












 , Bi-He Cai 7,*
, Bi-He Cai 7,*1 Department of Medical Laboratory Science, I-Shou University, 82445 Kaohsiung, Taiwan
2 Department of Biomedical Engineering, I-Shou University, I-Shou University, 82445 Kaohsiung, Taiwan
3 Department of Medical Science and Biotechnology, I-Shou University, 82445 Kaohsiung, Taiwan
4 Department of Pathology, E-Da Hospital, 82445 Kaohsiung, Taiwan
5 Department of Physical Therapy, I-Shou University, 82445 Kaohsiung, Taiwan
6 Department of Occupational therapy, I-Shou University, 82445 Kaohsiung, Taiwan
7 School of Medicine, I-Shou University, 82445 Kaohsiung, Taiwan
†These authors contributed equally.
Abstract
TP53 gene mutations are common in breast cancer and are linked to chemoresistance. p63, a p53 family member, can induce apoptosis independently of p53, representing a potential therapeutic target in TP53-mutant tumors. This study evaluated the synergistic effects of SB431542, a TGF-β type I receptor inhibitor, and doxorubicin in TP53-mutant breast cancer cells.
Isoform-specific RT-PCR was used to assess TAp63 and ΔNp63 expression following SB431542 treatment in T47D, MDA-MB-231, and MDA-MB-468 cells. Cell viability was assessed using the CCK8 assay. Synergistic interaction was quantified using the Coefficient of Drug Interaction (CDI). Caspase-3/7 activity assays and immunocytochemistry analyses were performed to evaluate apoptotic signaling and p63 expression. Inhibition studies using PETα, a p53-family inhibitor, and a pan-caspase inhibitor were conducted to determine the pathway dependency of the observed effects.
SB431542 selectively increased TAp63 but not ΔNp63 expression in all three TP53-mutant breast cancer cells. GAS6, a TAp63 target, was also upregulated by SB431542. Treatment with SB431542 and doxorubicin used in combination significantly reduced cell viability (CDI 0.54–0.63), increased caspase activity, and enhanced p63 expression. The anticancer effect was significantly reduced by co-treatment with either the p53-family inhibitor or the pan-caspase inhibitor, confirming that the cytotoxic response was mediated through TAp63 and caspase activation.
SB431542 potentiates doxorubicin-induced apoptosis in TP53-mutant breast cancer cells by upregulating TAp63 and activating caspase-dependent pathways. These findings suggest that targeting the TGF-β/TAp63 signaling axis may offer a novel therapeutic approach to overcome chemoresistance in aggressive, TP53-mutant breast cancers.
Keywords
- SB431542
- doxorubicin
- p63
- tumor suppressor protein p53
- mutation
- breast cancer
- drug interactions
SB431542 is a selective inhibitor of the TGF-
p63 exists in multiple isoforms, some with transactivation domains
(TAp63) and others without (
In this research, we investigated the potential anticancer effects of SB431542,
a selective ALK5 inhibitor, in combination with doxorubicin in mutant p53 breast
cancer cells. SB431542 effectively inhibits TGF
Human breast cancer cell lines T47D, MDA-MB-231 and BT-474 were obtained from
the Bioresource Collection and Research Center (BCRC, Hsinchu, Taiwan), while
MDA-MB-468 cells were acquired from the American Type Culture Collection (ATCC,
Manassas, VA, USA). Cells were cultured at 37 °C in a humidified
atmosphere containing 5% CO2. MDA-MB-231 and MDA-MB-468 cells were
maintained in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen, Carlsbad, CA,
USA), whereas T47D cells were grown in RPMI-1640 medium (Invitrogen, Carlsbad,
CA, USA). BT-474 were maintained in Hybri-Care Medium (ATCC, Catalog No. 46-X).
All media were supplemented with 10% fetal bovine serum (FBS; Invitrogen), 100
U/mL penicillin, and 100 µg/mL streptomycin (Invitrogen). All cell
lines were validated by STR profiling and tested negative for mycoplasma. Drug
treatments included: vehicle control (DMSO), SB431542 (20 µM;
MedChemExpress, Monmouth Junction, NJ, USA), doxorubicin (1 µM;
Sigma-Aldrich, St. Louis, MO, USA), PET
Cell viability was determined using the CCK-8 assay (Invitrogen). CCK-8 reagent
(10 µL) was added to each well of a 96-well plate, and the plate was
incubated at 37 °C in a CO2 incubator for 1 h. Absorbance was
measured at 450 nm using a SpectraMax iD3 microplate reader (Molecular Devices,
Silicon Valley, CA, USA). Wells containing only medium (no cells) were used as
blanks. Cells treated with DMSO served as the reference control and were set as
100% viability for normalization. Cell viability (%) was calculated using the
formula: [(OD(Drug) – OD(Blank))/(OD(control) – OD(Blank))]
Drug interaction was assessed using the coefficient of drug interaction (CDI),
calculated as: CDI = AB/(A
Total RNA was extracted using TRIzol reagent (Invitrogen). For cDNA synthesis, 1
µg of RNA was reverse transcribed with the QuantiNova Reverse
Transcription Kit (Qiagen, Hilden, Germany). Quantitative PCR was performed using
HOT FIREPol EvaGreen qPCR Mix Plus (Omics Bio, New Taipei City, Taiwan) and
gene-specific primers on a CFX Duet Real-Time PCR System (Bio-Rad, Hercules, CA,
USA). Relative gene expression was calculated using the
2-ΔΔCT method, with GAPDH as the internal control. Primer
sequences were: GAPDH: F 5′-GTCTCCTCTGACTTCAACAGCG-3′, R
5′-ACCACCCTGTTGCTGTAGCCAA-3′; TAp63: F
5′-TGTATCCGCATGCAGGACT-3′, R 5′-CTGTGTTATAGGGACTGGTGGAC-3′;
Cells were washed twice with PBS and fixed in 4% paraformaldehyde (100
µL per well) for 20 min. After two washes with wash buffer (1% BSA
in PBS), cells were blocked with 1% BSA and 0.2% Triton X-100 in PBS for 30
min. Primary antibody against p63
Caspase-3/7 activity was measured using the CellEvent Caspase-3/7 Red Detection Reagent (Invitrogen). The reagent was diluted in PBS to a final concentration of 5 µM and added to cells (100 µL per well) after removal of culture medium. Cells were incubated at 37 °C for 30 min in the dark. Nuclei were counterstained with Hoechst 33342 (1 µg/mL). Fluorescent images were captured using an ECLIPSE Ts2 fluorescence microscope (Nikon).
Cells cultured in 6-well plates were transfected with 25 nM (final concentration) of either control siRNA (sc-37007) or p63 siRNA (sc-36161) using 7.5 µL of TransIT-X2 Transfection Reagent (Mirus Bio, Madison, WI, USA; #MIR6000) for 48 hours. Following transfection, cells were harvested for RNA isolation and subsequently analyzed for p63 expression using quantitative real-time RT-PCR.
