



 , Wenhui Wang 1,†, Guiyan Liu 1, Lili Wang 1, Xinkang Zhang 2, Jie Mei 2,*
, Wenhui Wang 1,†, Guiyan Liu 1, Lili Wang 1, Xinkang Zhang 2, Jie Mei 2,* , Kai Yang 2,*
, Kai Yang 2,*1 Department of General Surgery, The Affiliated Jianhu Hospital of Xinglin College, Nantong University, Jianhu People’s Hospital, 224700 Yancheng, Jiangsu, China
2 Department of Oncology, The First Clinical Medicine College, Nanjing Medical University, 210029 Nanjing, Jiangsu, China
†These authors contributed equally.
Abstract
Collagen, the primary structural protein of the extracellular matrix, shows marked structure–function duality in the tumor immune microenvironment (TIME). Biomechanical and biophysical alterations—matrix stiffening, viscoelastic energy dissipation, fiber alignment and tumor-associated collagen signatures, reduced pore size, and solid stress—create migration tracks, sequester signaling cues that regulate proliferation and metabolism, and from dense barriers that impede immune infiltration. These changes are sensed and transduced via mechanosensing and mechanotransduction pathways, thereby reinforcing malignant behavior and immune exclusion. Given its dynamic, spatiotemporally regulated roles, collagen is a promising therapeutic target to overcome immunotherapy resistance. This review examines the structural features, biological functions, and regulatory pathways of collagen within the TIME. Based on these insights, several clinical strategies were highlighted: targeting cancerassociated fibroblasts and profibrotic signaling to reduce fibrosis; remodeling the matrix enzymatically or physically; and inhibiting collagen receptor signaling. These approaches are often combined with immune checkpoint inhibition. Future directions will emphasize biomarker-guided stratification of collagen status; combining therapies informed by mechanobiology; and using scalable, noninvasive monitoring to optimize immunotherapy outcomes.
Keywords
- tumor immune microenvironment
- collagen
- extracellular matrix remodeling
- mechanosensing
- therapeutic target
- immunotherapy
Collagen, comprising approximately 30% of the total protein mass in mammals, represents the most abundant structural component of the extracellular matrix (ECM). The collagen superfamily encompasses 28 distinct types (I–XXVIII) that collectively orchestrate a sophisticated three-dimensional scaffold essential for tissue architecture, biomechanical properties, and cellular homeostasis [1]. Under physiological conditions, the synthesis, deposition, and degradation of collagen are tightly regulated by a complex interplay of matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), and lysyl oxidases (LOX), maintaining a precise dynamic equilibrium. However, within the tumor immune microenvironment (TIME), this exquisite balance undergoes profound dysregulation, transforming collagen from a passive structural element into an active driver of malignant progression and therapeutic resistance [2, 3].
The pathological hallmark of many solid tumors is a pronounced desmoplastic
reaction, characterized by excessive deposition of fibrillar collagens
(predominantly types I, III, and V), aberrant cross-linking mediated by the LOX
family of enzymes, and architectural remodeling of the ECM [4]. Tumors
characterized by dense, collagen-rich tissue include pancreatic ductal
adenocarcinoma, cholangiocarcinoma, and triple-negative breast cancer [5]. By
contrast, tumors with relatively low collagen content include clear cell renal
cell carcinoma and glioma. This fibrotic response is primarily orchestrated by
cancer-associated fibroblasts (CAFs), which are activated through multiple
mechanisms, including profibrotic signaling pathways such as transforming growth
factor beta (TGF-
Critically, remodeled collagen matrix acts as a powerful immunosuppressive
barrier through multiple interconnected mechanisms. Dense, linearized collagen
fibers physically exclude cytotoxic T lymphocytes (CTLs) and other immune
effectors from the tumor parenchyma, a phenomenon quantified by reduced CD8+ T
cell/regulatory T cell (Treg) ratios in collagen-rich regions [9]. Beyond
physical exclusion, collagen fragments and specific collagen receptors (e.g.,
integrins) deliver inhibitory signals that directly suppress T-cell activation
and promote the polarization of tumor-associated macrophages (TAMs) toward an
M2-like phenotype [10]. This creates a self-reinforcing cycle, as inflammatory
cytokines and reactive oxygen species generated during frustrated immune
responses paradoxically enhance CAF activation and collagen production through
signal transducer and activator of transcription 3 (STAT3) and nuclear factor
kappa B (NF-
In addition to protumor roles, collagenous stroma can exert context‑ and stage‑dependent antitumor effects. Intact desmoplastic encapsulation may mechanically contain the lesion, stabilize perfusion, and transiently limit dissemination or antigen spillover [12]; however, chronic inflammation, fibroblast reprogramming, LOX‑mediated crosslinking, hypoxia signaling, and the immune system all work to break down the natural barriers, restructuring the tissue matrix into a dense, aligned network that promotes tumor invasion and excludes immune cells [13].
The clinical significance of this collagen-mediated immunosuppression cannot be overstated. Recent analyses of immunotherapy trials have identified high collagen density and specific collagen gene signatures as robust predictors of a poor response to checkpoint inhibitors across multiple cancer types [9]. Therefore, targeting the collagenous ECM has emerged as a crucial strategy for overcoming resistance to immunotherapy.
In physiological settings, tropocollagen, the basic building block of collagen, consists of three polypeptide chains coiled into a canonical right-handed triple helix. These monomers spontaneously self-assemble into staggered arrays, forming collagen fibrils. Subsequently, these fibrils are stabilized and endowed with tensile strength through covalent cross-linking. This porous and biomechanically compliant matrix provides critical tensional support and adhesion cues for maintaining cellular quiescence, while ensuring low-resistance pathways for nutrient perfusion and immune surveillance [1]. However, the collagen architecture within the TIME undergoes profound pathological remodeling (Fig. 1).
