










 , JiaLi C. Huang 1, Ziqi V. Wang 1, Angela L. Ferguson 1, Jasmine Minh Hang Nguyen 1, MingChang Zhang 1, Katharine A. Osborne 2, Badwi B. Boumelhem 1, Geoffrey W. McCaughan 1,3, Ken Liu 1,3, Mark D. Gorrell 1,*
, JiaLi C. Huang 1, Ziqi V. Wang 1, Angela L. Ferguson 1, Jasmine Minh Hang Nguyen 1, MingChang Zhang 1, Katharine A. Osborne 2, Badwi B. Boumelhem 1, Geoffrey W. McCaughan 1,3, Ken Liu 1,3, Mark D. Gorrell 1,*
1 Centenary Institute, Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia
2 NSW Health Pathology, Lidcombe, NSW 2141, Australia
3 AW Morrow Gastroenterology and Liver Centre, Royal Prince Alfred Hospital, Camperdown, NSW 2050, Australia
Abstract
Tumours contain many fibroblasts, endothelial cells, and leukocytes that are emerging as therapeutic targets complementary to targeting genetically unstable cancer cells. Immunotherapies directed towards this tumour microenvironment (TME) are increasingly effective. Targeting the endothelium has shown success, particularly in hepatocellular carcinoma (HCC). Cancer-associated fibroblasts (CAFs) are also attracting novel nascent therapeutic approaches, and fibroblast activation protein (FAP), which is specific to activated mesenchyme, is prominent amongst CAF markers. This review places emphasis upon FAP, human HCC, and FAP-targeting approaches for therapeutic benefit, including FAP inhibitors, radioligand therapy, T cell and antibody-dependent cytotoxicity/immunotherapy, and FAP-activated prodrugs.
Keywords
- FAP
- fibroblast activation protein
- radioligand therapy
- prodrug
- cancer-associated fibroblast
- tumour microenvironment
- hepatocellular carcinoma
- biliary tract cancer
The incidence of cancer is rising. Whilst 5-year relative cancer survival rates are also increasing, cancer remains a major contributor to disease burden and a leading cause of death globally. Therefore, understanding tumour pathogenesis in order to improve therapy and early detection is crucial. In addition to cancer cells, tumours contain many fibroblasts, endothelial cells, and leukocytes that interact with each other within the tumour microenvironment (TME).
Cancer cells are often genetically variable and unstable, whilst the composition of the TME can vary between tumours but its genetic stability is an advantage for therapeutic targeting. Although tumours are increasingly classified by molecular markers, the liver TME has unique features, and hepatocellular carcinoma (HCC) and biliary tract cancers (BTCs) need focused consideration. This review describes the TME and explores human liver cancer and the fibroblastic component of tumours, then approaches cancer-associated fibroblast (CAF) targeting in depth. The emphasis is a rationale for fibroblast activation protein (FAP)-directed CAF targeting, and for each of the FAP-targeting approaches for therapeutic benefit, including FAP inhibitors, antibody-based and CAR-T immunotherapies, and FAP-activated prodrugs.
The high specificity of FAP to activated fibroblasts is attracting a variety of approaches and the safety in humans of removing FAP+ fibroblasts has been established. Some efficacy data are emerging. The future of this field will focus on the question of which CAF-targeted approaches have the greatest efficacy. CAFs influence all other TME components, which are introduced here.
Tumour formation intertwines multiple intracellular, intercellular, and extracellular processes that drive abnormal cell growth and provide a favourable environment for cancer cell survival. Neoplasia is associated with chronic inflammation and a microenvironment likened to a non-healing wound. The TME consists of immune cells, blood vessels, extracellular matrix (ECM), fibroblasts, lymphocytes, bone marrow-derived inflammatory cells, and signalling molecules [1, 2]. The TME has both common features and heterogeneity, but is increasingly recognized as a therapeutic target that is more stable and less mutable than tumour cells.
Immune cells have vital roles in preventing tumor growth via secreted proteins and direct cell lysis. The immune component of the TME mainly consists of mononuclear macrophages, dendritic cells (DCs), T-cells, natural killer (NK) cells, and other myeloid cells.
Tumour-associated macrophages (TAMs) often colocalize with CAFs [3]. TAM are activated via different types of cytokines into two subtypes in the TME [4, 5]. The TAM M1 subtype performs pro-inflammatory anti-tumour roles, whereas M2 TAM generate opposing functions, having a role in tumour progression [5, 6]. The TAM M2 phenotype expresses CD206 and can release arginase-1, the anti-inflammatory cytokine interleukin-10 (IL-10), C–C motif chemokine ligand (CCL) 17, and CCL22. Understanding M1 and M2 is crucial and the transformation from M2 to M1 is a potential immune-stimulating target [7].
The DC is crucial in surveillance for tumour cells because, as antigen-presenting cells (APCs), DCs activate and recruit T-cells to the TME. Conventional DC type 1 (cDC1), expressing CD141, Clec9A, and X-C motif chemokine receptor 1 (XCR1), mainly drives cytotoxic CD8 T cell activation and attraction via the production of CXCL9, CXCL10, and IL-12 [8, 9]. CD1b/c+ cDC2 interact with CD4 T-helper cells, inducing IL-2 secretion and downstream activation of CD8 T-cells [9, 10].
CD8 T-cells are the major effector cells in tumour clearance, by secreting the cytotoxic enzymes granzymes or perforin, or activating caspase-dependent apoptosis via Fas/APO ligand binding [11, 12]. Via CD40 stimulation, CD4 cells can induce DCs to increase IL-12 and interferon (IFN) production to increase downstream CD8 T-cell activation, and can also induce B-cell maturation to initiate antitumor antibody production [13, 14, 15]. However, overactivation of CD8 T-cells can result in constant tissue damage and T-cell exhaustion. Tumour cells can also evade detection by T-cells by major histocompatibility complex (MHC) downregulation, or by expressing programmed death-ligand 1 (PD-L1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG3), or similar immune checkpoints to inhibit T-cell activation.
NK cells are a unique lymphocyte subset that lacks both CD3 and T-cell antigen receptors. Making up only 5–10% of the immune cell population, these CD56+ cells target MHC-I-deficient cells and are activated in the presence of cytokines such as IL-2, IL-12, IL-15, IL-18, and type-I IFN [16, 17, 18]. Upon activation, NK cells release granzymes and perforin to induce apoptosis, or release IFN-gamma to enhance T-cell priming [17, 18]. NK cells have less anti-tumour potency than T-cells, but NK cells are increasingly harnessed for immunotherapy.
Bone marrow-derived myeloid cells primarily differentiate into myeloid-derived suppressor cells (MDSCs) when recruited to the TME. MDSC abundance is associated with poorer cancer prognosis. MDSCs suppress the host’s anti-tumour immune system, so they are important in progression, proliferation, and immune evasion by cancer cells [19, 20]. Human monocyte-MDSCs express CD14 and have low to negative MHC-II and CD15. Human granulocyte-MDSCs lack CD14 but are positive for the granulocyte activation markers CD15 and CD66b [21, 22].
MDSCs act via diverse mechanisms. Enriched with nitric oxide synthase (NOS),
MDSCs can produce NO that inhibits antigen presentation to DC and induces
apoptosis [23]. Moreover, MDSCs can prevent T-cell activation [21]. Additionally,
MDSCs can recruit regulatory T-cells (Treg) to the TME to increase
immunosuppression, via the secretion of IL-10 and transforming growth factor
Understanding tumour perfusion is crucial for designing and delivering cancer therapies. Blood vessels supply nutrients and oxygen and remove waste products. Tumour growth requires angiogenesis to satisfy huge demands for oxygen and nutrients. In the 1970s, Folkman hypothesized that solid tumours require angiogenesis for their growth and metastasis, so blocking angiogenesis in tumours can be an effective therapy [24]. Indeed, anti-angiogenic therapy is an effective cotherapy in HCC [25, 26, 27, 28]. However, it is not a panacea because the tumour vasculature is heterogeneous. Compared to the normal vasculature, blood vessels in TME are unorganized, leaky, and hyperpermeable, so improving perfusion can improve treatment outcomes [28]. Thus, normalizing tumour vasculature is a potential therapeutic paradigm [26, 28].