Data are presented as mean
To investigate the effect of SB431542 on the expression of p63 isoforms in
p53-mutant breast cancer cells, isoform-specific RT-PCR was performed. In T47D
cells harboring the p53 L194F mutation, SB431542 treatment selectively increased
TAp63 expression without affecting
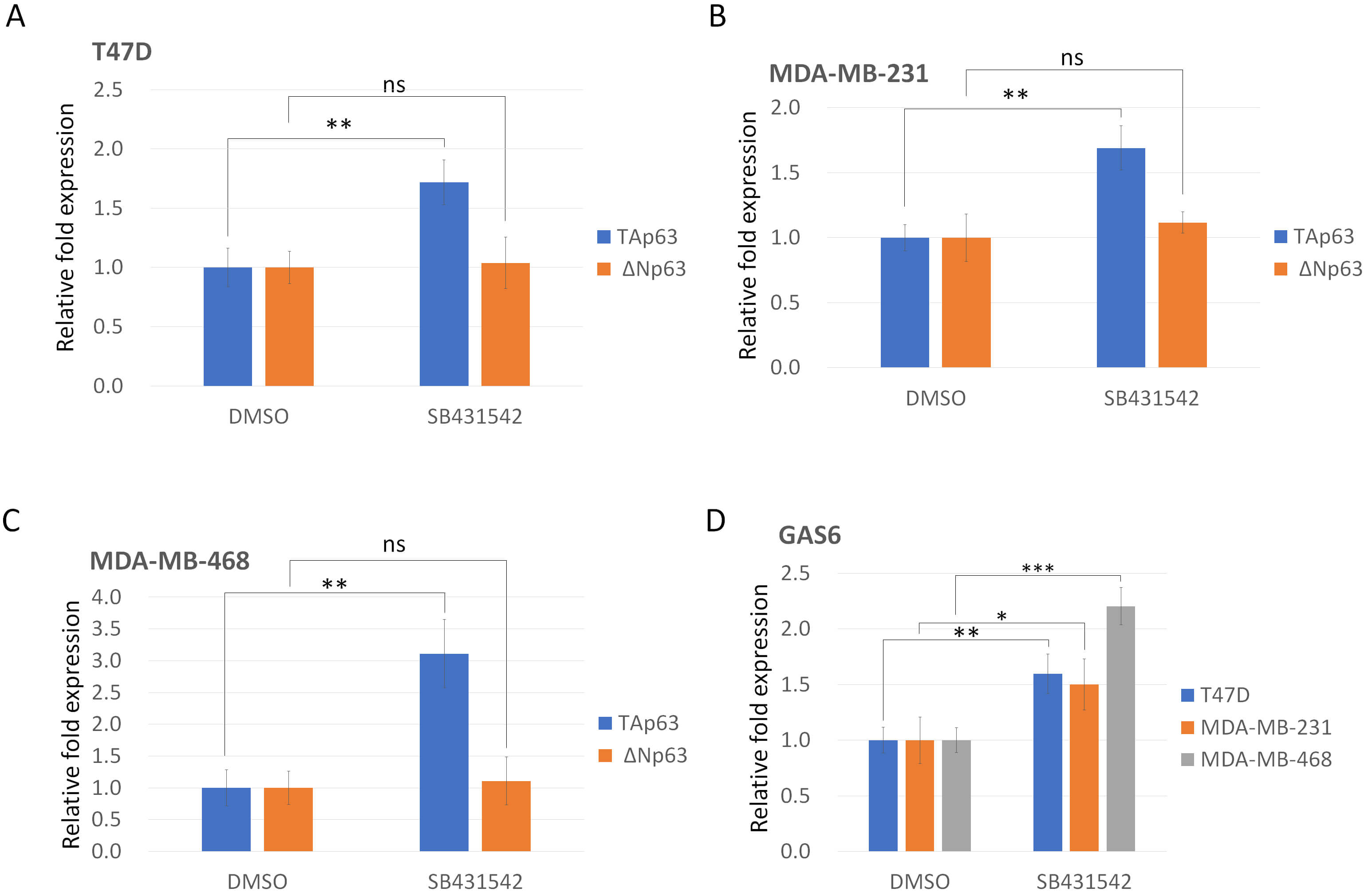 Fig. 1.
Fig. 1.
SB431542 increases TAp63 and its downstream target gene GAS6
expression in p53-mutant breast cancer cells. Breast cancer cells were treated
with SB431542 (20 µM) for 48 hours, after which cells were harvested for
RNA isolation and analyzed for mRNA expression using quantitative real-time
RT-PCR. (A) In T47D cells (harboring the p53 L194F mutation), isoform-specific
primers were used to detect the expression of TAp63 and
We first evaluated a range of SB431542 concentrations in T47D cells while co-treating with a fixed dose of doxorubicin to assess their combined cytotoxic effects (Supplementary Fig. 1). From this dose–response analysis, we determined that SB431542 had an IC50 value of 37.1 µM under these conditions (Supplementary Fig. 2). Notably, among the various concentration combinations, treatment with SB431542 at 20 µM together with doxorubicin at 1 µM resulted in the most pronounced synergistic reduction in cell viability (Supplementary Table 1). To determine whether SB431542 sensitizes p53-mutant cells to chemotherapy, T47D, MDA-MB-231, MDA-MB-468, and BT-474 (harboring the p53 E285K mutation) cells were treated with SB431542 (20 µM), doxorubicin (1 µM), or their combination for 48 hours. Cell viability was assessed using the CCK-8 assay (Fig. 2). In all four cell lines, the combination of SB431542 and doxorubicin significantly reduced cell viability compared to either agent alone, indicating a synergistic cytotoxic effect.
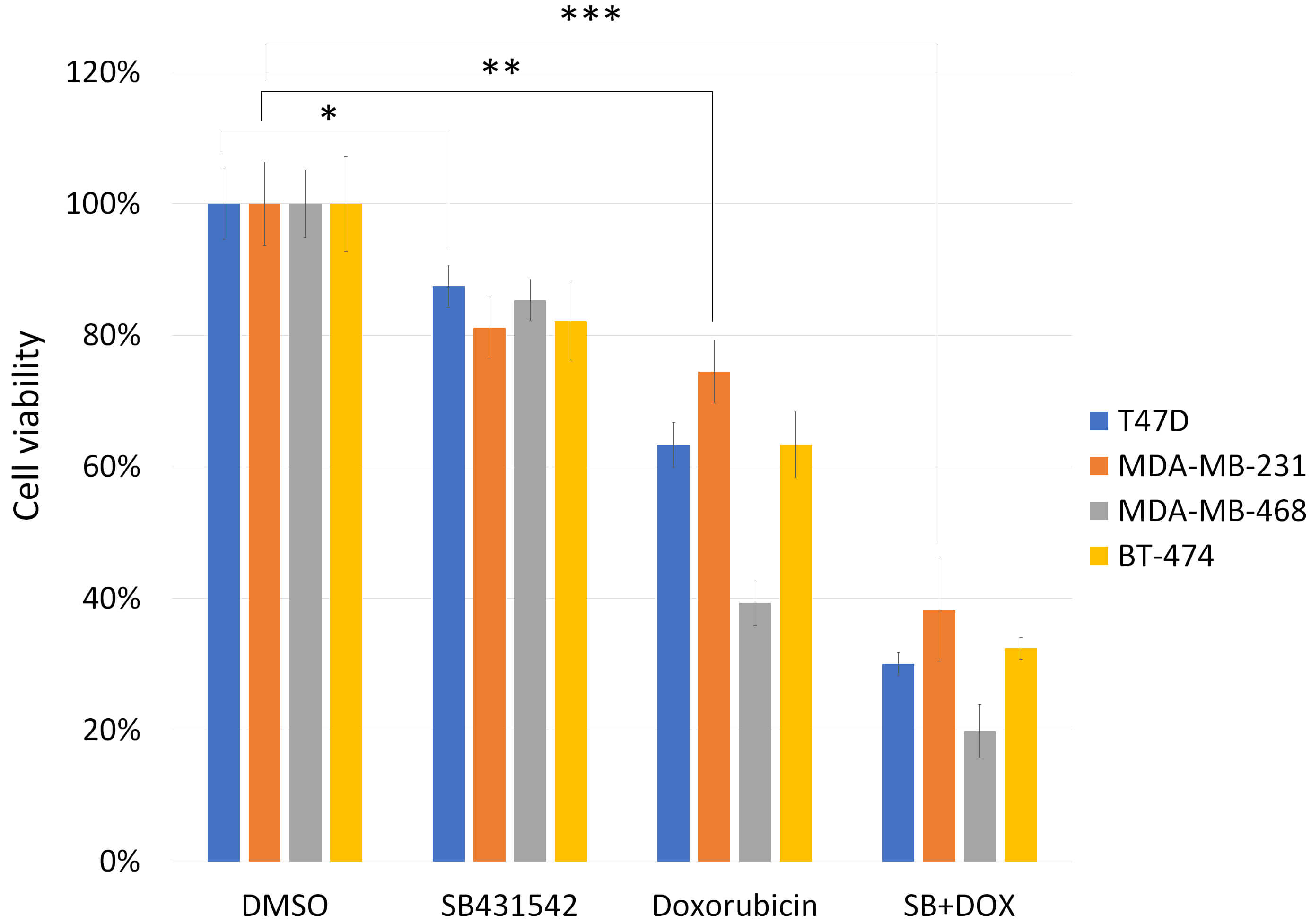 Fig. 2.
Fig. 2.