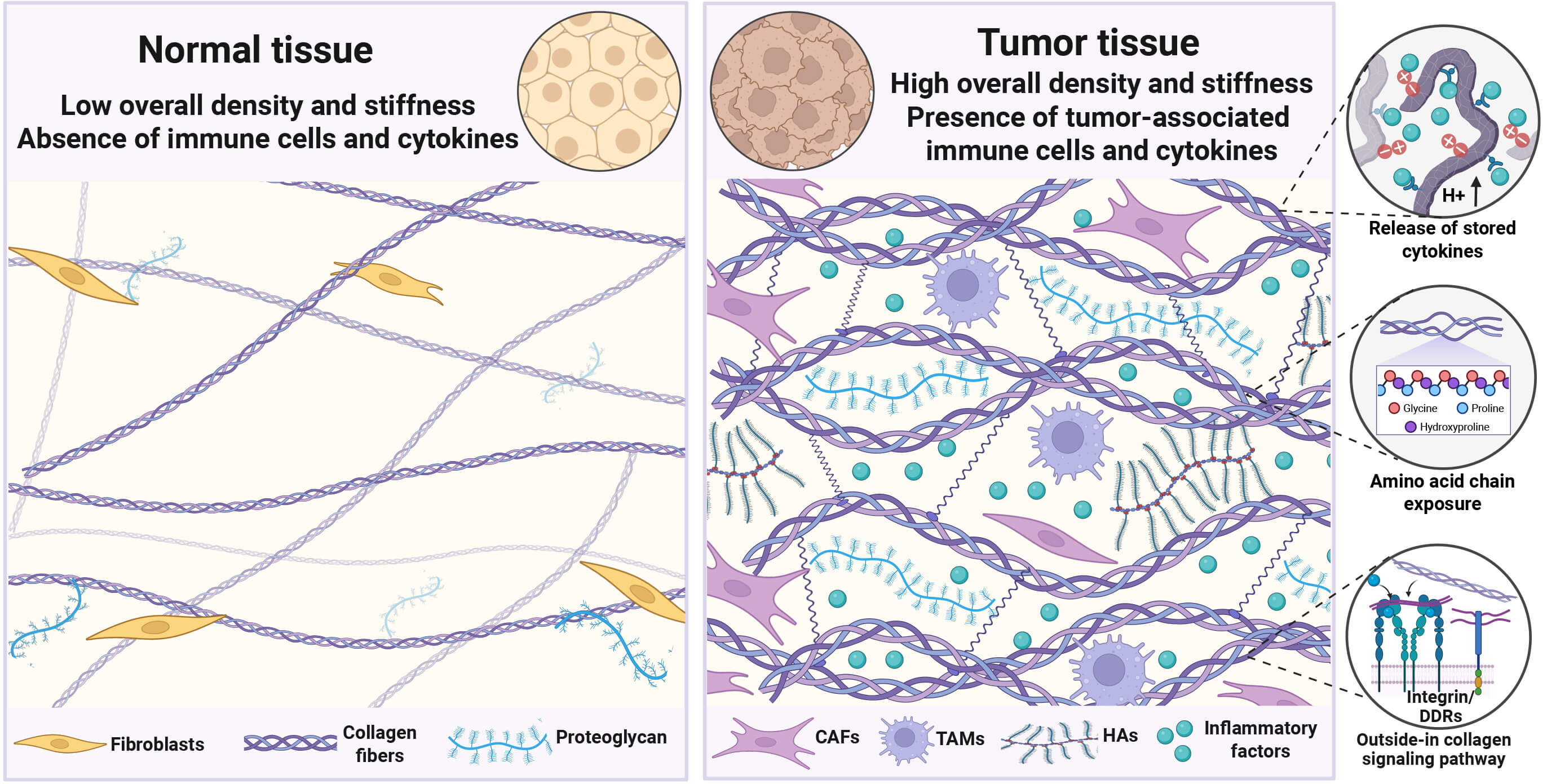 Fig. 1.
Fig. 1.
Remodeling of collagen in the tumor immune microenvironment (TIME). Unlike normal tissues, collagen fibers in the TIME are characterized by increased diameter, greater density, and extensive cross-linking, leading to formation of the unique tumor ECM that is both highly dense and rigid. Collagen interacts with tumor and immune cells by releasing inflammatory factors, exposing binding sites, and recognizing collagen receptors. CAFs, Cancer-associated fibroblasts; TAMs, Tumor-associated macrophages; HAs, Hyaluronic acids.
The changes in collagen in terms of stiffness, spatial structure, and fiber morphology jointly promote tumor progression.
A primary alteration is the dramatic stiffening of the tumor stroma, a process driven by both the aberrant deposition of fibrillar collagens and their extensive cross-linking. Tumors frequently show higher levels and enzymatic activity of LOX enzymes, catalyzing the formation of covalent bonds that dramatically increases tissue tension [14]. Clinically, this is evidenced by the elastic modulus of breast cancer tissue, which can reach 4–12 kPa, starkly contrasting with the 0.4–2 kPa of normal mammary tissue [15]. This heightened matrix stiffness is not a passive feature but is a potent protumorigenic signal.
Concurrently, the matrix undergoes profound architectural reorganization. The physiological, porous, and randomly oriented collagen meshwork transforms into a dense, linearized architecture, resulting in a significant reduction in interstitial pore size from the normal range of 10–50 µm down to 1–10 µm in tumors [16]. This densified structure forms a formidable physical barrier that severely impedes the intratumoral trafficking of immune effector cells, such as CTLs, as well as the perfusion of therapeutic agents. This phenomenon, widely termed “immune exclusion”, is an important mechanism contributing to resistance against both chemotherapy and immunotherapy [5].
On a microscopic scale, the morphology of individual collagen fibers is also remodeled. Tumor-associated collagen fibers become significantly thicker and longer, with diameters increasing from 50–500 nm in healthy tissue to 200–2000 nm [17]. Critically, at the tumor’s invasive front, these fibers are often realigned and bundled into taut, linear tracks oriented perpendicularly to the tumor boundary. These unique topological features are known as TACS [18].
