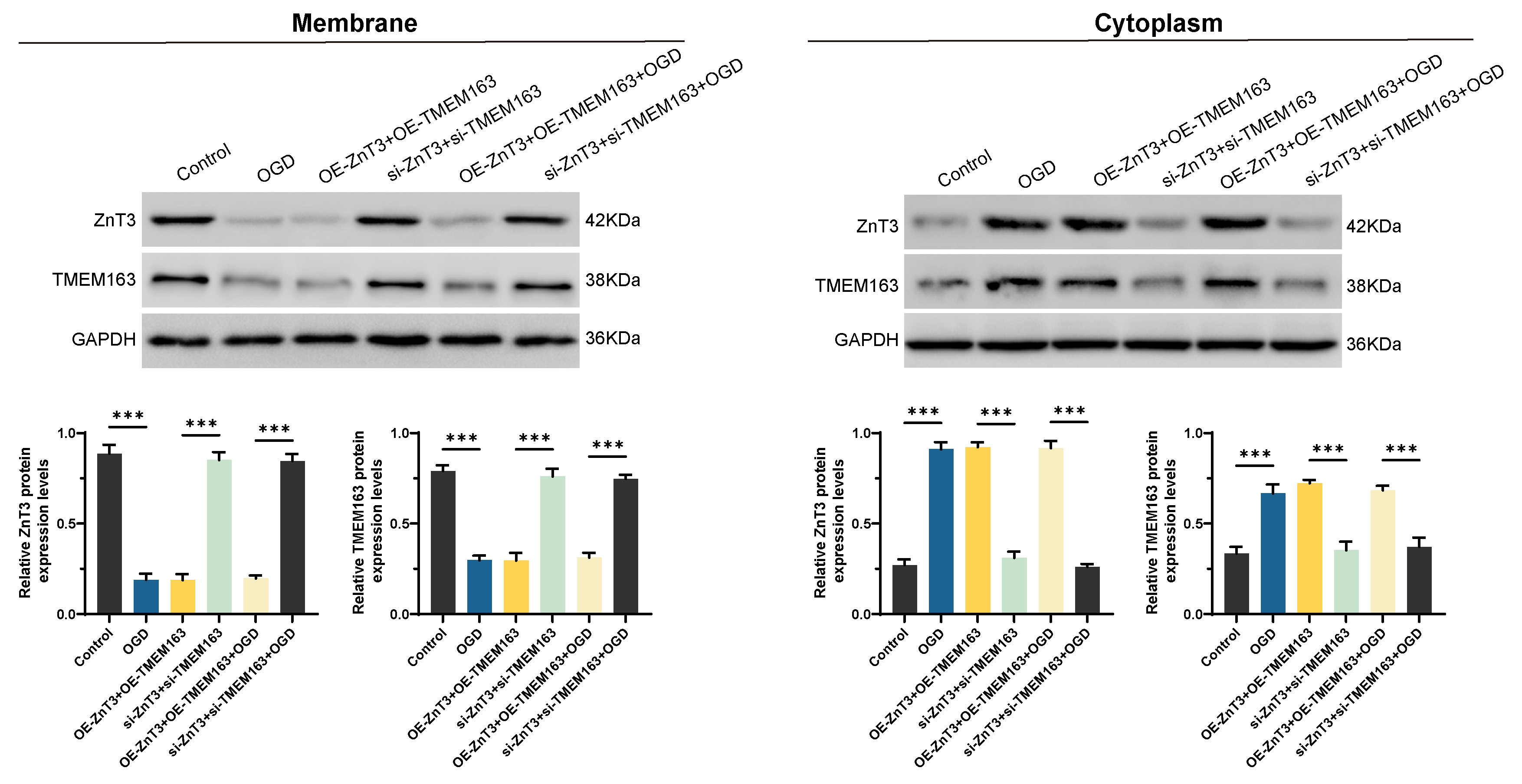 Fig. 4.
Fig. 4.
OGD promotes cytoplasmic translocation of ZnT3 and TMEM163. Under OGD, membrane-localized ZnT3/TMEM163 translocated to the cytoplasm.