Cytotoxic effects of SB431542, doxorubicin, or a
combination of both on p53-mutated breast cancer cells. T47D, MDA-MB-231,
MDA-MB-468 and BT474 breast cancer cells were treated with SB431542 (20
µM), doxorubicin (1 µM), or a combination of both for 48 hours. Cell
viability was assessed using the CCK-8 assay. The combination treatment
significantly reduced cell viability compared to either agent alone, indicating
enhanced cytotoxicity. Cells treated with DMSO alone were used as the baseline
control and defined as 100% viability, serving as the reference point for
normalizing responses to other drug treatment conditions. Data are presented as
the mean
Drug interactions were quantitatively evaluated by calculating the Coefficient of Drug Interaction (CDI). CDI values of 0.54 in T47D cells, 0.63 in MDA-MB-231 cells, 0.59 in MDA-MB-468 cells, and 0.62 in BT-474 (Table 1) all fell below 1, further confirming a synergistic interaction between SB431542 and doxorubicin.
| T47D | DMSO | SB431542 | Doxorubicin | SB+DOX |
| 20 μM | 1 μM | |||
| 100.00 (%) | 87.45 (%) | 63.32 (%) | 30.03 (%) | |
| CDI = [ survival% (Drug A + DrugB) ] / [survival% (Drug A) * survival% (Drug B) ] | CDI | |||
| 0.54 | ||||
| MDA-MB-231 | DMSO | SB431542 | Doxorubicin | SB+DOX |
| 20 μM | 1 μM | |||
| 100.00 (%) | 81.14 (%) | 74.51 (%) | 38.27 (%) | |
| CDI = [ survival% (Drug A + DrugB) ] / [survival% (Drug A) * survival% (Drug B) ] | CDI | |||
| 0.63 | ||||
| MDA-MB-468 | DMSO | SB431542 | Doxorubicin | SB+DOX |
| 20 μM | 1 μM | |||
| 100.00 (%) | 85.36 (%) | 39.36 (%) | 19.87 (%) | |
| CDI = [ survival% (Drug A + DrugB) ] / [survival% (Drug A) * survival% (Drug B) ] | CDI | |||
| 0.59 | ||||
| BT-474 | DMSO | SB431542 | Doxorubicin | SB+DOX |
| 20 μM | 1 μM | |||
| 100.00 (%) | 82.15 (%) | 63.39 (%) | 32.40 (%) | |
| CDI = [ survival% (Drug A + DrugB) ] / [survival% (Drug A) * survival% (Drug B) ] | CDI | |||
| 0.62 | ||||
The drug interaction between SB431542 and doxorubicin was evaluated using the
Coefficient of Drug Interaction (CDI), calculated as: CDI = AB/(A
To assess apoptosis induction, caspase-3/7 activity was examined by fluorescent staining. In MDA-MB-231 cells, SB431542 induced a mild increase in caspase-3/7 activation, whereas doxorubicin caused a more substantial effect. The combination treatment led to a pronounced increase in red fluorescence, indicating enhanced apoptotic signaling (Fig. 3). Similarly, in T47D cells, SB431542 alone modestly increased caspase activity, whereas doxorubicin induced a strong response. The combination further amplified both the intensity and frequency of caspase-positive cells (Fig. 4), consistent with a synergistic apoptotic effect.
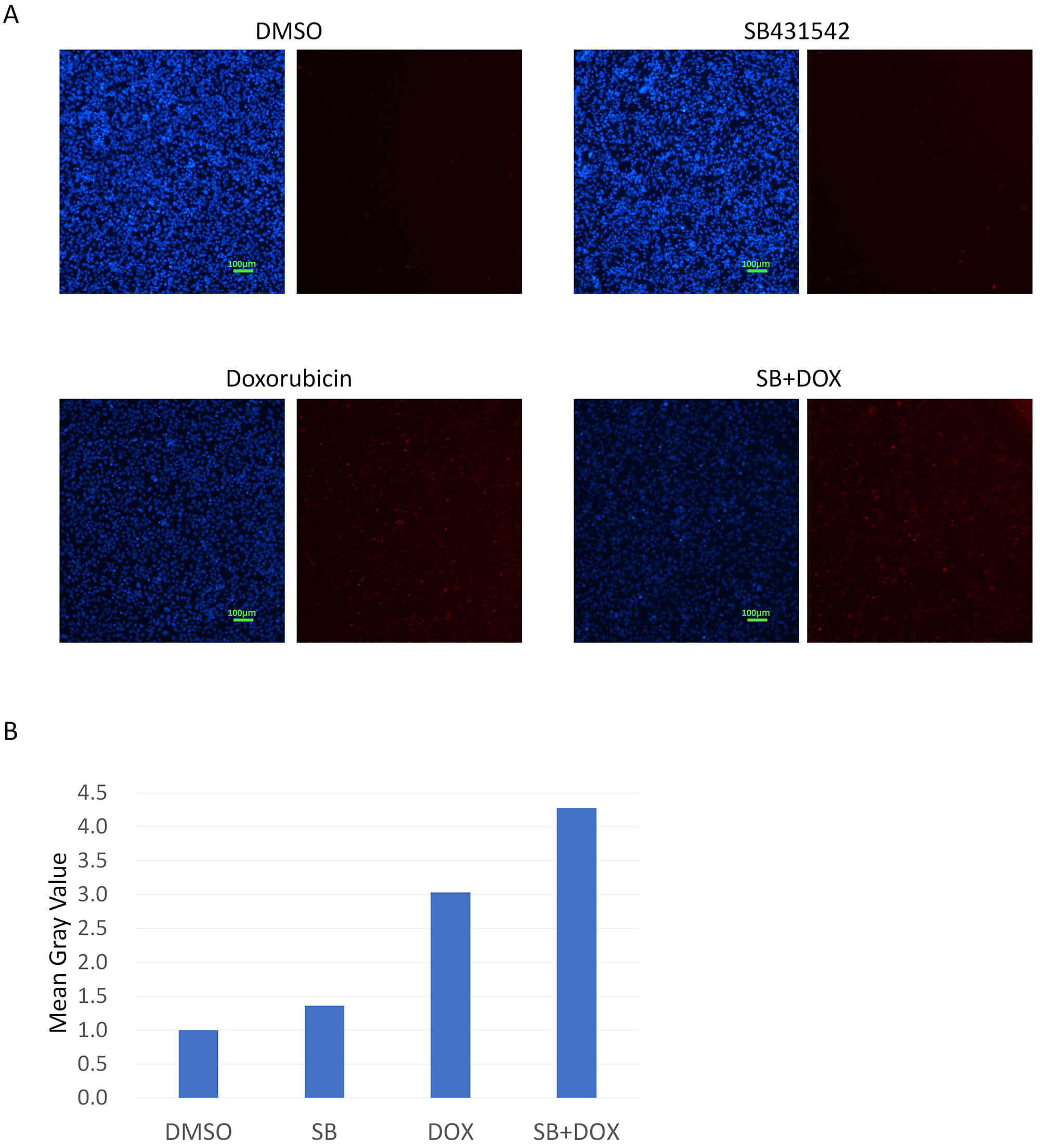 Fig. 3.
Fig. 3.
Caspase-3/7 activation in MDA-MB-231 cells treated with
SB431542, doxorubicin, or a combination of both. (A) MDA-MB-231 cells were
treated with SB431542, doxorubicin, or a combination of both, followed by direct
staining for Caspase-3/7 activity (red fluorescence). Compared to the DMSO
control, SB431542 alone caused a slight increase in Caspase-3/7 activation.