Beyond its role as a physical scaffold, the collagen matrix operates as a
dynamic and potent biochemical signaling hub. On the one hand, the TME’s collagen
network functions as a crucial reservoir, concentrating and modulating the
bioavailability of growth factors, cytokines, and chemokines. The high density of
positively charged residues on collagen fibers facilitates the electrostatic
sequestration of numerous anionic signaling molecules, including TGF-
On the other hand, the collagen fibers themselves serve as solid-phase ligands, directly initiating intracellular signaling cascades upon binding to cell surface receptors on both cancer and immune cells. On cancer cells, specific cryptic binding motifs within the collagen triple helix, such as the GFOGER (for Gly-Phe-Hyp-Gly-Glu-Arg) and RGD (Arg-Gly-Asp) sequences, become exposed during matrix remodeling [19]. These motifs are recognized by integrin heterodimers and discoidin domain receptors 1 and 2 (DDR1/2), which are unique receptor tyrosine kinases [20]. This engagement establishes a pernicious feed-forward loop: activated cancer cells increase their secretion of MMPs, which further remodel the collagen matrix, exposing more binding sites and thus amplifying these protumorigenic signals. Additionally, collagen can bind to inhibitory receptors on immune cells (e.g., leukocyte-associated immunoglobulin-like receptor-1 [LAIR-1]) [21].
Structural remodeling of the collagen matrix is not just a sign of disease but is the mechanistic basis for changes to its function. Thus, within the TIME, collagen is not a passive architectural bystander but is a dynamic orchestrator of tumor malignancy and immunity (Fig. 2).
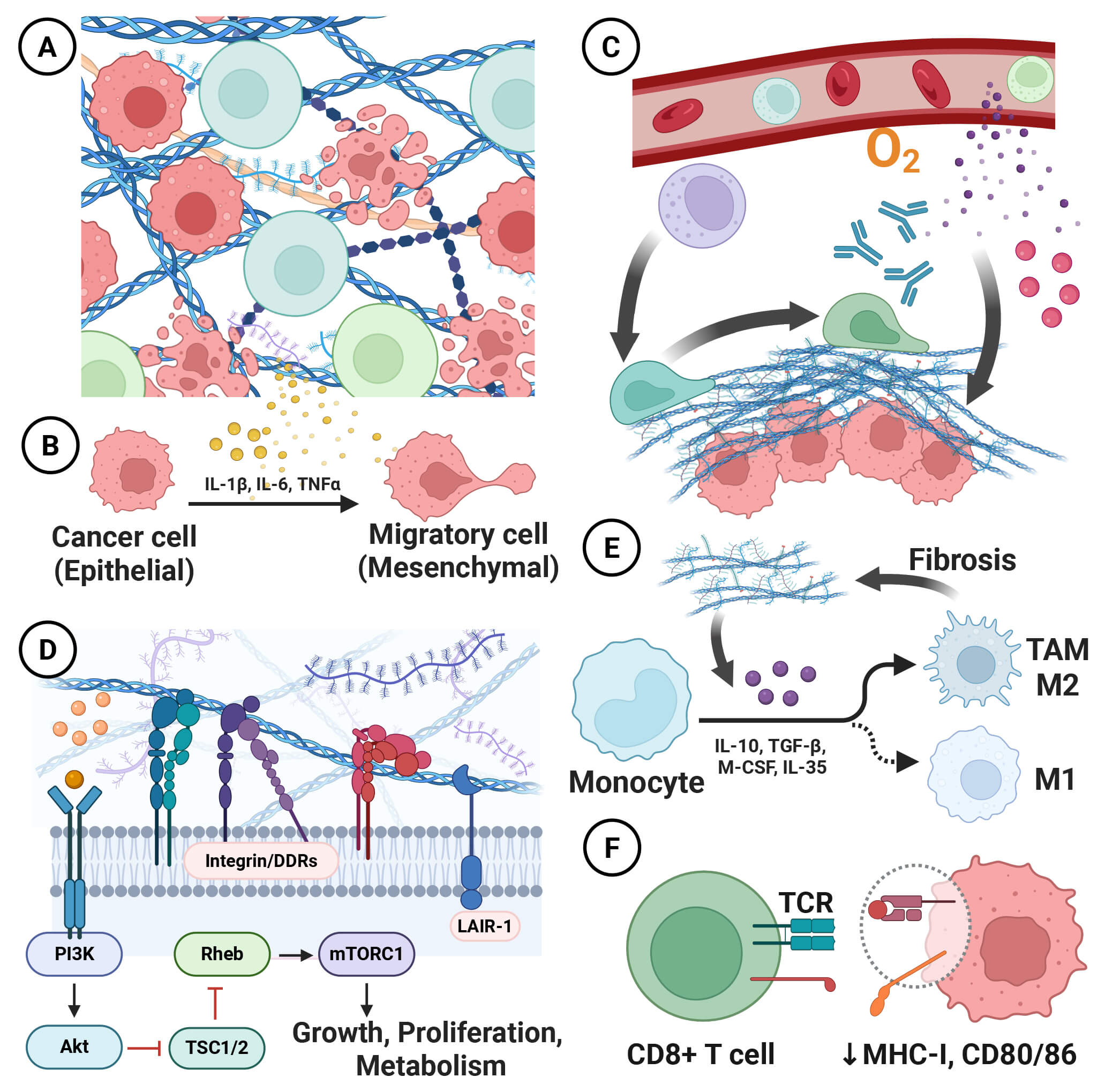 Fig. 2.
Fig. 2.
Functions of collagen in the TIME. Collagen is not merely a passive bystander but is an active participant in malignancy, playing a dynamic role through its intricate “structure–function” continuum: (A) Within the tumor, the collagen fiber network provides cells with spatial guidance and movement tracks. (B) Collagen-derived cytokines promote the tumor epithelial–mesenchymal transition (EMT) and subsequently facilitate tumor metastasis. (C) At the tumor margin, the collagen barrier restricts the infiltration of immune cells, antitumor drugs, and nutrients. (D) Collagen mediates extracellular-to-intracellular signal transduction by releasing stored cytokines or binding to collagen receptors. (E,F) Collagen inhibits tumor immune responses by inducing the polarization of anti-inflammatory cell phenotypes and downregulating major histocompatibility complex class II (MHC-II) and co-stimulatory molecules.
The primary physical function of collagen is to provide a critical three-dimensional structural support framework for tumor tissues. Tumor cells tend to proliferate preferentially in regions with higher collagen density. These areas not only offer stable anchor points necessary for cells to resist mechanical stress and maintain their shape but also function as migration tracks [22]. Cells can migrate along these collagen fibers through contact guidance, at speeds up to 5 to 10 times faster than free migration in the matrix. Live imaging techniques have confirmed that tumor cells preferentially invade outward along collagen fibers perpendicular to the tumor boundary (TACS-3) [18]. Notably, different types of immune cells have varying abilities to utilize these collagen tracks, which may be related to their deformability: dendritic cells and macrophages can efficiently use these tracks for migration, while T-cell migration is more dependent on the interstitial network’s gaps [23].