Silencing reduced this translocation, while overexpression did not further
enhance it. Data represent mean
Zinc transport in neurons represents a complex physiological process that requires precise regulation by a series of transporter proteins, including ZnT family members and TMEM163 [9, 16]. Our comprehensive study has demonstrated that intracellular and extracellular zinc levels in hippocampal neurons are dynamically regulated through the coordinated actions of ZnT3 and TMEM163 proteins. Importantly, we found that OGD can induce significant abnormalities in both the distribution patterns and functional states of ZnT3-TMEM163 complexes on neuronal cell membranes and within the cytoplasm. These pathological changes ultimately lead to extracellular zinc overload and subsequent neuronal degeneration (Fig. 5). Notably, our experimental results show that targeted modulation of ZnT3-TMEM163 expression can effectively reverse OGD-induced structural and functional damage to neurons. The current findings provide a solid theoretical foundation for future research focusing on VaD animal models, while also highlighting the crucial importance of zinc-centered therapeutic targets for VaD treatment.
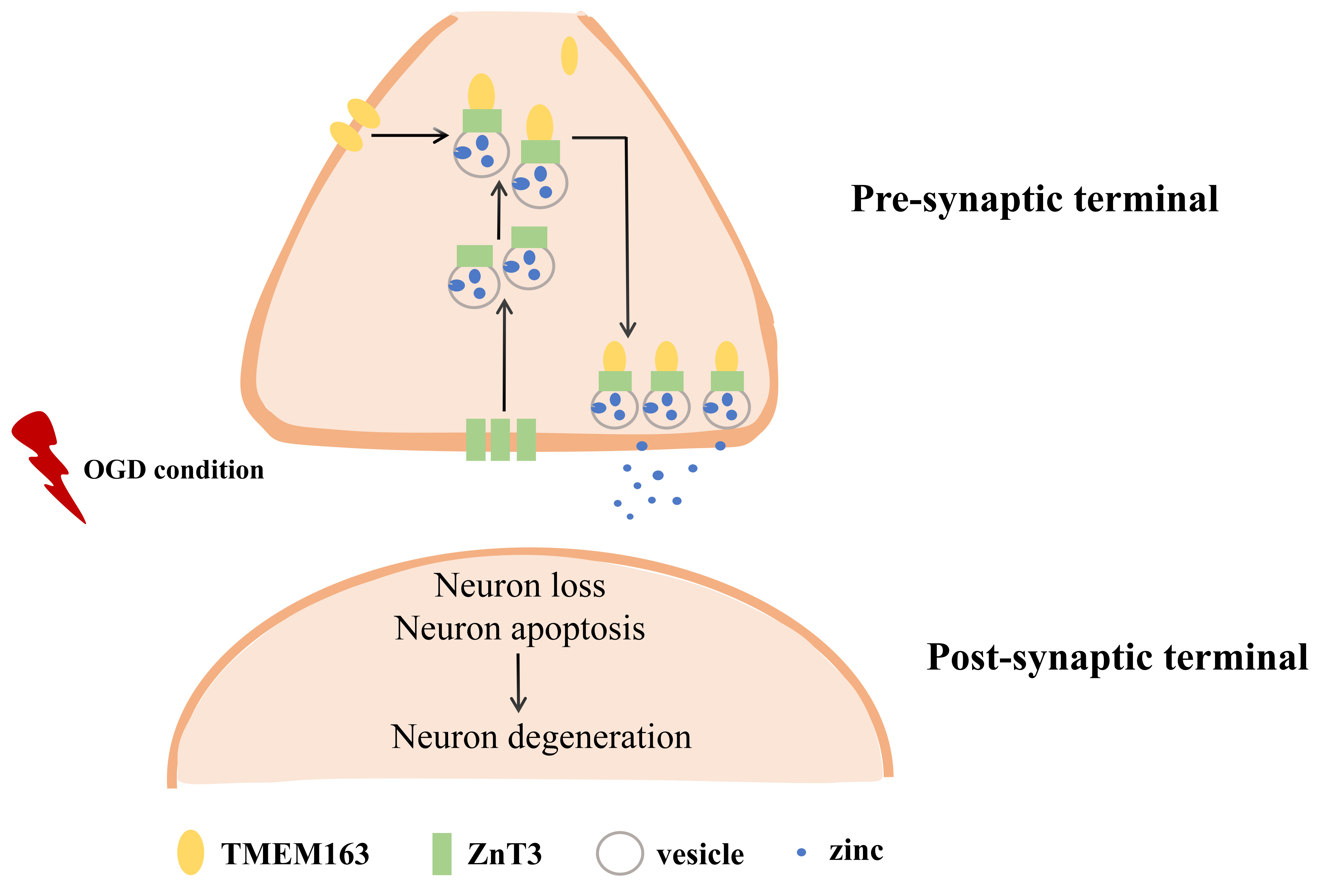 Fig. 5.