Doxorubicin alone induced a marked increase in Caspase-3/7 activity. The
combination of SB431542 and doxorubicin further enhanced this activation,
suggesting a synergistic induction of apoptosis. Hoechst 33342 was used as a
nuclear counterstain (blue). Scale bar, 100 µm (10
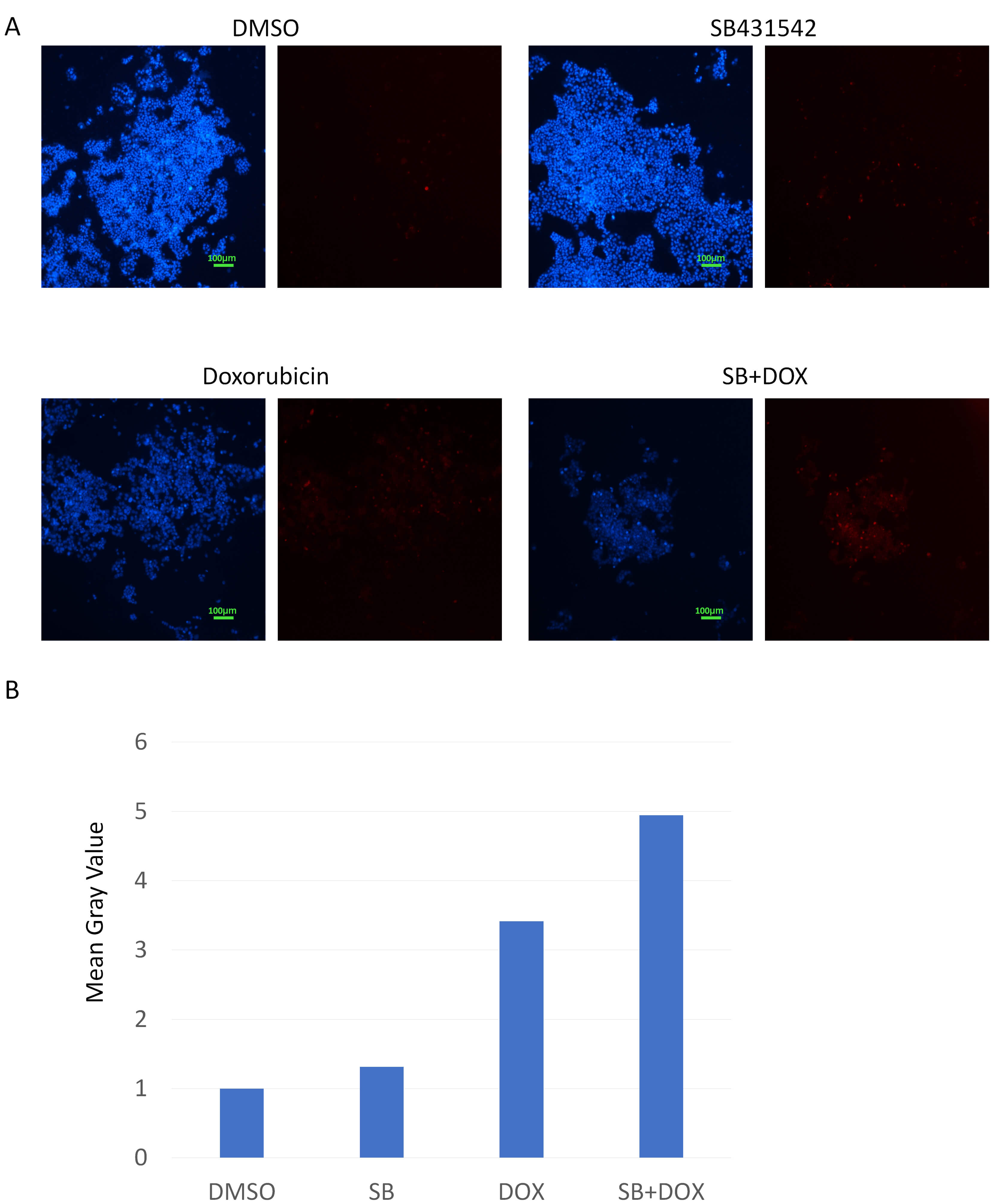 Fig. 4.
Fig. 4.
Caspase-3/7 activation in T47D cells treated with
SB431542, doxorubicin, or a combination of both. (A) T47D cells were treated and
stained for Caspase-3/7 activity. SB431542 alone resulted in a moderate increase
in caspase activation. Doxorubicin significantly induced Caspase-3/7 activity
(red). The combination treatment further enhanced both the number and intensity
of red fluorescent signals, indicating pronounced apoptotic induction. Hoechst
33342 was used as a nuclear counterstain (blue). Scale bar, 100 µm
(10
Immunofluorescence staining revealed cell line-specific effects on p63 protein expression. In MDA-MB-231 cells, SB431542 alone had minimal impact on p63 levels, while doxorubicin induced a clear upregulation. The combination treatment further increased the proportion of p63-positive nuclei, despite a reduction in overall cell number (Fig. 5). In T47D cells, SB431542 alone significantly elevated p63 expression, which was further enhanced by doxorubicin (Fig. 6). SB431542 and doxorubicin elicited comparable p63 induction in MDA-MB-468 and BT-474 cells (Supplementary Figs. 3,4). Notably, nearly all surviving cells exhibited strong nuclear p63 signals under combination treatment, suggesting a link between p63 activation and apoptosis (Figs. 5,6, Supplementary Figs. 3,4).
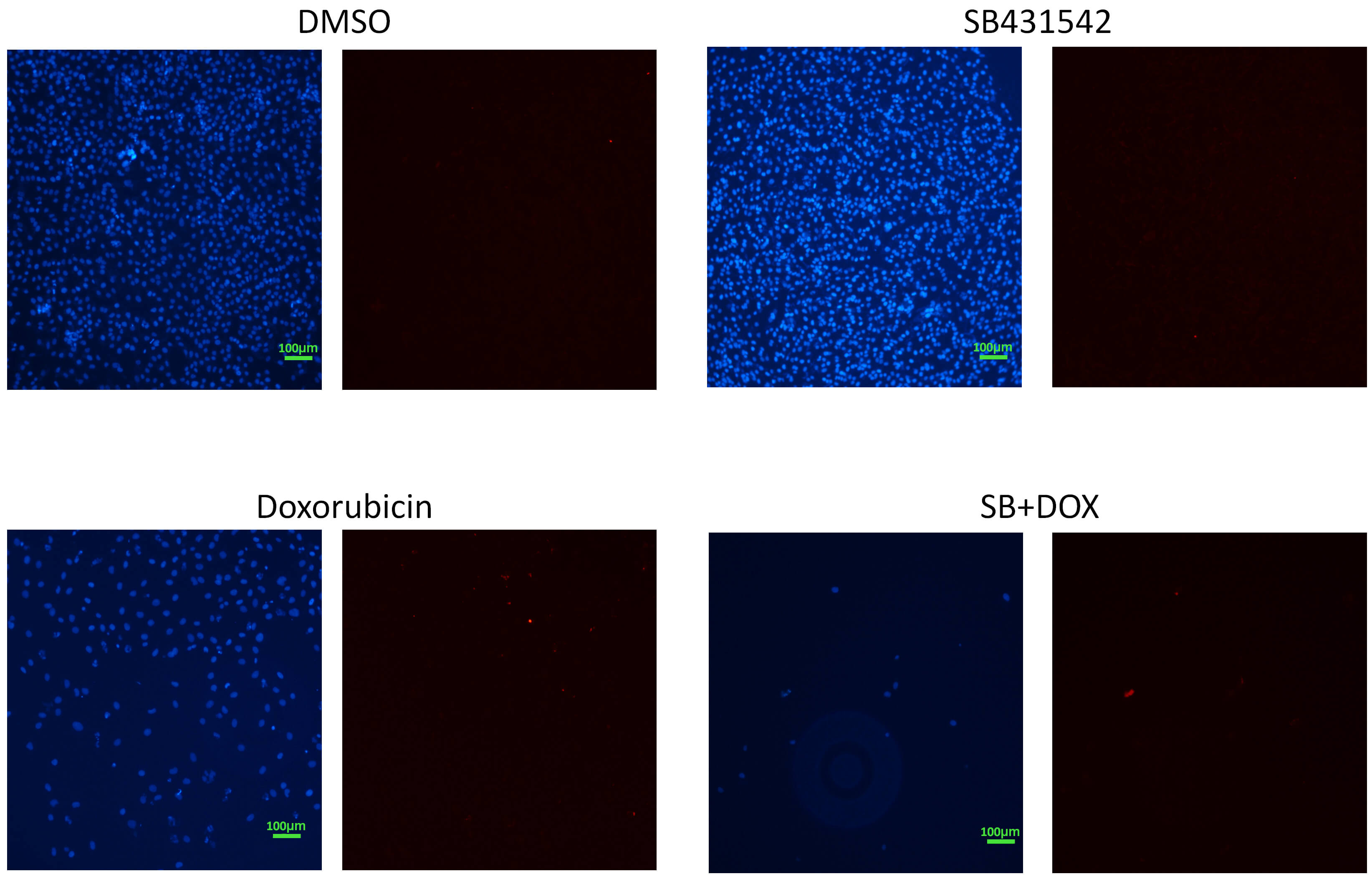 Fig. 5.