High-density collagen deposition can form a fibrous capsule around the tumor, serving a protective function. On the one hand, this dense barrier severely hinders the effective penetration of chemotherapy drugs and antibody drugs, which is a significant cause of treatment resistance. For instance, in pancreatic cancer characterized by dense stroma, the penetration distance of drugs is often limited to 50–100 micrometers around blood vessels, far below the range required to achieve an effective therapeutic concentration [24]. On the other hand, it significantly restricts the infiltration of immune effector cells. Studies have shown that for every doubling of collagen density, the infiltration depth of CD8+ T cells decreases by approximately 30% [11]. Notably, some immune cells also have regulatory roles in tumor fibrosis. Research has found that TAMs, through mannose receptor-mediated mechanisms, upregulate inducible nitric oxide synthase, thereby generating reactive nitrogen species to promote tumor fibrosis [25].
In the TIME, the physical functions of collagen and its biochemical functions are not independent of each other but form a continuous spectrum through “mechano-chemical transduction”: extracellular mechanical stress is first integrated into intracellular signals, which then cascade to affect cell proliferation, metabolism, and immune regulation [26].
Mechanical cues of collagen are sensed by cell-surface receptors (integrins
These mechanosignals further orchestrate a profound metabolic reprogramming. In
response to matrix stiffness, the Hippo–YAP/TAZ signaling axis is activated,
which, in concert with the hypoxia common in poorly vascularized fibrotic
regions, stabilizes hypoxia-inducible factor 1 alpha (HIF-1
In parallel, collagen provides precise directional guidance to immune cells by forming stable concentration gradients through its degradation products and chemokines bound to it. For instance, the degradation product PGP (Pro-Gly-Pro) of type I collagen can specifically recruit neutrophils through the C-X-C motif chemokine receptor 2 receptor, while the NC1 domain fragments of type IV collagen (e.g., tumstatin and canstatin) have anti-angiogenic activity [31]. Particularly in the TIME, the chemotactic effect of collagen tends to recruit immunosuppressive cells, such as Tregs and myeloid-derived suppressor cells. For the few effector cells that do infiltrate, the dense, stiff matrix impairs the maturation and antigen-presentation capacity of dendritic cells, evidenced by a marked reduction in MHC-II and co-stimulatory molecule (cluster of differentiation 80 [CD80]/CD86) expression [32]. Furthermore, matrix stiffness induces cytoskeletal tension in T cells, physically disrupting the formation of a stable immunological synapse. Crucially, studies have found that collagen can directly transmit strong inhibitory signals to various immune cells (including T cells and natural killer [NK] cells) by binding to LAIR-1 [8].
The collagenous matrix of the TIME is the dynamically, on-demand, and precisely regulated output of a complex interplay among synthesis, enzymatic remodeling, and degradation. These processes collectively manifest as profound spatiotemporal heterogeneity that evolves with and contributes to malignancy (Fig. 3).
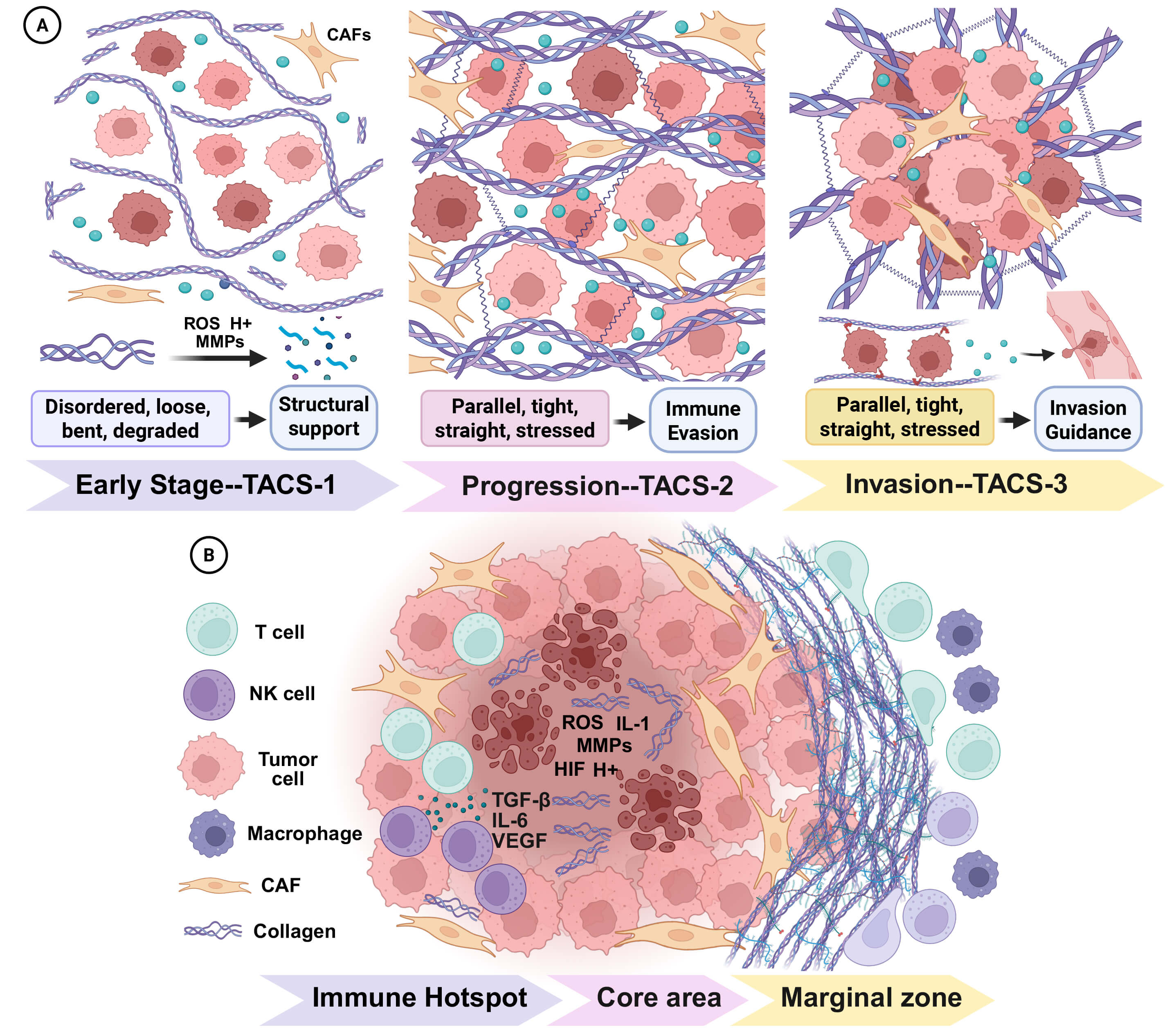 Fig. 3.