Tumour endothelial cells (TECs) have abnormal morphology and molecular and signalling profiles and interact with other TME components, including angiogenic cytokines, CAFs, and immune cells [28, 29, 30]. Tumour vasculature can act against leucocyte extravasation, antigen presentation, DC maturation, and T cell activation [31, 32, 33]. Dysregulated tumour angiogenesis can lead to hypoxia and acidosis in the TME [34]. Hypoxia can induce angiogenic factors that can suppress cell adhesion molecule expression on TECs, which can suppress immune cell migration into a tumour [35]. Moreover, TECs have lower MHC expression [36]. In these ways, tumour angiogenesis engenders an immunosuppressive environment that favours cancer development.
ECM is primarily made by activated fibroblasts that also make ECM-regulating enzymes, including families of lysyl oxidase (LOX), matrix metalloproteinase (MMP), disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), and ADAM enzymes. ECM is primarily composed of collagens, proteoglycans, elastin, fibronectin, and laminins [37]. The ECM has vital roles in cell interaction and communication via reacting to or releasing growth factors. The ECM is dynamic as it is continuously remodelled during normal and pathological conditions [38, 39], but the rigidity of crosslinked collagens provides 3-dimensional physical support for cells and tissues.
Fibroblasts were first defined in the 19th century as non-vascular,
non-epithelial, non-inflammatory cells found in connective tissues and all organs
[40]. Normal fibroblast functions are crucial as they secrete many molecules,
like type I, III, and IV collagens, proteoglycans, fibronectin, laminins,
glycosaminoglycans, MMPs, MMP inhibitors, and prostaglandins, to maintain ECM
homeostasis [40]. In adult organs, fibroblasts are relatively quiescent but can
be activated by molecules including transforming growth factor
CAFs arise from normal fibroblasts, mesenchymal stromal cells, adipocytes,
pericytes, hepatic stellate cells, epithelial cells, and endothelial cells (Fig. 1, Ref. [42]). Epithelial to mesenchymal transition (EMT) is a dominant mechanism
[43]. Endothelial to mesenchymal transition (EndMT) can be induced by
TGF-
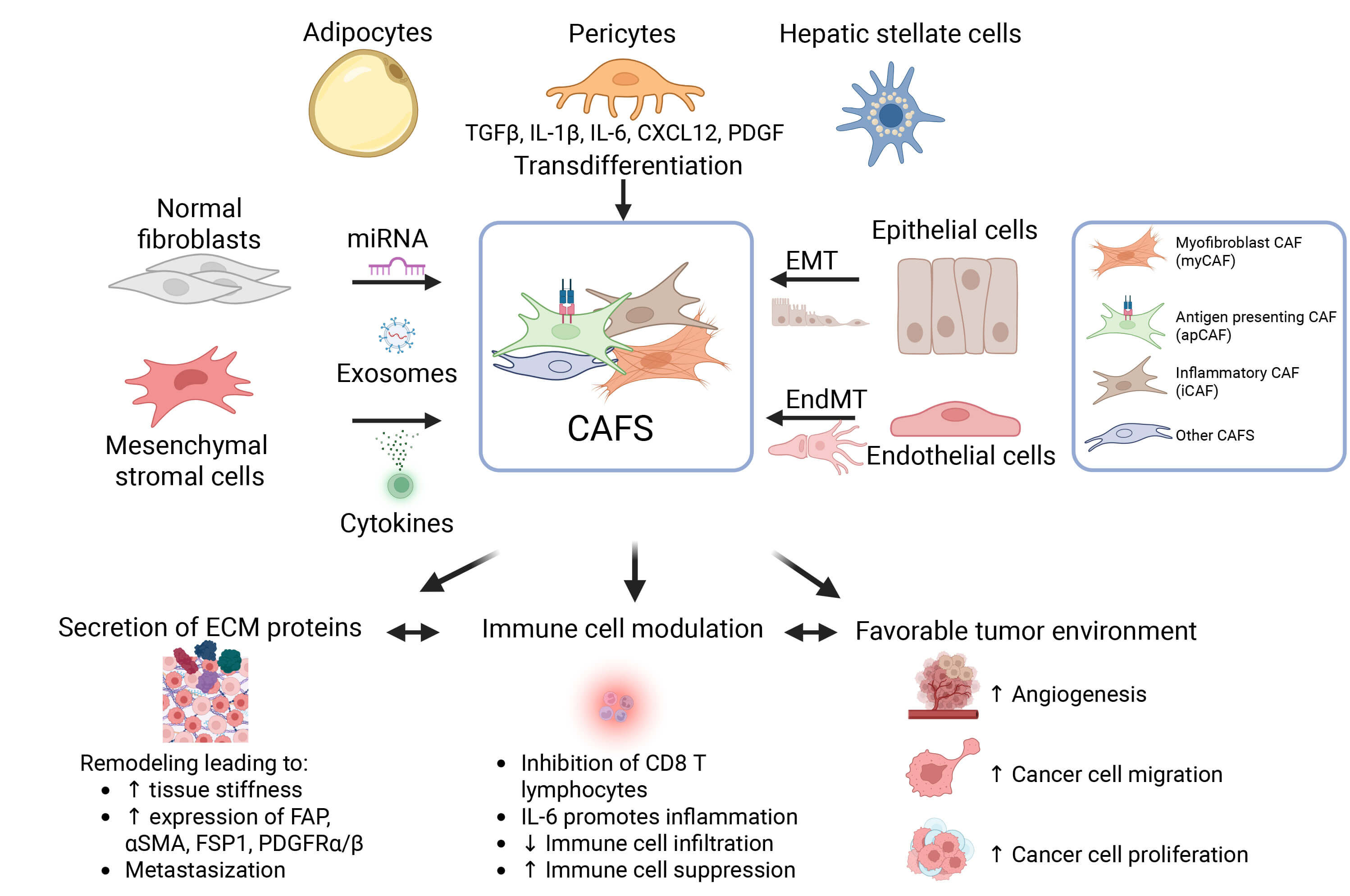 Fig. 1.
Fig. 1.
Origin, heterogeneity, and roles of cancer-associated fibroblasts (CAFs) in the tumour microenvironment (TME). CAFs arise from multiple cellular sources, including normal fibroblasts, mesenchymal stromal cells, adipocytes, pericytes, hepatic stellate cells, epithelial cells via epithelial-to-mesenchymal transition (EMT), and endothelial cells via endothelial-to-mesenchymal transition (EndMT). Various molecular signals, including miRNAs, exosomes, and cytokines, drive these processes. CAF subtypes include myofibroblastic, antigen-presenting, and inflammatory. CAFs exhibit diverse functions. ECM, extracellular matrix. Created in BioRender. Boumelhem, B. (2025) https://BioRender.com/1xhq7rj and modified from [42].