Fig. 5.
p63 expression in MDA-MB-231 cells detected by
immunofluorescence. MDA-MB-231 cells were stained for p63 (red). SB431542
treatment did not markedly alter p63 levels. In contrast, doxorubicin increased
p63 expression. Combination treatment with SB431542 and doxorubicin, although
reducing overall cell number, resulted in a nearly 1:1 ratio of red fluorescent
signals to nuclei, indicating an increased p63 expression per cell. Hoechst 33342
was used as a nuclear counterstain (blue). Scale bar, 100 µm
(10
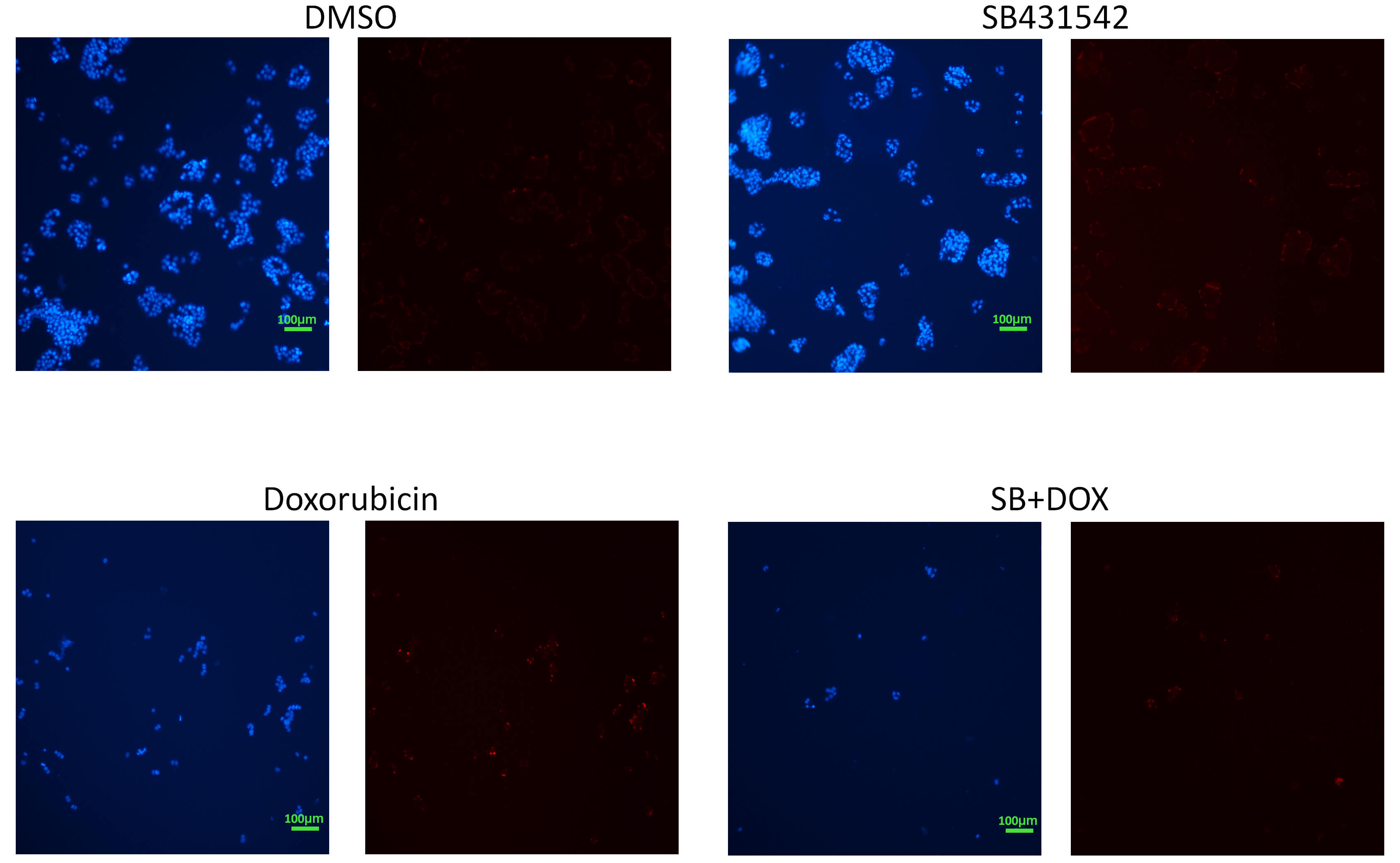 Fig. 6.
Fig. 6.
p63 expression in T47D cells detected by
immunofluorescence. T47D cells showed a substantial increase in p63 expression
(red) upon treatment with SB431542 alone. Doxorubicin similarly induced p63
expression. The combination treatment resulted in a reduced number of cells, but
with nearly all nuclei showing p63 positivity, further confirming increased p63
expression and suggesting a link to apoptotic activity. Hoechst 33342 was used as
a nuclear counterstain (blue). Scale bar, 100 µm (10
To determine whether the cytotoxic effect of the combination treatment is
dependent on p53 family signaling, breast cancer cells were treated with SB431542
and doxorubicin in the presence or absence of PET
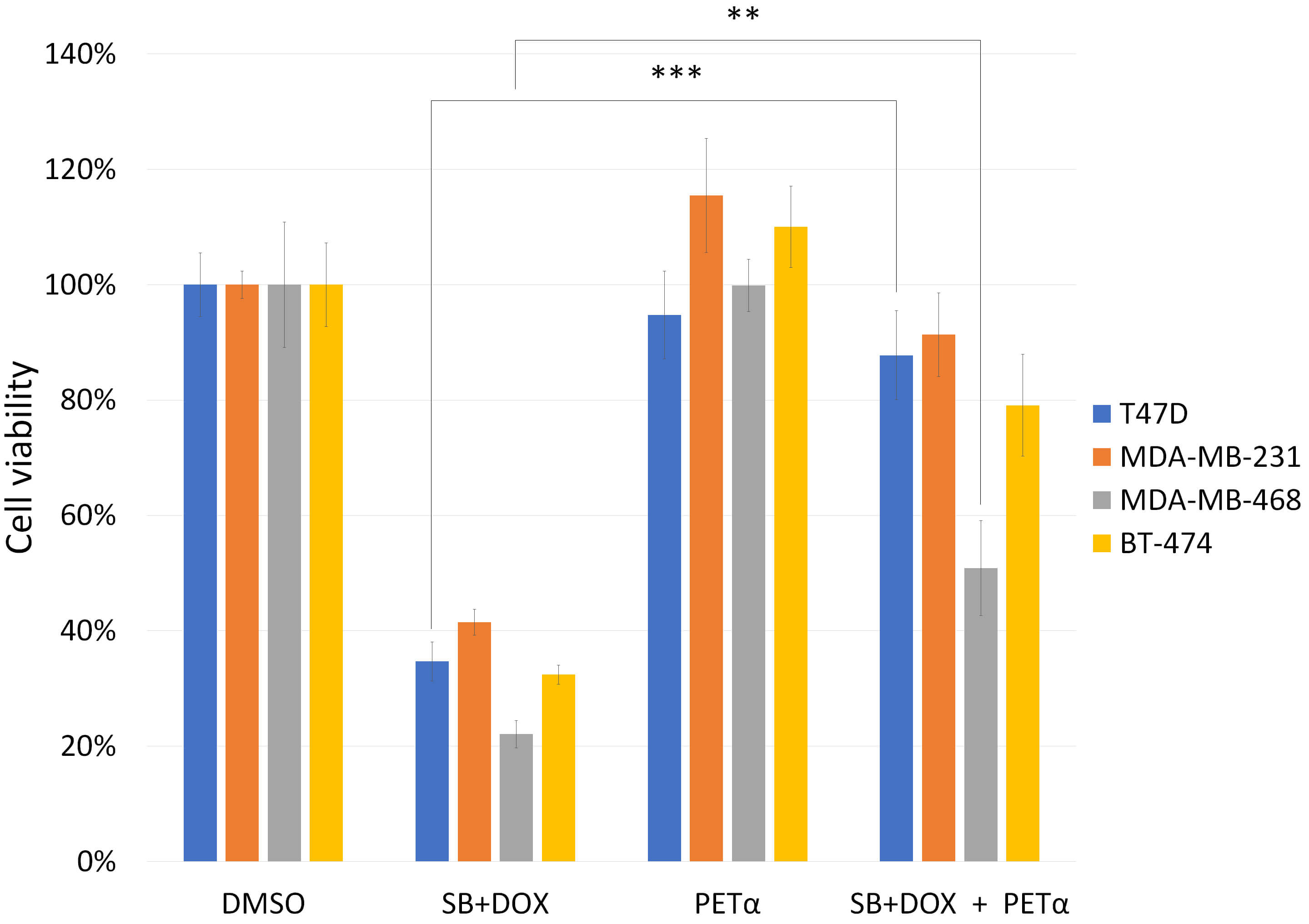 Fig. 7.
Fig. 7.
Effect of p53 family inhibition on SB431542 and
doxorubicin cytotoxicity in breast cancer cells. T47D, MDA-MB-231,
MDA-MB-468 and BT-474 cells were treated with SB431542,
doxorubicin, and a combination of both with or without the p53 family inhibitor
PET
Furthermore, the pan-caspase inhibitor (Z-VAD-FMK) reversed the reduction in cell viability caused by the combination treatment (Fig. 8), demonstrating that caspase activity is required for the observed cell death, and confirming the apoptotic nature of the cytotoxic response.