Fig. 3.
Spatiotemporal heterogeneity of collagen. The physical and biochemical properties of collagen exhibit dynamic and stage-specific changes during tumor development, both temporally and spatially. These alterations are closely associated with the biological behavior of tumors: (A) In the early stages, loose collagen structures provide sufficient space for tumor cell expansion. As the tumor progresses, increased collagen density facilitates immune evasion. (B) Within the tumor core and immune hotspot regions, collagen undergoes localized degradation under the influence of inflammatory factors. By contrast, at the tumor periphery, collagen forms a dense barrier that restricts immune cell infiltration.
First, the acidic pH of the TIME (typically 6.5–6.8) can affect the collagen network by modulating charge changes. Under acidic conditions, the protonation of key amino acid residues on collagen (e.g., lysine and arginine) increases, raising the surface positive charge density by approximately 30%. This not only alters the electrostatic binding ability of growth factors but also influences the interaction between collagen fibers, potentially leading to relaxation of the fiber network. Additionally, acidic pH can directly activate pH-sensitive proteases such as cathepsins B and L, accelerating collagen degradation [33].
Second, the MMP family is the main enzymatic driver of collagen turnover. The TIME is characterized by the aberrant expression and activity of specific MMPs, including interstitial collagenases (MMP-1, MMP-8, MMP-13) that execute the initial cleavage of the native collagen triple helix, and gelatinases (MMP-2, MMP-9) that degrade the denatured products and associated basement membrane structures [34]. Secreted predominantly by CAFs and tumor cells, these MMPs form highly active “hotspot regions” at the tumor invasion front. This localized degradation not only carves out pathways for cellular invasion but also liberates bioactive collagen fragments and releases matrix-sequestered signaling molecules, which in turn regulate angiogenesis and inflammation [35].
Furthermore, oxidative stress profoundly affects the integrity of collagen. Elevated reactive oxygen species (ROS) levels in the TIME can directly oxidize proline and lysine residues in collagen, destabilizing the triple-helical structure and even causing peptide chain breaks. Additionally, ROS can activate latent MMPs, indirectly accelerating collagen degradation. This oxidative damage is highly spatially specific, mainly occurring in the hypoxia-reoxygenation junction areas within the TIME [34].
Finally, the tumor-associated inflammatory cytokine network regulates collagen
synthesis and degradation. For instance, tumor necrosis factor alpha and
interleukin 1 beta (IL-1
The dynamic remodeling of collagen architecture has been systematically characterized into distinct TACS, which serve as structural indicators of tumor invasiveness and potential biomarkers for evaluating treatment responses [7].
In the early stage of tumor occurrence (precancerous lesions and early in situ carcinoma), TACS-1 is defined by a locally disorganized and wavy collagen configuration. These fibers, showing only a modest increase in density, loosely encircle nascent tumor foci. This signature reflects a period of localized matrix degradation, primarily mediated by tumor cell-secreted MMPs, which creates the necessary space for initial tumor expansion and has relatively immune accessibility [38]. Early, less‑aligned matrices can sequester antigens and chemokines within peritumoral niches and support local immune surveillance, acting as a transient barrier to systemic spread; this immunoprotective compartmentalization diminishes as fibers straighten, pores narrow, and inhibitory collagen–immune receptor pathways intensify [39].
As the tumor progresses and its volume increases, it exerts significant mechanical pressure on the surrounding stroma, leading to the TACS-2 signature. This stage is characterized by collagen fibers that become linearized, bundled, and aligned tangentially (parallel) to the tumor boundary [38]. This architectural shift is dominated by the robust activation of CAFs, which become the primary source of de novo collagen synthesis, dramatically increasing overall matrix density and stiffness [40].
When tumor enters the invasion stage, it is marked by the emergence of TACS-3. These fibers are reorganized into thick, straightened bundles oriented perpendicularly to the tumor margin. This radial alignment creates veritable ‘highways’ that facilitate directed cell migration into the surrounding tissue. At this stage, the matrix reaches its peak density and degree of cross-linking, providing stable, pre-established conduits for invasion [13]. Critically, intravital imaging has confirmed that the vast majority of motile, invasive tumor cells migrate along these TACS-3 fibers [18]. In addition, TACS-3 also features dual physical–ligand barriers and T-cell rejection characteristics, including collagen–LAIR-1 signaling and DDR1/DDR2-directed invasion [41].
Notably, the tumor immune response itself can also have a reverse effect on the
evolution of collagen. In the early stage of immune activation, local degradation
of collagen occurs by effector T cells and NK cells, forming “hot zones” where
immune cells aggregate [42]. However, as chronic inflammation is established, the
elevated levels of T helper 2-type cytokines and TGF-
The collagenous stroma within the TIME is organized into distinct spatial regions, from the peritumoral capsule to the invasive front and perivascular niches. This structural heterogeneity has functional consequences.
The central regions of many tumors are often characterized by a sparse and fragmented collagen landscape. This is typically a consequence of localized hypoxia and necrosis, which limit the metabolic activity of stromal cells and favor a degradative enzymatic environment [14]. However, in some dense tumors (e.g., pancreatic cancer), even the central region can maintain a high collagen content, which is mainly related to the tumor type and distribution pattern of CAFs [24].