Cadherin switching, with less E-cadherin and more N-cadherin, is a hallmark of EMT [51]. The abnormal expression of cadherin affects cell–cell attachment, which is central to cancer cell invasiveness. The normal fibroblast is crucial for connective tissue, which is indispensable for cell progression and organ growth and structural integrity (Table 1, Ref. [46, 52, 53, 54, 55, 56, 57, 58, 59, 60]). However, fibroblasts can be activated by certain stimuli, such as HtrA serine peptidase 1 [61], TANK-binding kinase 1, TGF, galectin-1 via TGF [60], pappadin [62], and WD repeat domain 5 [63, 64]. WD repeat domain 5 is an important soluble factor, up-regulated by CXCL-8, that can induce EMT in cervical squamous cell carcinoma [65]. Pancreatic ductal adenocarcinoma (PDAC) generally involves extensive fibrotic stroma and ECM deposition by CAFs [66].
| Cell Type | Trigger | Linked Pathway | Cancer Cell Type | Reference |
| Normal fibroblast | Galectin-1 | TGF- |
Gastric cancer cell lines | [60] |
| Long non-coding RNA (LncRNA) | Not known | Mouse melanoma cell line B16F0 | [52] | |
| Hepatocyte growth factor (HGF) | Induction of HGF secretion | Gastric cancer cell line MKN45 | [53] | |
| Bone marrow mesenchymal stem cell | Tumour-derived secreted GRP78 (glucose-regulated protein 78) | TGF- |
Human colon cancer cell lines | [54] |
| Mesenchymal stem cell | Type 2 diabetes mellitus | TGF- |
Triple negative breast cancer (TNBC) | [55] |
| CXCR4 | CXCR4/TGF- |
Human CRC cell lines | [46] | |
| Adipocyte | Wnt3a | Wnt/ |
Murine 4T1 breast cancer cell line | [56] |
| Pericytes | BMP2 | PI3K/AKT and MEK/ERK | Gastric cancer cell line | [57] |
| Epithelial | EMT | TGF- |
[58] | |
| Endothelial | EndoMT | TGF- |
[59] |
Table footnote: BMP2, bone marrow protein 2; HGF, hepatocyte growth factor;
TGF-
In HCC, Egf7 can trigger liver fibroblast activation. Egf7 could recruit liver
fibroblasts, then induce the
Extracellular vesicles (EVs) are small, membrane-derived vesicles or exosomes. EVs are biologically active and can mediate cancer-stroma communication [71, 72]. Some specific molecules of EVs, such as microRNA (miR) and protein transported via EVs to fibroblasts, may activate the latter [73] (Table 2, Ref. [74, 75, 76, 77, 78, 79, 80]). MicroRNA is a subgroup of non-coding RNA that can modulate many cell functions and cancer-derived EV-mediated stimulation is an established mechanism for fibroblast activation in tumours [81, 82, 83, 84]. Therefore, microRNAs are important in transdifferentiation to CAF [85].
| Cancer Type | Related miRNA | Pathway or Related Factors | References |
| Lung cancer | miR-1247-3p | [74] | |
| Breast cancer | miRNA-146a | TXNIP | [76] |
| miRNA-221 | A20/c-Rel/CTGF signaling | [77] | |
| Liver cancer | miRNA-21 | PDK1/AKT signaling | [75] |
| Colorectal cancer | miRNA-200 | EMT | [78] |
| miR-92a-3p | miR-92a-3p/KLF4/CH25H axis | [79] | |
| Lung cancer | miRNA-196a | AnnexinA1 and CCL2 | [80] |
Table footnote: CTGF/CCN2, connective tissue growth factor; KLF, Kruppel-like factor; PDK1, pyruvate dehydrogenase kinase 1; TP53INP1, tumour protein p53 induced nuclear protein 1; TXNIP, thioredoxin interacting protein.
A great deal more could be understood on the mechanisms of transformation and differentiation from normal cell types to CAF, which may vary with cancer genotype [48].
Functionally, CAFs contribute to the TME through (1) secretion of ECM proteins, increasing tissue stiffness and a barrier; (2) modulating immune responses by inhibiting CD8+ T cell activity, promoting IL-6–mediated inflammation, reducing immune infiltration, and enhancing immune suppression; and (3) supporting a pro-tumorigenic environment and metastasis by stimulating angiogenesis, cancer cell migration, and proliferation.
In the liver, CAFs support the development of intrahepatic cholangiocarcinoma (ICC), in which CAFs and particularly FAP+ CAFs are abundant [86, 87, 88, 89, 90, 91]. In a mouse model, iCAF can trigger tumour-expressed MET via HGF to promote ICC growth, and myCAFs also promote ICC [86].
Metabolic interactions between cancer cells and CAFs in TME are an important
influence on tumour progression. For example, Capan-1 PDAC cells co-cultured with
CAFs produce glutamic acid and lactic acid, while citric acid and isocitric acid
are released from iCAFs [92]. Cellular metabolism can be targeted by novel cancer
therapies. Non-toxic short interfering (si)MFAP-5/desloratatine (DES), which is
in biocompatible polypept(o)ide-based polyion complex micelles constructed with a
triblock copolymer composed of a cationic poly(l-lysine) complexed with
anti-MFAP-5 siRNA (siMFAP-5) via electrostatic interaction, and a
poly(
MicroRNAs can drive EMT and can influence cancer progression and metastasis [84, 94, 95, 96]. Exosome miRNA-139 inhibits MMP-11 expression to influence tumour invasion in gastric cancer [97]. In contrast, the absence of miR-200 in lung cancer can promote cancer metastasis [98]. Exosomes, proteases, growth factors, and cytokines of CAFs interact with cancer cells. Fibroblast growth factor 2 (FGF-2) secreted by CAFs is upregulated by serine protease 23 (PRSS-23). Consistently, both FGF-2 and PRSS-23 are closely involved in EMT and expressed by CAFs and mesenchymal cells [99]. Amphiregulin is an epidermal growth factor (EGF) often overexpressed in human cancer [100]. Amphiregulin secretion can be stimulated by lysophosphatidic acid (LPA) and, through an LPAR1 and LPAR3/Gi/Rho signaling cascade, contributes to cancer cell aggressiveness [101].
CAFs can produce chemokines and cytokines that regulate immune cells and cancer
cell migration. IL1R1+ CAFs in breast cancer influence CD4+ and
CD8+ T cells as well as Treg cells, as shown by LIANA (ligand–receptor
analysis), a comprehensive computational framework designed to infer cell–cell
communication by analyzing single-cell RNA sequencing (scRNA-seq) data [102].
CCL2, CXCL12, and IL6 upregulation in IL1R1+ iCAFs can exert
macrophage/monocyte chemotaxis to suppress immune responses [102]. Also, M2
macrophages in TME can stimulate the expression of PD-L1 by cancer cells [103, 104]. Moreover, increased TAM and CAF densities in neuroblastoma or CRC,
with macrophages localized adjacent to CAFs, associate with cancer progression
[3, 72, 105, 106]. When co-cultured with cancer cells, human monocytes and
mesenchymal stromal cells can activate into TAM and CAF, respectively, then
produce the pro-tumorigenic cytokines TGF-
CAFs and “educated” fibroblasts can promote cancer cell growth and
immunosuppression in the TME [107]. CAFs can “educate” normal fibroblasts to
further promote cancer cell growth. Specifically, in diffuse-type gastric
cancers, CAF-educated fibroblasts can trigger NF-
Mesenchymal cell biomarkers are used in identifying CAFs, in particular FAP,
The complexity of CAFs has only recently been probed using advanced technologies
to classify their different phenotypes and functions in situ within human
tumours. Recently, pan-cancer classifications of human CAFs have defined numerous
phenotypes, including matrix-, inflammatory-, vascular-, tumour-like-,
antigen-presenting-, interferon response-, reticular-like-, and dividing CAFs,
with good concordance across multiple cancers [113]. Integrative analyses of
multiple in situ proteomic and spatial transcriptomic platforms propose four CAF
subtypes conserved across eight cancer types [114]. These CAF subtypes are based
on phenotypic markers: CAF1 has TGFB, ACTA2, CXCL8; CAF2 has FAP, PDPN, MMP2;
CAF3 has PDGFR
Despite these insights, cancers such as glioblastoma, PDAC, and HCC have unique microenvironments and only partially align with these classifications of CAFs [113] or immune cells [115]. Indeed, a unique CAF subset in PDAC is CD144+ CAF (endo-CAF) that negatively correlates with recurrence-free survival and overall survival in PDAC [116]. In addition, it is likely that fibroblast phenotypes change or fluctuate over time due to fibroblast plasticity. To date, pan-cancer CAF classification is useful but limited by TME variability, and so lacks histopathological or molecular clarity.