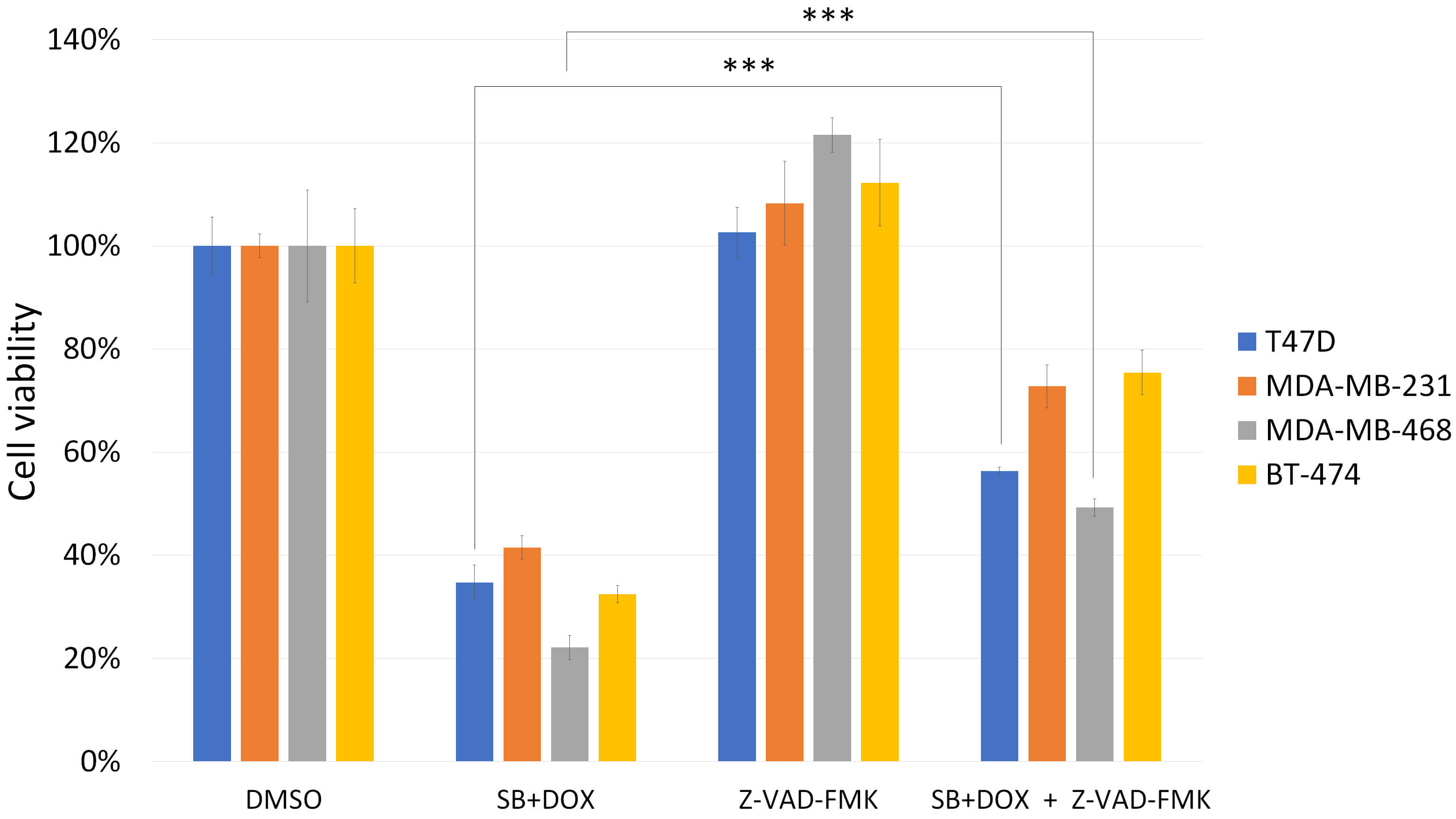 Fig. 8.
Fig. 8.
Effect of caspase inhibition on SB431542 and doxorubicin
cytotoxicity in breast cancer cells. T47D, MDA-MB-231, MDA-MB-468 and
BT-474 cells were treated as above, with or without the pan-caspase
inhibitor (Z-VAD-FMK). Similar to Fig. 7, the combination of SB431542 and
doxorubicin significantly reduced cell viability after 48 hours. Co-treatment
with Z-VAD-FMK reversed this effect, indicating that the cell death induced by
SB431542 and doxorubicin is caspase-dependent and likely apoptotic in nature.
Cells treated with DMSO alone were used as a 100% viability control, serving as
the reference for normalizing responses to drug treatments. Data are presented as
mean
To determine whether p63 mediates the cytotoxic effects of SB431542 and doxorubicin, we performed p63 knockdown using siRNA. Both SB431542 and doxorubicin induced TAp63 expression, and this induction was abolished by p63 siRNA (Fig. 9A). The combination of SB431542 and doxorubicin further enhanced p63 expression, which was likewise reversed by p63 knockdown. Consistently, treatment with SB431542, doxorubicin, or a combination of both significantly reduced cell viability; however, these cytotoxic effects were markedly rescued in cells transfected with p63 siRNA (Fig. 9B). These findings indicate that p63 plays a critical role in mediating the cytotoxicity induced by SB431542 and doxorubicin.
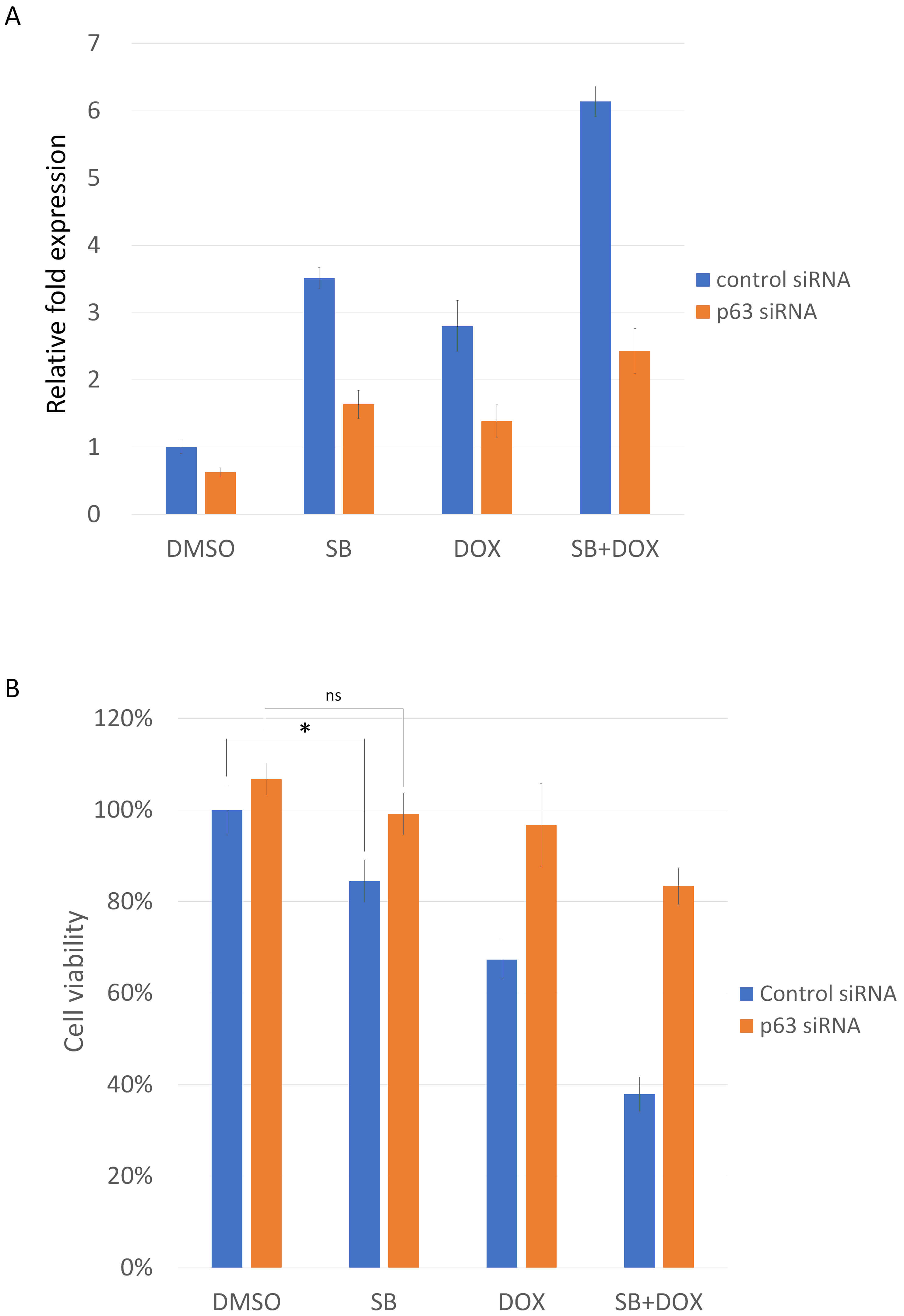 Fig. 9.