Encapsulating the tumor mass is the peritumoral region or invasive front, which typically represents the zone of maximal collagen deposition. Here, a dense, highly cross-linked fibrous capsule (often 50–200 µm thick) forms a strong physical barrier. This structure can achieve a mechanical stiffness 10- to 20-fold greater than that of adjacent normal tissue [14]. Spatially resolved transcriptomic studies have confirmed that CAFs in this area express the highest levels of collagen synthesis genes (e.g., COL1A1) and cross-linking enzyme genes (e.g., LOX). This marginal barrier is a critical determinant of both immune cell exclusion and drug penetration [9].
At the junction between tumors and normal tissues, there is a dynamic transition zone with a width of approximately 100–500 micrometers. Real-time imaging shows that the collagen in this area is in a highly dynamic remodeling state, with both synthesis and degradation actively occurring simultaneously [17]. This interface represents a critical arena for tumor–immune interactions. Thus, the collagen density and structure in this area directly affect the efficiency of immune surveillance [14].
Finally, a special pattern of collagen tissue is present around the tumor neovasculature. This collagen is frequently arranged in concentric circles, forming a vascular sheath [43]. This structure, on the one hand, provides structural support for the fragile tumor blood vessels, and on the other hand, forms another barrier for the extravasation of immune cells from the blood vessels. A study has shown that a higher density of collagen density surrounding blood vessels is statistically linked to lower levels of immune cell infiltration [44].
The pivotal role of collagen in orchestrating tumor progression and immune evasion has established it as a critical therapeutic target. Preclinical and clinical efforts are largely focused on two complementary axes. The first involves anti-fibrotic approaches aimed at inhibiting excessive collagen synthesis, primarily by targeting CAFs or their activation pathways [45]. The second involves matrix remodeling, which seeks to degrade the established collagen barrier or neutralize the downstream protumorigenic signaling it elicits via cell–matrix receptors [46] (Table 1, Ref. [47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63]).
| Mechanism | Target | Drug | Medication regimen | Cancer type(s) | Phase | Outcome | Trial no. | Reference |
| Inhibit collagen biosynthesis | CAF | Talabostat mesylate | Single-agent vs. carboplatin or temozolomide | Brain and central nervous system, childhood germ cell, and kidney cancer | I | At the dose level of 600 µg/m², serum DPP-4 inhibition reached 85% at 24 h. | NCT00303940 | [47] |
| Simlukafusp- |
FAP-IL2v q3w/qw-q2w + atezolizumab | Cervical squamous cell carcinoma | II | n = 48. Of the 45 evaluable patients, 12 (27%; 95% CI: 16.0–41.0) had OR, including 3 CR and 9 PR. | NCT03386721 | [48] | ||
| TGF- |
Galunisertib (LY2157299) | 80 or 150 mg bid LY2157299 | Pancreatic and lung cancer | I | n = 12. 7 patients (80 mg: n = 2; 150 mg: n = 5) had TEAEs: increased brain natriuretic peptide (n = 2), leukopenia (n = 2), and rash (n = 2) | NCT01722825 | [49] | |
| 160 or 300 mg bid LY2157299 + sorafenib or ramucirumab | Carcinoma, hepatocellular | II | Median OS was 7.3 months (95% CI: 4.9–10.5) in A and 16.8 months (95% CI: 10.5–24.4) in B. (A: increased AFP, 160/300 mg; B: normal AFP, 300 mg) | NCT01246986 | [50] | |||
| Fresolimumab | 1 or 10 mg/kg q3w fresolimumab + radiotherapy | Metastatic breast cancer | II | Patients with high dose of fresolimumab had a better OS compared to patients with low dose (HR = 2.73, p = 0.039) | NCT01401062 | [51] | ||
| PDGF/FGF | Pirfenidone | 500 mg q8h Pirfenidone vs placebo from Tipifarnib trial | Plexiform neurofibroma | II | Median TTP for pirfenidone was 13.2 months compared to 10.6 months for the placebo (two-tailed p = 0.92; one-tailed p = 0.46) | NCT00076102 | [52] | |
| Nintedanib | 200 mg bid po Nintedanib | Advanced non-small cell lung cancer | II | n = 20. ORR = 15% with 3 PR, 12 SD. 13 (65%) |
NCT02299141 | [53] | ||
| Imatinib | 400 mg bid po Imatinib | Advanced melanoma | II | median TTP was 54 days and median OS was 200 days. No patient was free of progression at 6 months | NCT00027586 | [54] | ||
| Enhance collagen degradation | MMP | S-3304 | 800, 1600, 2400, 3200 mg bid po | Solid tumors | I | n = 32. Toxicities were mostly gastrointestinal. Maximum tolerated dose was not reached | NCT00033215 | [55] |
| HA | PEGPH20 | PEGPH20 + nab-paclitaxel/gemcitabine (PAG) vs. nab-paclitaxel/gemcitabine (AG) | Metastatic pancreatic ductal adenocarcinoma | II | PFS in PAG (HR = 0.73; 95% CI = 0.53–1.00; p = 0.049). HA-high (0.51; 0.26–1.00; p = 0.048). HA-high PAG vs. AG: ORR = 45% vs. 31% and median OS = 11.5 vs. 8.5 months (0.96; 95% CI = 0.57–1.61) | NCT01839487 | [56] | |
| lysyl-oxidase | Simtuzumab | iv gemcitabine, 1000 mg/m2 + 200 or 700 mg simtuzumab or placebo | Metastatic pancreatic adenocarcinoma | II | Gemcitabine + simtuzumab 700 mg/200 mg/placebo: median PFS = 3.7 months (HR = 1.09; 95% CI = 0.74–1.61; p = 0.73), 3.5 months (1.13; 0.76–1.66; 0.61), and 3.7 months. Median OS = 7.6 months (0.83; 0.57–1.22; 0.28), 5.9 months (1.07; 0.73–1.55; 0.69), and 5.7 months. ORR = 13.9%, 14.5%, and 23.5% | NCT01472198 | [57] | |
| Interfere with signal transduction | DDR1/2 | Nilotinib (DDR1 antagonist) | 400 mg bid nilotinib | Melanoma with KIT mutation (A) or brain metastasis (B) | II | 3/11 in A (27%) and 1/8 in B (12.5%) achieved 4-month disease control. 2 PR in A (18.2%). Median TTP and OS was 3.3 (90% CI = 2.1–3.9 months) and 9.1 months (90% CI = 4.3–14.2 months) | NCT00788775 | [58] |
| Integrin | Cilengitide | Cetuximab + platinum-based chemotherapy (control) or Cilengitide 2000 mg qw (CIL-once)/q2w (CIL-twice) | Advanced non-small-cell lung cancer | II | PFS = 6.2 vs. 5.0 months for CIL-once vs. control (HR = 0.72; p = 0.085). EGFR |
NCT00842712 | [59] | |
| CEND-1 (targets |