Fig. 5.
Schematic illustration of ZnT3-TMEM163 on zinc efflux and neuronal degeneration in OGD condition.
Synaptic zinc serves as a pivotal element for normal neural transmission and neurogenesis processes. The disruption of zinc homeostasis has been well documented in various neurological disorders including AD, multiple sclerosis, epilepsy, and traumatic brain injury [3, 20, 21, 22, 23]. In chronic cerebral ischemia models, elevated levels of extracellular zinc coupled with ZnT3 overexpression have been shown to significantly exacerbate blood-brain barrier damage [24]. This pathological phenomenon was similarly observed in acute ischemic cell and rat models, where zinc leakage into microvasculature activates superoxide production and increases metalloproteinase 2/9 activity, ultimately resulting in the loss of tight junction proteins and endothelial cell death [25]. Our study extends these important findings by demonstrating that zinc efflux and extracellular overload are associated not only with ZnT3 overexpression but also with increased TMEM163 expression levels. Most remarkably, even under severe OGD conditions, simultaneous gene silencing of both ZnT3 and TMEM163 could effectively reverse zinc-mediated neuronal damage. These groundbreaking results strongly suggest a previously unrecognized role of TMEM163 in zinc-related pathological mechanisms during cerebral ischemia.
Previous studies have established that TMEM163 can physically co-localize and functionally interact with TRPML1 and P2X receptors [26, 27]. A particularly relevant recent study conducted in HEK293 cells observed a strong functional correlation between TMEM163 and ZnT proteins, providing direct evidence that TMEM163 can form heterodimers with ZnT3 [19]. Intriguingly, these TMEM163-ZnT3 heterodimers exhibited similar zinc efflux capabilities as TMEM163 homodimers. These important findings suggested the intriguing possibility that disruption of the structural and functional synergy between TMEM163 and ZnT3 might represent a fundamental mechanism underlying neural injury during hypoxic conditions. In our current study, we systematically tested this hypothesis and made several key discoveries: First, we confirmed that TMEM163 and ZnT3 do indeed form immunoprecipitable complexes in neurons. Second, we found that this co-expression was significantly elevated under OGD conditions. Third, we observed that the interaction showed synchronous changes with both overexpression and silencing of the proteins. Furthermore, given the established expression synergy between ZnT3 and TMEM163 in regulating zinc homeostasis, we conducted detailed subcellular localization studies. Our results revealed a fascinating redistribution pattern: under pathological conditions, both proteins showed decreased membrane localization but increased cytoplasmic accumulation, suggesting their active translocation to facilitate zinc transport. Most importantly, when both proteins were overexpressed, we observed dramatically increased zinc outflow and corresponding neurotoxic effects. These comprehensive findings support a novel conceptual framework in which the combined dysfunction of ZnT3 and TMEM163 represents a central pathological mechanism driving zinc overload, neuronal damage, and consequent cognitive impairment in OGD model of cerebral ischemia.
Further investigation is required to elucidate the downstream signalling pathways that bridge ZnT3/TMEM163-mediated zinc efflux to neuronal apoptosis. The present study demonstrates a clear sequence of events: OGD, upregulation and dysregulation of ZnT3/TMEM163, extracellular zinc overload, neuronal apoptosis. However, the precise molecular machineries transducing zinc toxicity into activation of the apoptotic cascade are not fully delineated here. Based on existing literature, several plausible mechanisms can be proposed. Firstly, zinc overload is a potent inducer of mitochondrial dysfunction. Excessive zinc can permeate into mitochondria, leading to loss of mitochondrial membrane potential, cytochrome c release, and activation of the intrinsic (mitochondrial) apoptotic pathway [28, 29, 30]. Secondly, disruptions in zinc homeostasis are closely linked to Endoplasmic Reticulum (ER) stress. Zinc deficiency within the ER lumen, potentially exacerbated by efflux transporters like ZnT3 and TMEM163, can impair the function of ER-resident zinc-dependent enzymes, leading to the accumulation of misfolded proteins and triggering the Unfolded Protein Response (UPR), which can ultimately culminate in apoptosis [31, 32]. Furthermore, the interplay between zinc and oxidative stress is well-established. The disruption of zinc homeostasis can foster the generation of reactive oxygen species (ROS), which in turn can damage lipids, proteins, and DNA, activating stress kinases and apoptotic signaling [33, 34]. Intriguingly, our co-immunoprecipitation data confirms a physical interaction between ZnT3 and TMEM163. It is tempting to speculate that this complex may not only regulate zinc flux but also serve as a scaffold to recruit downstream signaling partners, thereby directly influencing cell death pathways. Future studies employing transcriptomic and proteomic approaches in our model will be essential to map the entire signaling network from zinc efflux to apoptosis, with a specific focus on mitochondrial integrity, ER stress markers, and oxidative stress endpoints.