Fig. 9.
p63 mediates the cytotoxic effects of SB431542 and doxorubicin.
(A) Real-Time RT-PCR analysis showing TAp63 expression following treatment with
SB431542, doxorubicin, or a combination of both, with or without p63 siRNA.
Drug-induced TAp63 upregulation is reversed by p63 knockdown. Cells transfected
with control siRNA and subsequently treated with DMSO alone were used as the
baseline control and set to a relative value of 1. (B) Cell viability assays
demonstrating reduced viability after SB431542, doxorubicin, or combination
treatment. The decrease in viability is rescued by p63 knockdown, indicating
p63-dependent cytotoxicity. Cells transfected with control siRNA and subsequently
treated with DMSO alone served as the baseline control and were designated as
100% viable. Data are presented as the mean
To further investigate the role of p63 in breast cancer, we analyzed its
expression across cancer types and clinical subgroups. According to the UALCAN
database [29], the most pronounced reduction in p63 expression occurs
specifically in breast cancer and prostate adenocarcinoma, a pattern not observed
in most other cancer types (Fig. 10A). Consistent with this finding, p63
expression was markedly lower in breast cancer tissue compared with normal breast
tissue (Fig. 10B). Within breast cancer subtypes, p63 expression follows the
trend: normal
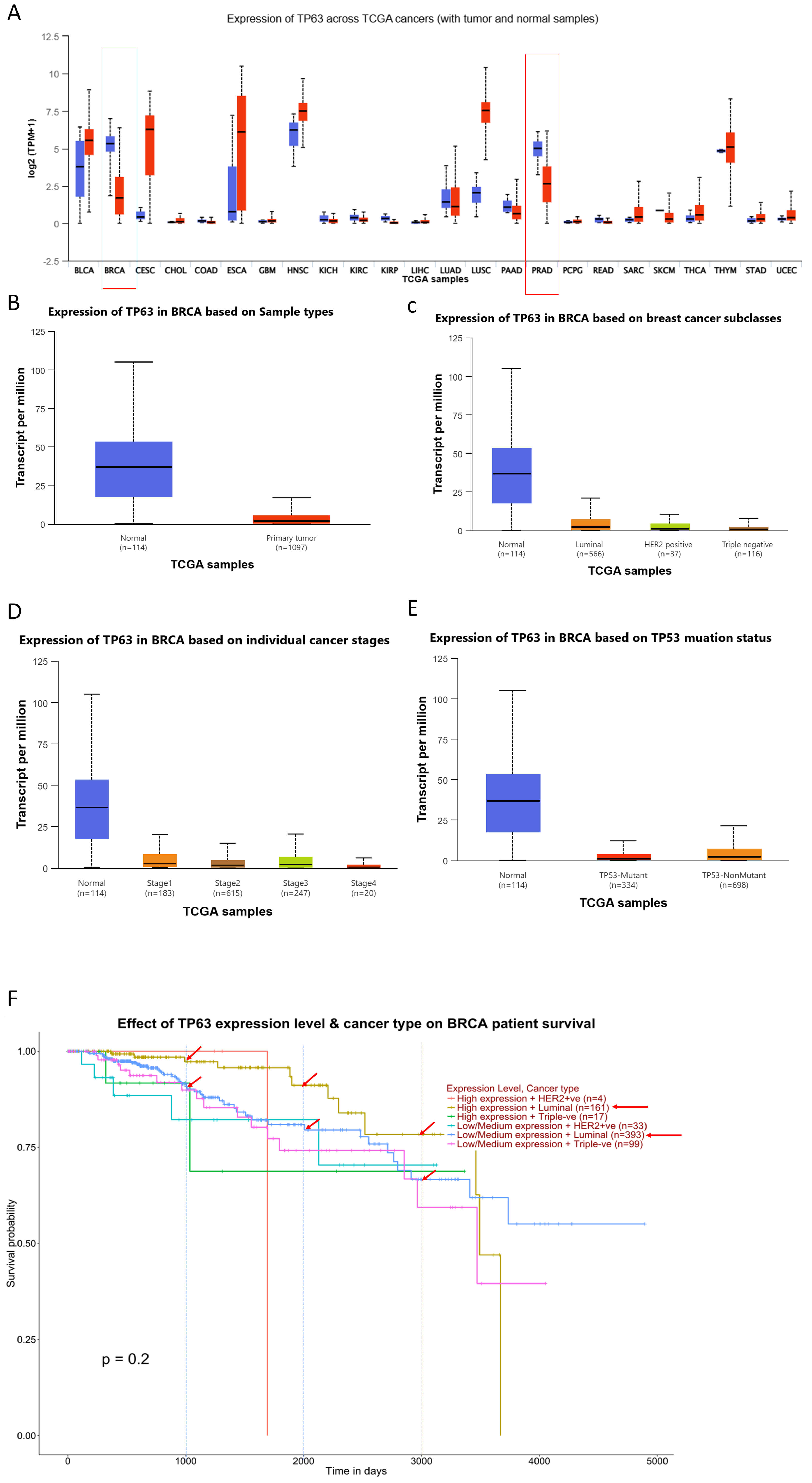 Fig. 10.
Fig. 10.
p63 expression patterns and clinical significance in breast
cancer. (A) Pan-cancer analysis of p63 expression across multiple tumor types. A
marked downregulation of p63 is observed specifically in breast cancer and
prostate adenocarcinoma (red columns), whereas most other cancers do not show
significant changes. (B) Comparison of p63 expression between normal breast
tissue and breast cancer samples. Breast cancer shows significantly reduced p63
expression. (C) p63 expression across breast cancer subtypes. Expression levels
follow the trend: normal
Our findings demonstrate that SB431542 enhances the cytotoxic and apoptotic
effects of doxorubicin in breast cancer cells harboring mutant p53, particularly
through mechanisms involving TAp63 activation. In p53-mutated breast cancer
cells, combination treatment with SB431542 and doxorubicin significantly reduced
cell viability, as evidenced by CCK-8 assay results (Fig. 2), and yielded a CDI
value of 0.54–0.63 (Table 1), indicating a synergistic interaction. Apoptosis
was markedly increased in both MDA-MB-231 and T47D cells upon combination
treatment, as indicated by enhanced caspase-3/7 activity (Figs. 3,4). These
results are consistent with prior reports that TGF-
Mechanistically, our data suggest that the enhanced apoptosis induced by
SB431542 and doxorubicin is at least partially mediated through the activation of
p63. Immunofluorescence staining revealed that p63 expression was upregulated in
T47D and MDA-MB-231 cells treated with either SB431542 or doxorubicin, with
further enhancement observed upon combination treatment (Figs. 5,6). Since TAp63
has been shown to promote apoptosis, cell cycle arrest, and DNA damage response
in p53-deficient contexts [16, 31], these findings support the hypothesis that
SB431542 may enhance doxorubicin-induced cytotoxicity by upregulating TAp63. This
is further supported by the reversal of cytotoxicity upon co-treatment with
PET
Our results also indicate that apoptosis is the primary mode of cell death induced by the combination treatment, as evidenced by caspase dependency. Co-treatment with a pan-caspase inhibitor (Z-VAD-FMK) significantly attenuated the cytotoxic effect of SB431542 and doxorubicin in both T47D and MDA-MB-468 cells (Fig. 8), consistent with caspase-3/7 activation data. This suggests that the observed cell death is caspase-mediated and apoptotic in nature. Previous studies have demonstrated that activation of TAp63 can trigger mitochondrial apoptosis pathways, particularly in the context of p53 mutation or loss [16]. Our findings align with these observations and extend them by identifying a novel combination—SB431542 plus doxorubicin—that effectively activates this alternative apoptotic pathway in mutant p53 breast cancer models.