CEND-1 + nab-paclitaxel + gemcitabine | Metastatic pancreatic ductal adenocarcinoma | I | no dose-limiting toxicities were observed (n = 31). The most common grade 3 or 4 events were neutropenia (17, 55%), anemia (8, 26%), leukopenia (5, 16%), and pulmonary embolism (4, 13%). 22 (71%) occurred serious adverse events | NCT03517176 | [60] | ||
| Indirect anti-collagen | B7-H3 | YL201 (B7-H3 ADC) | Dose-escalation and expansion | Nasopharyngeal carcinoma, lung cancer, esophageal squamous cell carcinoma | I | extensive-stage small cell lung cancer (ORR = 63.9%), nasopharyngeal carcinoma (ORR = 48.6%), lung adenocarcinoma (ORR = 28.6%) and lymphoepithelioma-like carcinoma (ORR = 54.2%) | NCT0543423, NCT06057922 | [61] |
| Angiogenesis | Sunitinib | Sunitinib (A) vs. no therapy (B) after taxane-based chemotherapy | Metastatic breast cancer | II | PFS |
NCT00270413 | [62] | |
| Diabetes-related | Metformin | Docetaxel/prednisone + metformin 850 mg bid (D+M) or placebo (D+P) | Metastatic castration-resistant prostate cancer | II | in D+M and D+P, ORR was 28% and 24%, median PFS was 7.8 and 6.0 months and the median OS was 27 and 20 months | NCT01796028 | [63] |
Abbreviations: bid, twice daily; qw, once weekly; q2w, every 2 weeks; q3w, every 3 weeks; po, by mouth (oral); iv, intravenous; OR, objective response; ORR, objective response rate; CR, complete response; PR, partial response; PFS, progressionfree survival; OS, overall survival; HR, hazard ratio; CI, confidence interval; TTP, time to progression; TEAEs, treatmentemergent adverse events; HA, hyaluronic acid; DDR, discoidin domain receptor; ADC, antibodydrug conjugate; EGFR, epidermal growth factor receptor; FAP, fibroblast activation protein; IL2v, interleukin 2 variant comple; DPP-4, dipeptidyl peptidase-4; SD, stable disease; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; FGF, fibroblast growth factor.
Direct ablation or functional modulation of CAFs represents a foundational strategy in antifibrotic tumor therapy. Talabostat (PT-100) [47], a small molecule inhibitor of fibroblast activation protein (FAP), exemplifies this approach. By targeting FAP—a serine protease highly expressed on activated CAFs that facilitates ECM remodeling—Talabostat disrupts the proteolytic activity essential for collagen turnover and CAF maintenance [64]. However, clinical translation has revealed modest single-agent efficacy, likely due to the remarkable plasticity of the stromal compartment and compensatory activation of alternative fibrogenic pathways. This limitation has catalyzed the development of combination therapies, such as combining ICIs with FAP-directed chimeric antigen receptor (CAR) T cells or antibody-drug conjugates (ADCs) [5, 48]. Early-phase trials have shown encouraging tumor regression and stromal depletion, suggesting that cellular immunotherapy may overcome the barriers that hampered small molecule approaches [65].
A complementary strategy is to inhibit the signaling pathways that drive the
profibrotic phenotype in CAFs. The TGF-
This concept of pathway inhibition extends to leveraging clinically approved
antifibrotic agents. Drugs such as pirfenidone [52] and nintedanib [53],
standard-of-care for idiopathic pulmonary fibrosis, exert pleiotropic effects by
simultaneously inhibiting multiple fibrogenic pathways including TGF-
Therapeutic strategies for matrix degradation have undergone a significant paradigm shift, moving away from early, crude approaches towards highly specific and targeted interventions.
Initial forays with broad-spectrum MMPs inhibitors were largely unsuccessful in clinical trials. This failure stemmed from significant dose-limiting toxicities, and more critically, a flawed underlying premise, namely, these inhibitors failed to discriminate between protumorigenic MMPs (e.g., MMP-2, MMP-9) and those with antitumorigenic or homeostatic functions (e.g., MMP-8) [70]. Small molecule agonists and antibody fragments that enhance MMP-8 activity have been shown to accelerate collagen turnover, curb metastatic spread, and boost intratumoral CD8+ T-cell density in breast- and pancreatic-cancer models [55, 71]. Moreover, studies have exploited tumor-specific proteases (e.g., FAP or MMP-14) to activate otherwise quiescent pro-MMP-8 or pro-MMP-9 zymogens, thereby confining collagenolysis to the malignant compartment [72].
Another more direct and precise but technically challenging strategy involves the exogenous delivery of matrix-degrading enzymes. Local or intratumoral delivery of recombinant collagenase (Clostridial class I/II) formulated in thermosensitive hydrogels or lipid nanoparticles can liquefy dense type I collagen. In mouse models of pancreatic cancer, a single intratumoral dose followed by gemcitabine doubled intratumoral drug concentrations and extended median survival by 40%. Similar gains have been reproduced with doxorubicin-loaded liposomes and ICIs, indicating that mechanical decompression, rather than enzyme-drug synergy per se, underlies the therapeutic benefit [73, 74]. The primary obstacle remains the safe and effective translation of this powerful local effect into a viable clinical therapy.
Beyond direct collagenolysis, an alternative strategy involves degrading other
critical structural components of the ECM. Hyaluronic acid (HA) is a large
glycosaminoglycan that creates immense interstitial fluid pressure and organizes
the stroma. The enzymatic agent PEGPH20 (pegvorhyaluronidase) [56] works by
systemically degrading HA. It melts the gel-like component of the matrix, which
not only improves vascular perfusion but also relieves the mechanical constraints
on the collagen fibers. While this approach showed significant promise in
clinical trials for patient subgroups with “HA-high” tumors, its development
has unfortunately faced setbacks [75]. Besides, inhibition of LOX-mediated
cross-linking (e.g.,
Beyond physically degrading the matrix, a more nuanced therapeutic strategy aims to sever the protumorigenic communication lines between malignant cells and the collagen scaffold.