The HCC microenvironment is characterized by structurally and functionally abnormal immune cells, tumour vasculature and stroma, including CAFs [117]. Technological advances in simultaneously localizing multiple cell phenotypes have enabled the description of unique cellular neighbourhoods in liver cancer, such as organization into perivascular immune neighbourhoods [118], to better understand disease progression.
By combining data from spatial proteomics, spatial transcriptomics, and multiplex imaging, HCC-specific FAP+ CAFs and CAF-C7 (complement-7) CAF subsets have been identified [119]. These HCC CAF subsets are mainly located in intratumoral and marginal zones, respectively, with roles in inflammation and tumour progression and in wound healing, respectively. Thus, these HCC-specific CAF subsets might have some opposing functions. FAP+ CAFs’ inflammatory hubs involving PD1+ CD8+ T cells with reduced cytotoxic capabilities imply a contribution to immune suppression. Dense ECM fibres in FAP+ CAFs stroma may also physically restrict anti-tumour immune capabilities [119]. Such studies demonstrate that high-plex multi-modal technologies are advancing our understanding of CAF diversity and roles in cancer development and progression. Further such analyses are essential to move forward the understanding of TME for better targeting and discovery of TME-directed therapies.
Malignant cells can have very abnormal morphology, or can appear nearly identical to a normally functioning cell nearby, and so are categorized histopathologically as well as poorly differentiated. In HCC, hepatocytes lose polarity, particularly near portal tracts and fibrotic septa, and can exhibit ballooning that associates with altered gene expression. Chronic inflammation consequent upon hepatocyte damage can lead to fibrosis, but all these processes are reversible. Fibroblast location is an important component of the TME, and their presence and intercellular signaling can modify the TME.
CAFs have been extensively studied in mice and there is promising data from human tumours of types other than liver, despite fibroblast heterogeneity. Currently, treatment of the underlying liver disease seems the most effective antifibrotic, and subsequently anti-cancer strategy [120, 121]. However, the known critical interactions of CAF in the TME make them a prospective target for developing therapies for HCC.
Dense fibroblastic banding or encapsulation commonly surrounds tumours in murine models, but encapsulation is not typical in immunocompetent primary human tumours [120, 122]. Some subtypes of HCC differ, which needs more immunophenotype mapping in primary human tissue to elucidate. Overall, patterns of fibroblast localization vary markedly (Fig. 2), and include fibroblastic cores in tumours, fibroblasts in semi-lunar configurations semi-circumferentially in the tumour, and near circumferential fibrous “walls” [123]. Fibroblasts and fibrosis are usually denser around the edge of HCC tumours as a broad category. It is uncommon in HCC for fibroblasts to be infiltrative, or nesting in the tumour space. The architecture and location of TME and cancer cells in the tissue are also not necessarily static, responding to cellular signaling and changing location and function accordingly. Spindled cells can be stained to determine lineage, as not all spindle cells are fibroblasts. Thus, architectural aspects of the TME require consideration in CAF targeted therapies, especially whether such therapy might enhance embolization.
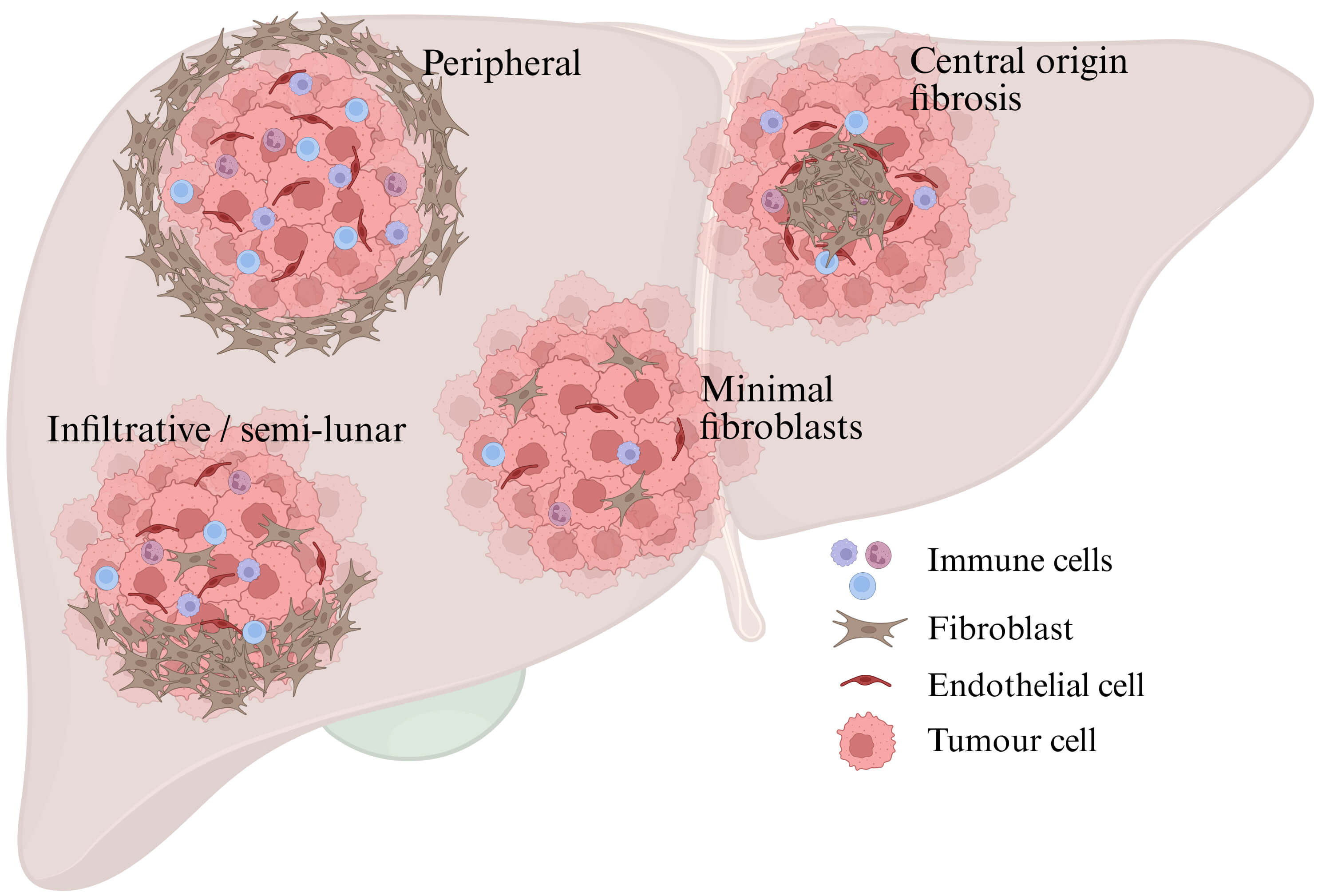 Fig. 2.
Fig. 2.
Stromal architectures in hepatocellular carcinoma (HCC) vary regarding fibroblast location and density. In HCC tumours, stromal fibroblasts are generally in lower abundance than in the surrounding liver but location and abundance vary. Created in BioRender. Boumelhem, B. (2025) https://BioRender.com/mrbznhb.
CAFs across different tumour types, typed by molecular marker or organ system, display remarkable heterogeneity and plasticity [48, 123]. Animal models are not able to fully mirror the human immune system, cancer microenvironment, and potential for off-target effects, and this fundamentally limits their utility for the development of useful molecules for human treatment [48, 123, 124].