This research provides compelling evidence that SB431542 enhances the antitumor efficacy of doxorubicin in breast cancer cells with mutant p53, through a mechanism that involves activation of TAp63 and caspase-mediated apoptosis. This is of clinical interest as TP53 mutations are present in over 30% of breast cancers, particularly in aggressive subtypes such as triple-negative breast cancer (TNBC), and are often associated with poor response to chemotherapy and increased recurrence [32]. Targeting the function of mutant p53 may pave the way for novel treatments in aggressive TNBC [33]. In this study, two cell lines—MDA-MB-231, and MDA-MB-468—were TNBC cells. This means our findings are particularly relevant to TNBC, a breast cancer subtype that currently lacks targeted therapies and is often resistant to conventional chemotherapy. The observed enhancement of doxorubicin efficacy by SB431542 suggests that this combination could represent a promising therapeutic strategy specifically for TNBC patients harboring TP53 mutations. By activating alternative tumor-suppressive pathways such as TAp63 and promoting caspase-mediated apoptosis, this approach may help overcome the limitations imposed by mutant p53 and improve clinical outcomes in this challenging breast cancer subtype.
A key mechanistic insight from our work is the observed upregulation of p63
expression, both at baseline in SB431542-treated cells and in combination with
doxorubicin (Figs. 5,6). TAp63, a structural homolog of p53, has been shown to
compensate for p53 loss by activating similar downstream pro-apoptotic targets
such as BAX, PUMA, and NOXA [34, 35]. The reversal of cytotoxicity upon treatment
with the p53 family inhibitor PET
T47D (harboring the p53 L194F mutation) and MDA-MB-231 (with the p53 R280K mutation) breast cancer cell lines carry aggregated forms of p53 mutations [36]. Both cell lines exhibited higher resistance to the chemotherapeutic drug doxorubicin than MDA-MB-468 cells, which carry the non-aggregated p53 R273H mutation and demonstrated a better response to doxorubicin [37]. Our combination treatment produced significantly synergistic effects in all three cell lines, as indicated by coefficient of drug interaction (CDI) values below 0.7. This suggests a synergistic interaction regardless of the p53 mutation aggregation status. Therefore, this drug pairing shows potential therapeutic benefit in breast cancers with either aggregated or non-aggregated types of p53 mutations.
In addition, emerging evidence suggests that TGF-
Further analysis shows that p63 expression is consistently and markedly reduced in breast cancer compared with normal tissue (Fig. 10B). Interestingly, this pronounced downregulation was highly cancer-type specific, occurring prominently in breast cancer and prostate adenocarcinoma but not in most other malignant tissues (Fig. 10A), indicating a context-dependent regulatory function.
Within breast cancer, lower p63 expression was strongly associated with more aggressive clinicopathological features, including HER2-positive and triple-negative subtypes (Fig. 10C). These subtypes are characterized by higher proliferative indices and poorer outcomes, suggesting that loss of p63 may contribute to tumor progression or reflect dedifferentiation of epithelial cells. Similarly, the decrease in p63 expression observed in stage IV disease supports the notion that p63 reduction may correlate with metastatic capability or advanced tumor biology (Fig. 10D).
A particularly notable finding is the association between p53 mutation and reduced p63 expression in breast cancer (Fig. 10E). We demonstrated that p63 expression can be successfully restored in TP53-mutant breast cancer cells using SB431542, particularly in combination with doxorubicin. This pharmacologic induction of p63 suggests that, despite the presence of mutant p53, the TAp63 pathway remains responsive to external modulation. The ability of SB431542 plus doxorubicin to elevate p63 levels and reactivate downstream apoptotic signaling highlights a potential therapeutic strategy to counteract the loss of p53 function and improve treatment responsiveness in these otherwise chemoresistant tumors.
Doxorubicin induces DNA damage and activates p53 family stress signaling [16, 41], leading to induction of TAp63 activation. In contrast, SB431542 blocks
TGF-
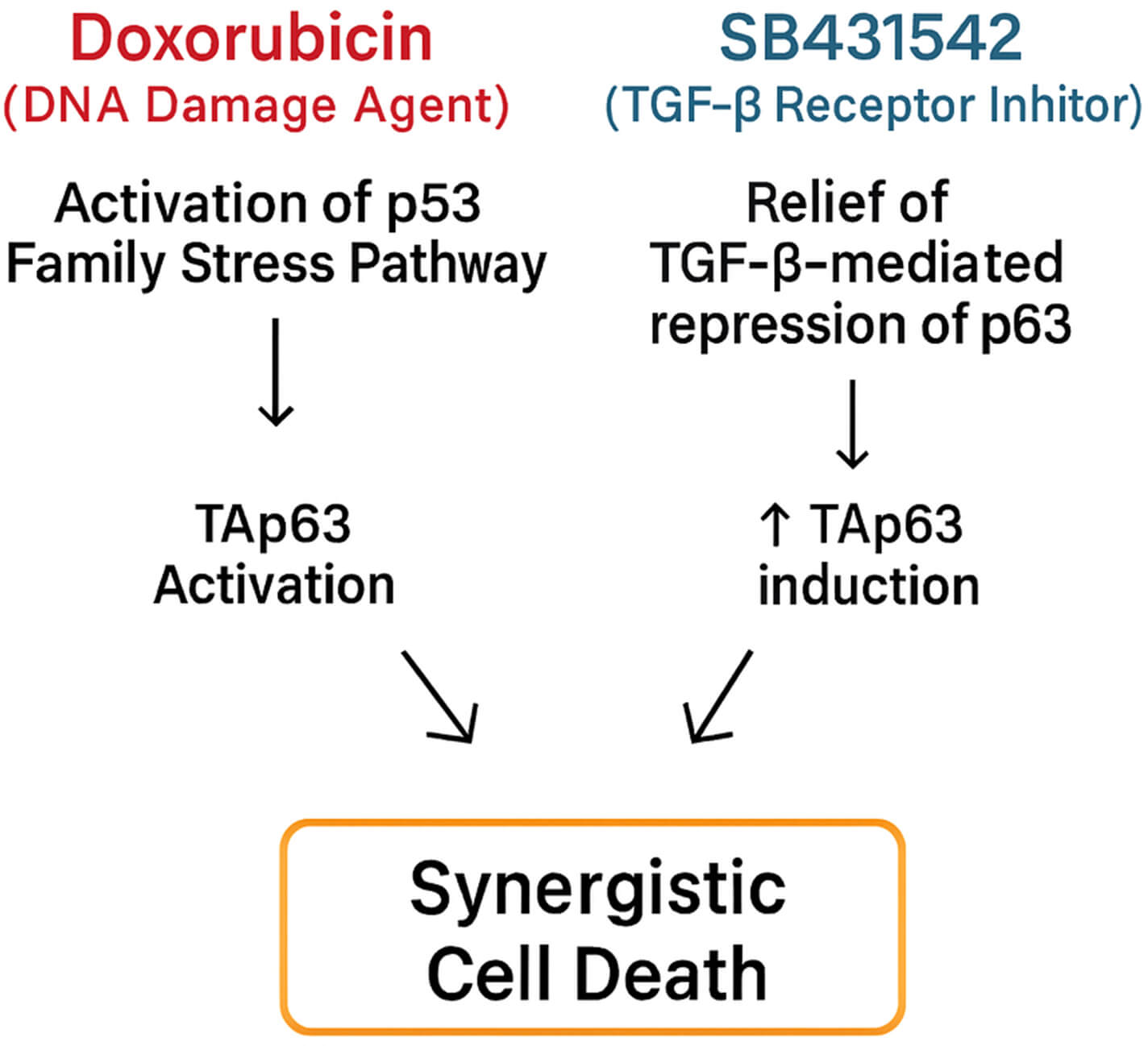 Fig. 11.
Fig. 11.
Proposed mechanism underlying the synergistic effects of
SB431542 and doxorubicin via convergent activation of the p63-dependent apoptotic
pathway. Doxorubicin induces DNA damage, leading to activation of p53-family
stress signaling and activation of TAp63. Concurrently, SB431542 inhibits
TGF-
Our findings provide a strong mechanistic rationale for combining the
TGF-
Our findings identify a novel drug combination—SB431542 plus
doxorubicin—that effectively activates an alternative apoptotic pathway in
mutant p53 breast cancer models. Our findings highlight a previously
underexplored therapeutic strategy: targeting the TGF-
The data are available from the corresponding authors on reasonable request.
YLK, CCC and BHC conceived and designed the study. YLK, YJL, TCH, KYW and STC performed experiments. JYC, KYL, HHW and YTC performed the statistical analysis. CCC and BHC received fundings to support research. BHC wrote the manuscript. CCC and BHC conducted review and editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to acknowledge the technical support given by the Basic Medical Core Laboratory, I-Shou University College of Medicine.
This research was supported by the Medical Student Research and Development Scholarship Program [EDAHS112038], and the grant of E-DA Hospital [EDAHJ114001].
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL45389.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.