DDR1/2 have emerged as a particularly compelling target class. As the only receptor tyrosine kinases activated directly by collagen, they initiate potent and sustained downstream signaling cascades. Strong preclinical data have shown that the selective small molecule inhibitor, nilotinib [58], effectively suppresses the activation of CAFS, as well as collagen-induced invasion and metastasis [58]. The validation of this concept has propelled several DDR inhibitor candidates into Phase I clinical trials [77].
Integrins, fundamental receptors governing cell–ECM adhesion, have been
explored as therapeutic targets for several decades. Early enthusiasm was
tempered by the high-profile clinical failure of broad-spectrum agents like
cilengitide (an RGD-mimetic targeting
A significant frontier in stroma-modulating therapy involves the strategic use of agents whose primary mechanisms of action are not directly antifibrotic, but that exert potent, indirect effects on collagen-producing CAFs and the broader TIME.
The ADC YL201 targets B7-H3 (CD276) [61], a molecule co-expressed on tumor cells, immune cells, and CAFs. This approach offers a dual benefit: directly blocking T-cell immune checkpoint while also weakening the CAF-mediated collagen network to remodel the immunosuppressive stroma [80, 81].
Angiotensin II receptor blockers such as Losartan, originally developed as antihypertensives, have been repurposed to inhibit the profibrotic RAS axis within tumors [82]. Blocking the angiotensin II type 1 receptor on CAFs directly suppresses their activation and collagen synthesis. Critically, this also alleviates solid stress, which decompresses intratumoral blood vessels, thereby improving perfusion and enhancing the drug delivery [83].
The impact of anti-angiogenic drugs on fibrosis is highly dependent on their
specific targets. Multikinase inhibitors targeting PDGF and FGF receptors (e.g.,
sunitinib [62]) can directly suppress CAF activation. By contrast, agents that
exclusively target the VEGF pathway (e.g., bevacizumab) can induce tumor hypoxia.
This, in turn, can paradoxically promote a more aggressive, profibrotic CAF
phenotype via HIF-1
The antidiabetic drug Metformin has demonstrated significant antifibrotic effects by activating the central metabolic sensor, AMP-activated protein kinase (AMPK). AMPK activation inhibits the anabolic processes required for CAF activation and ECM protein synthesis. This metabolic intervention effectively suppresses the fibrogenic function of CAFs [37, 63].
The multifaceted roles of collagen in the TIME represent a paradigm shift in our understanding of cancer biology and therapeutic resistance. This review systematically delineated how collagen transitions from a passive structural scaffold to an active orchestrator of malignancy, functioning through interconnected physical and biochemical mechanisms. Beyond its role as a physical impediment to immune cell infiltration and drug penetration, the collagenous matrix operates as a sophisticated signaling hub that actively promotes tumor progression through mechanotransduction pathways, metabolic reprogramming, and direct immunosuppressive signals [12]. The spatiotemporal heterogeneity of collagen, evolving dynamically throughout tumor progression and varying across distinct microanatomical regions, underscores the complexity of targeting this multifunctional component of the TIME [42].
Current therapeutic strategies targeting the collagenous matrix have yielded both promising advances and instructive failures: (i) a predominance of small, earlyphase and histologyheterogeneous cohorts with largely exploratory endpoints; (ii) suboptimal target–disease alignment and minimal enrichment for specific collagen states (density, alignment, crosslinking, and hyaluronan load)—with the HAhigh signal seen with PEGPH20 illustrating the need for biomarkerguided selection; (iii) limited incorporation of mechanistic pharmacodynamic assessments of matrix remodeling (e.g., TACS, pore size, stiffness, and circulating collagen fragments) and few paired biopsies; (iv) insufficient testing and suboptimal sequencing of rational combinations with immunotherapy or mechanotransduction inhibitors. These constraints likely contribute to mixed or marginal efficacy signals (e.g., negative outcomes with LOX/LOXL2 or integrin strategies) and complicate attribution to the collagen–immune axis; and (v) the dual role of collagen in tumors argue for stage‑aware stromal interventions: dismantling a protective capsule too early may facilitate spread, whereas targeted de‑alignment or de‑crosslinking after the loss of containment can improve immune access and therapeutic efficacy [11, 46].
In conclusion, re-engineering the collagenous matrix represents one of the most promising frontiers in modern oncology. In addition, we argue for prospectively defined biomarkers that classify tumor collagen states and immune accessibility, such as the pan-cancer level immunocollagenic subtypes that we have already established [5]. Success will require continued interdisciplinary collaboration, bridging matrix biology, immunology, bioengineering, and clinical oncology to translate these mechanistic insights into transformative therapies that can overcome the stromal barriers to cure.
ADC, antibody-drug conjugate; AFP, alpha-fetoprotein; ARB, angiotensin II receptor blocker; AMPK, AMP-activated protein kinase; CAR, chimeric antigen receptor; CAF, cancer-associated fibroblast; CD80, cluster of differentiation 80; CD86, cluster of differentiation 86; CTL, cytotoxic T lymphocyte; CR, complete response; CI, confidence interval; DDR, discoidin domain receptor; RGD, Arg-Gly-Asp; EMT, Epithelial–mesenchymal transition; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; FAP, fibroblast activation protein; FGF, fibroblast growth factor; GFOGER, Gly-Phe-Hyp-Gly-Glu-Arg; HA, hyaluronic acid; HR, hazard ratio; HIF, hypoxia-inducible factor; HSPG, heparan sulfate proteoglycan; ICI, immune checkpoint inhibitor; IL-1
YZ, WW, and KY conceived the structure of the review, performed the literature search and analysis, and wrote the initial draft of the manuscript. XZ and LW contributed to the key sections, critically revised the manuscript for important intellectual content, and were specifically involved in the preparation of the figures and table. JM, KY and GL provided the conceptual framework, supervised the project, secured funding, and revised and finalized the manuscript. All authors contributed to editorial changes in the manuscript. All authors have read and approved the final submitted version. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.