Staining fibroblasts would visualize the cellular components of the TME to better understand TME structure, cellular interactions, and whether an ECM barrier forms. Reticulin stain is used regularly in anatomical pathology to visualize the non-cellular network in HCC for diagnostic purposes; however, CAF research would benefit from more frequent visualization of the cellular TME, which is not routine in diagnostic pathology, but would be useful for translating CAF-targeted therapies. Some understanding of CAF interactions with malignant cells, the immune system, and benign tissues has been elucidated in human organ systems [40, 125]. However, in humans, the fibroblast subtypes and CAF-associated cellular patterns in primary HCC are not well understood [118]. Moreover, the types of fibroblasts present in histopathological subtypes of HCC, location in relation to the tumour cells, and changes during tumour development and the influences of changes in leukocyte subsets are needed in large numbers of tumours for improved CAF targeting in cancer therapies.
A variety of compounds have been found to influence CAFs by non-immunological mechanisms (Table 3, Ref. [126, 127, 128]).
| Drug Name | Chemical Structure | Pathway or Biomarker | Reference |
| Chelerythrine chloride | 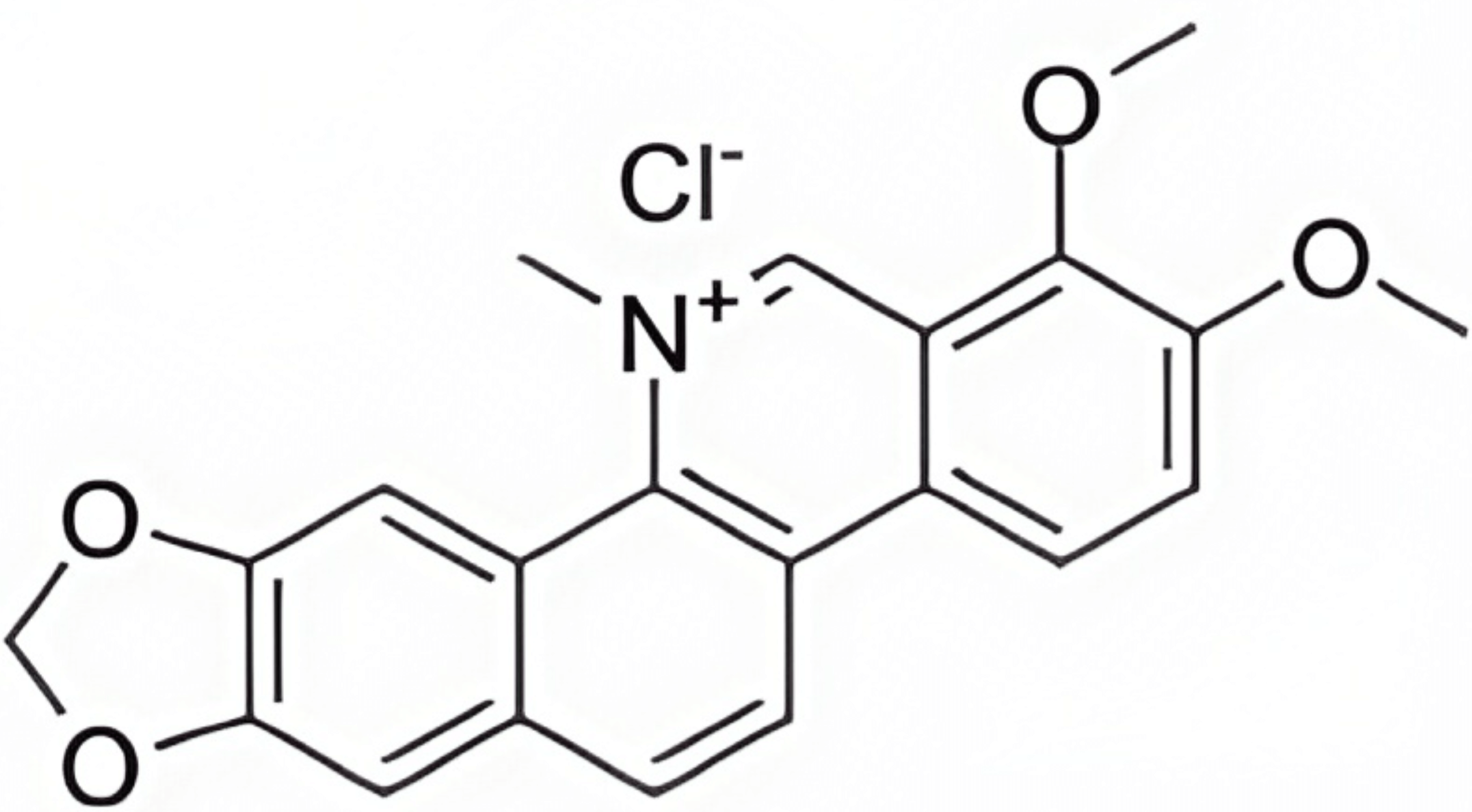 |
WNT10B/ |
[126] |
| Mirogabalin | 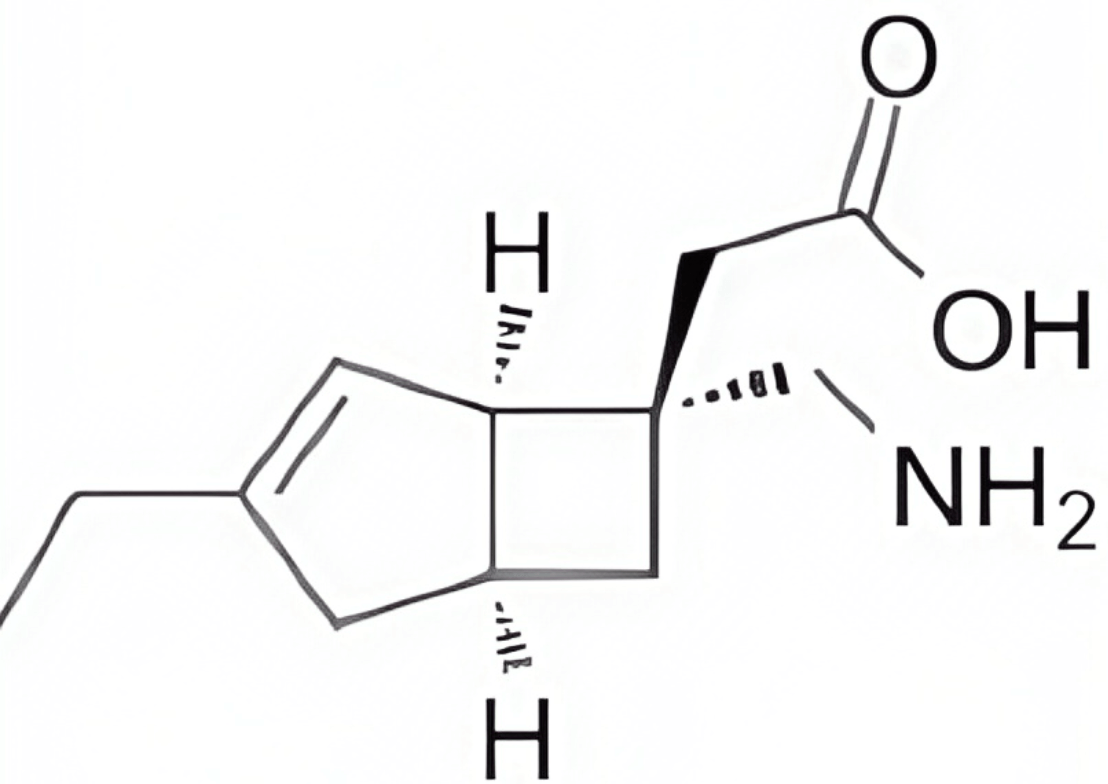 |
Decreased CAF and cancer-associated pain | [127] |
| Triptonide | 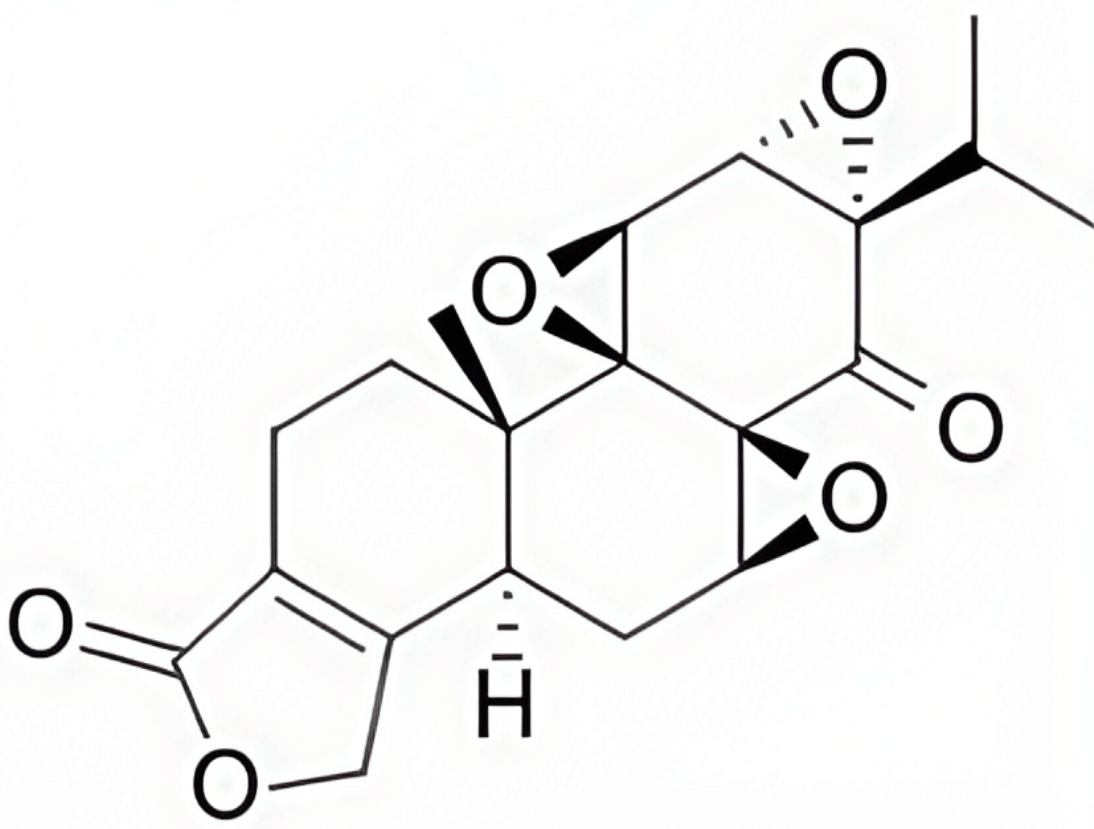 |
microRNA-301a suppression, and microRNA-149 upregulation in CAFs | [128] |
Immunotherapies based on CAF targeting are expanding, based on a rationale that
CAFs can secrete barrier collagens and suppress T cell infiltration into TME
while being far less genetically and phenotypically variable than tumour cells
[129]. Such approaches target biomarkers of CAFs. Pharmacological inhibition of
NADPH oxidase 4 (NOX4) with Setanaxib promotes intra-tumoral CD8+ T cell
infiltration [130]. Inhibition of TGF-
FAP-targeted DNA vaccination can weaken chemotherapy resistance [135], perhaps implying that any method of depleting CAFs might weaken chemotherapy resistance. To leverage immunotherapy, combining FAP-targeted cancer vaccines, such as CpVR-FAP or MVA-FAP, with cyclophosphamide can inhibit tumor growth [135].
A decade ago, a potential safety concern involved possible FAP expression in skeletal muscles and bone marrow [136], but recent safety evaluations of FAP-targeted therapies have set aside this concern [137, 138, 139, 140, 141, 142, 143, 144, 145].
As FAP occurs in soluble form in plasma, some methods of FAP targeting of CAFs might be misdirected by soluble circulating FAP (see section 4.3). This possibility should be and generally has been considered for each approach, while allowing for the 20-fold greater concentration of circulating FAP in mice than in humans [141, 146].
Increasingly, combination therapies of different modes of action (MOAs), such as chemotherapy and immune checkpoint or other immunotherapy, show promising, potentially synergistic, efficacy increases [147]. Combining nanobody–paclitaxel (Nab-PTX) with soluble TRAIL/Apo2L (tumor necrosis factor-related apoptosis-inducing ligand; TNFSF10) has shown improved efficacy and less resistance, compared to a single agent in a PDAC model [147]. Both mesenchymal stromal cells and cancer cells die in this treatment regimen [147]. Also, a multi-target nanoparticle delivery system using docetaxel-containing gold nanoparticles (GNPs) has shown promise for targeting both CAFs and cancer cells in vitro [148].