It is imperative to reconcile our findings with reports that present an apparently contradictory role for ZnT3. On the one hand, in present study and others, upregulation of ZnT3 contributes to zinc efflux and neuronal injury in conditions of ischemia [12, 24]. On the other hand, as demonstrated by the preceding study, in chronic neurodegenerative models such as AD, loss of ZnT3 and vesicular zinc have been shown to accelerate amyloid plaque pathology and exacerbate cognitive decline [35, 36]. Additionally, ZnT3 plays a crucial role in maintaining adult hippocampal neurogenesis by regulating cell proliferation and neuronal differentiation in hippocampal cells [10]. The temporal and spatial dynamics of zinc release are critical. In conditions of acute ischemia, a substantial and unregulated efflux of zinc may be triggered by the upregulation of ZnT3/TMEM163, resulting in rapid excitotoxic and metabolic injury. Conversely, in chronic conditions, the physiological, activity-dependent release of zinc from synapses via ZnT3 may serve a regulatory function in modulating neuronal circuits and preventing pathological protein aggregation. Consequently, the net effect of ZnT3 activity is likely to be determined by the disease context, the magnitude and duration of zinc dyshomeostasis, and the specific cellular compartments involved.
While our study provides significant insights into the role of ZnT3-TMEM163 in OGD-induced injury, it has certain limitations that point to future research directions. First, it must be noted that a large number of zinc transporter proteins participate in the complex regulation of zinc homeostasis. Beyond ZnT3, other family members such as ZnT1 have been implicated in amyloid plaque accumulation in AD mouse models, with elevated hippocampal ZnT1 levels observed in early-stage AD patients [37]. Similarly, ZnT6 expression in the hippocampus has been shown to increase significantly in subjects with amnestic mild cognitive impairment [38]. Therefore, the specific functional contributions of individual ZnT family members require systematic investigation in future studies. Second, while our in vitro cell culture system provided excellent control over experimental variables by minimizing confounding humoral factors, the critical findings of this work clearly need validation in appropriate VaD mouse models to provide in vivo evidence for the roles of ZnT3 and TMEM163 in VaD pathogenesis. Most importantly, as noted in the preceding discussion, the specific downstream signaling cascades that link ZnT3/TMEM163-mediated zinc dyshomeostasis to the execution of neuronal apoptosis are not fully elucidated here. The mechanisms we proposed, including mitochondrial dysfunction, ER stress, and oxidative stress, require direct experimental confirmation in future studies. Unraveling these detailed pathways will be crucial for identifying the most tractable molecular targets for therapeutic intervention downstream of zinc overload.
In conclusion, our study provides compelling evidence that ZnT3 and TMEM163 engage in a sophisticated functional partnership to maintain zinc homeostasis under both physiological and hypoxic conditions. The OGD-induced degeneration of primary hippocampal neurons, characterized by significant structural damage, decreased cell viability, and increased apoptosis, appears to be mechanistically linked to extracellular zinc overload mediated by ZnT3-TMEM163 dysregulation. Most importantly, our demonstration that targeted inhibition of ZnT3 and TMEM163 can effectively mitigate neuronal degeneration highlights their strong potential as novel therapeutic targets for treating VaD and other cerebral ischemia-related cognitive disorders. These findings open new avenues for developing zinc-centered neuroprotective strategies against vascular cognitive impairment.
All data reported in this paper will be shared by the correspondence author upon reasonable request.
Study design: WY, ZG and CZ; Conceptualization: ZG and CZ; Data curation: SQ and JX; Formal analysis: JW and NL; Investigation: ZG and CZ; Writing — original draft: WY, ZG and CZ; Writing — review and editing: WY. All authors contributed to editorial changes in the manuscript. All authors read and approved the final version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
During the preparation and revision of this work, the authors used ChatGPT and DeepSeek in order to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL45588.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.