GNPs can act in nanomedicine as a drug carrier for chemotherapy and a radiosensitizer in radiotherapy [149, 150, 151]. A novel multi-target delivery system using docetaxel and GNPs is promising in targeting both CAFs and cancer cells [148]. Docetaxel can block the cell cycle at G2/M phase by stabilizing microtubules to prevent spindle assembly during mitosis, which traps GNPs within the cancer cell. The quantity of intracellular GNPs is proportional to the effectiveness of chemical drug treatment and radiotherapy in both CAFs and cancer cells. This technology is being further developed to improve radiotherapy and potentially reduce adverse effects [148, 152, 153].
FAP was first discovered as a cell surface antigen that can be highly expressed in CAFs in many types of human epithelial tumours compared to its barely detectable level in non-diseased human tissues [146, 154, 155, 156]. FAP is a type II transmembrane serine protease and belongs to the prolyl oligopeptidase or S9 protease family, which also includes dipeptidyl peptidase (DPP) 4, DPP8, and DPP9 [157]. FAP is a unique protease as it contains both endopeptidase activity and dipeptidyl peptidase activity for hydrolyzing the post-proline bond [157, 158, 159].
FAP can promote pathological disease progression via its unique characteristics. Therefore, fully understanding interactions between its natural protein substrates and FAP is critically important for understanding the many types of diseases that involve activated fibroblasts.
The well-known substrates of FAP are FGF-21 [160, 161, 162], collagens [158, 162, 163, 164, 165],
and
FGF-21 is a key regulator of cell metabolism and has an important role in metabolic disorders. The cleavage site of FAP is at Pro-171, close to the C-terminus and in a site necessary for cognate receptor activation. Therefore, FGF-21 is an important substrate for FAP. FGF-21 is cleaved by FAP in primate plasma [160]. This cleavage of FGF-21 can limit the half-life of pharmacological doses of human FGF-21 [161], but all the forms of FGF-21 that are under development for metabolic dysfunction therapy are modified to prevent hydrolysis by FAP. There is a knowledge gap in understanding the concentrations of active FGF-21 in illnesses compared with healthy individuals [170].
FAP can also cleave some DPP4 substrates, including substance P, peptide YY, and neuropeptide Y. Neuropeptide Y was the first identified physiological substrate of FAP [171]. Examining many substrates indicates that FAP prefers a small hydrophobic amino acid to an acidic residue at the P1′, P3, and P4 sites. It has specificity for particular residues at P2, preferentially cutting after Gly-Pro [162, 168].
FAP is often strongly expressed on the cell surface of activated fibroblasts (membrane FAP; memFAP). A soluble form of FAP in the circulation (cFAP) [146] is an active derivative of memFAP but its mechanism of release from the cell surface, perhaps by a sheddase protease, is not understood. Mouse serum contains 10- to 20-fold more cFAP than human serum [146] and mouse cFAP is greatest before weaning, but does not associate with disease in adult mice, which indicates that mouse and human FAP protein abundances are regulated differently.
In humans, the quantity of the enzyme-active form of cFAP is proportional to total cFAP protein [172]. In humans, the many studies showing significant associations between cFAP and disease severity include fibrosis in metabolic dysfunction-associated steatosis [172, 173], inflammatory bowel disease [174, 175], cancer [176], and certain coronary disease events [177, 178, 179], but not obesity [172].
In inflammatory bowel disease, elevated cFAP is positively associated with endoscopically assessed mucosal remodeling and FAP overexpression by myofibroblasts [174] in diseased tissue. As in liver [180, 181], FAP expression probably reflects the intensity of tissue remodeling because it hydrolyzes a number of collagens and other ECM-associated proteins. However, the physiology of cFAP and memFAP in inflammatory bowel disease needs to be understood.
FAP is usually overexpressed by activated CAFs in tumour stroma and contributes to stroma formation and remodeling in epithelial carcinomas [154, 156, 182]. As in liver [180, 183], it is likely that FAP+ cells are associated with crosslinked collagen in tumours. However, the potential for cFAP as a serum biomarker more broadly than for liver fibrosis is unclear [172, 177, 178]. Possibly, a large well-perfused organ that makes most serum proteins is more likely than a tumour to contribute to cFAP from liver myofibroblasts. The few studies on this question have provided mixed outcomes [175, 184], including less cFAP in cancer patients [176, 185]. Serum cFAP levels in cancer patients with solid tumours are likely influenced by several diverse parameters, including vascular integrity, quality of blood perfusion, tumour location and size, and possible interference with mechanisms of shedding FAP from CAFs. Although total intratumoral FAP content generally correlates with tumor size (see section 4.6.3, below), there is no consistent association between cFAP levels and high FAPi PET signal intensity. Consequently, cFAP lacks utility for cancer detection/screening. However, we speculate that longitudinal studies of changes in cFAP following cancer therapy might be informative (Fig. 3, Ref. [172, 181]).
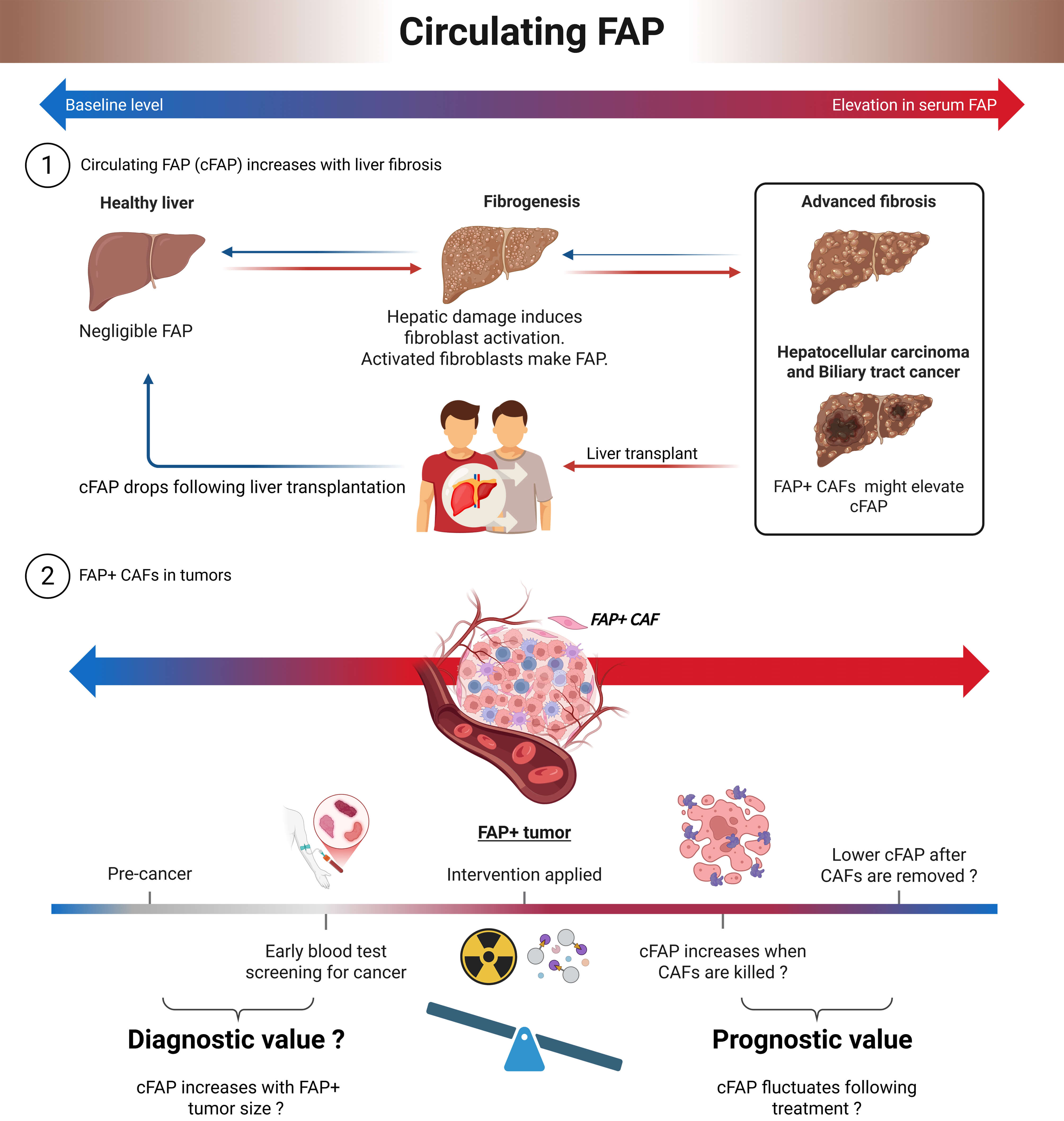 Fig. 3.
Fig. 3.
Circulating fibroblast activation protein (cFAP). Intrahepatic FAP occurs in activated myofibroblasts and CAFs. Part 1: Circulating FAP (cFAP) correlates with fibrosis severity in fatty liver disease [181], and drops soon after removal of a liver that has severe fibrosis [172]. Part 2: We speculate that although cFAP is not a reliable indicator of cancer, it might vary with increases and decreases in intratumoral FAP+ CAF numbers.
Greater understanding is needed regarding the relationship between circulating and cellular FAP, the mechanism of converting memFAP to cFAP, and the cellular origins of cFAP. Stromal fibroblasts in epithelial carcinoma, fibroblasts in fibrotic tissue during wound healing, such as cardiac fibrosis, cirrhosis, and arthritis, and embryogenic mesenchymal fibroblasts are identified origins of FAP expression [154, 186, 187], but the originating cell type of increased cFAP in diabetes is unclear [188].
Naturally, in a clinical setting, cFAP quantitation as a biomarker would be interpreted in context, especially for a patient with multiple conditions that might influence cFAP levels. Perhaps cFAP will be most useful at the time of onset of extensive fibroblast activation and at times of extensive removal of activated fibroblasts. The latter situation occurs following liver transplantation, and is also likely to occur following a CAF-targeted therapy. We have shown that cFAP falls following liver transplantation [172], so cFAP quantitation following any therapy that is designed to kill FAP+ cells in humans should be undertaken. However, cFAP may fall following tumour removal but perhaps not following radiation therapy [185]. Extensive death of FAP+ cells might not occur abruptly but rather be gradual over time, and sometimes radiation therapy results in fibrosis. When assessing the response to FAP-targeted cancer therapy, early and later blood test timing might detect dynamic fluctuations in cFAP levels (Fig. 3). Nevertheless, quantifying cFAP as a biomarker has exhibited some clinical potential in cancer and in hepatic and cardiac fibrosis [172, 173, 176, 177, 185, 189].
Several methods of measuring cFAP have been developed; primarily, enzyme assay and ELISA [172, 173, 176], which are very well correlated with each other [172]. Unlike MMPs, FAP does not have a zymogen form and so is constitutively active. ELISA can be influenced by proteases that degrade the antibody epitope recognized by an ELISA. FAP enzyme assay can be influenced by protein degradation and by natural inhibitors of FAP. Osteolectin, which is expressed in bone and is a serum biomarker for active bone remodeling, is a natural inhibitor of the enzyme activity of FAP [190, 191]. Thus, perhaps serum osteolectin is a potential modulator of cFAP activity. There is some, but limited, evidence that both intact and catalytically inert FAP protein have tumour-promoting activity, probably through protein–protein interactions [186, 192].
FAP exhibits a variety of roles in tumours [156, 193, 194]. These roles include a functional role in cancer cell migration and metastasis [182, 192, 195, 196, 197, 198, 199], which may occur when FAP is made by a cancer cell. Recombinant overexpression of FAP in the human breast cancer cell line MCF-7 can increase FAK phosphorylation, thereby enhancing cell growth, migration, and cell invasion [199]. This effect may be reversible by FAK inhibition. In addition, FAP has proteolytic actions upon ECM proteins [200, 201, 202] that may disrupt ECM structure to facilitate tumor cell migration and invasion [156]. FAP may also disrupt ECM formation by degrading LTBP, ECM1, and thrombospondins and disrupt ECM structure by degrading denatured collagens [162, 169]. In addition, FAP can promote the growth and invasion of cancer cells by regulating the activation of kinases ERK and AKT [200, 203]. ECM degradation is a complex process that typically involves proteases, including MMPs, MMP inhibitors (TIMPs), tyrosine kinases, and various hydrolases [204]. Thus, FAP-mediated ECM degradation could influence tumour progression by modulating cell migration, angiogenesis, immune response, and inflammation.
TGF-
FAP promotes angiogenesis via cellular and molecular cooperation [207, 208, 209]. Vascular endothelial growth factor-A (VEGF-A) can enhance endothelial tubule formation [210]. FAP can influence VEGF-A expression in osteosarcoma, renal carcinoma (RCC), and CRC cells via PI3K/AKT and ERK signaling pathways [203, 207, 209]. So, FAP+ mesenchymal cells or pericytes adjacent to blood vessels might drive vascularization. Angiogenesis-promoting molecules, such as Ang-2, endothelin-1, and insulin growth factor binding protein-2, are produced by FAP+ mesenchymal cells [208]. Moreover, in lung cancer and breast cancer models, FAP has been implicated in tumour angiogenesis [211, 212, 213].
Stromal FAP promotes cancer progression via EMT through the
Wnt/
FAP+ cells are discussed here because their functions can overlap with outcomes of FAP overexpression in vitro. However, FAP enzyme inhibition should not be conflated with the removal of FAP+ CAFs cells, because FAP+ cells exert both FAP-dependent and FAP-independent effects.
FAP+ fibroblasts dysregulate immune responses, with involvement in
migration and differentiation of TAMs, CD4+CD25+ Treg cells, and MDSCs
[136, 216, 217]. FAP+ cells make multiple chemokines, most prominently CCL2.
CCL2 is not a FAP substrate [162]. Instead, FAP induces STAT3 activation and CCL2
upregulation in CAFs [218]. This STAT3 signaling pathway is activated, causing
CCL2 release, thus recruiting MDSCs through a CCL2 gradient [219]. MDSCs in the
TME are immunosuppressive, mainly via TGF-
FAP+ CAFs secrete abundant fibronectin 1 (FN1) that engages integrin
CXCL12 primarily regulates cell migration via its cognate receptor, CXCR4 [221]. CXCL12 is typically distributed in a gradient within the ECM, guiding the migration of inflammatory cells and other cell types along specific paths [221]. CXCL12/SDF-1 is a FAP and DPP4 substrate [168, 222] made at high levels by FAP+ CAFs [136]. So, in the context of inflammation, FAP influences the CXCL12-CXCR4 signaling pathway [223], which involves PI3K-Akt, ERK-MAPK, and JAK-STAT [224]. In a pancreatic cancer model, targeting CXCL12 produced by FAP-expressing CAFs can enhance the therapeutic efficacy of anti-PD-L1 immunotherapy [136]. Similarly, immunomodulation induced from FAP+ fibroblasts through the CXCL12/CXCR4 axis occurs in oesophageal carcinoma [225]. Thus, several models demonstrate the association between FAP and the CXCL12/CXCR4 signaling pathway in the TME. Disrupting the CXCL12-CXCR4 pathway in tumours is desirable and might be achieved by removing FAP+ CAFs.
FAP is linked with the Th1 cytokine interleukin-17 (IL-17), reminiscent of the Th1/Th17 pro-inflammatory association with DPP4 [226, 227]. IL-17A is an important pro-inflammatory cytokine in chronic liver diseases and HCC [228, 229]. IL-17 is an independent trigger of FAP expression by hepatic stellate cells (HSCs) both in vitro and in vivo [230]. Moreover, IL-17RA and FAP are significantly positively correlated with HSC activation, with activation indicated by SOX9, cytokeratin7, and cytokeratin19 expression in human liver [230]. FAP overexpression changes cell behavior in HSC [192]. Furthermore, FAP expression in CAFs has been positively associated with the expressions of PDL-2, PD-1, CTLA4, and PDL-1 in HCC cells, in which cell proliferation and migration and inhibition of apoptosis increase [231]. Thus, like DPP4/CD26, intrahepatic FAP may be pro-inflammatory and foster tumours.
A challenge of antiangiogenic therapy in CRC liver metastasis models (CRCLMs) is blood vessel co-option in bevacizumab-resistant CRCLM [232], which is associated with FAP+ HSC density [233]. Depleting FAP+ HSC in a CRCLM xenograft tumour mouse model disrupted the co-opted sinusoidal blood vessels and increased mouse survival time by about one-third [233, 234]. Thus, removing FAP+ cells from mice can be efficacious and safe in tumour models.
In patients with BTC, such as ICC, highly expressed FAP is significantly associated with worse survival, which indicates the important role of FAP in promoting tumour growth in ICC and its potential as a diagnostic biomarker or therapeutic [89].
The literature reviewed above establishes FAP as a marker of pathogenic CAFs. FAP enzyme inhibition may confer anti-cancer outcomes but all therapeutics that remove FAP+ CAF are expected to deplete tumours of FAP activity, so may, in part, replicate outcomes of FAP inhibition. Both solid tumours and most fibrotic tissues contain high expression of FAP, making FAP also a potential target in fibrosis [156, 235, 236]. The specific distribution and specific proteolytic activity of FAP make it an ideal target for theranostic approaches to tumours and fibrotic conditions.
To achieve precision treatment and lowered risk of drug resistance, high specificity therapies are critical. Photodynamic therapy is being developed for both cancerous and non-cancerous diseases. The indispensable features of this therapy are the photosensitizer, with an appropriate emission wavelength, and intracellular oxygen. The principle is based on fluorescence resonance energy transfer (FRET), aggregation-induced emission, photo-induced electron transfer, and self-quenching of the photosensitizer [237, 238, 239, 240]. Excitation of a photosensitizer triggers energy transfer onto an oxygen molecule to generate cytotoxic reactive oxygen species. A FAP sensitive cleavage site is conjugated with a clinical photosensitizer, such as methylene blue (MB) [241]. The pro-photosensitizer FAP-MB derivatives are screened for maximum hydrolytic efficiency of activation by recombinant human FAP and relative fluorescence intensity of tumours to normal organs (Fig. 4, Ref. [242]).
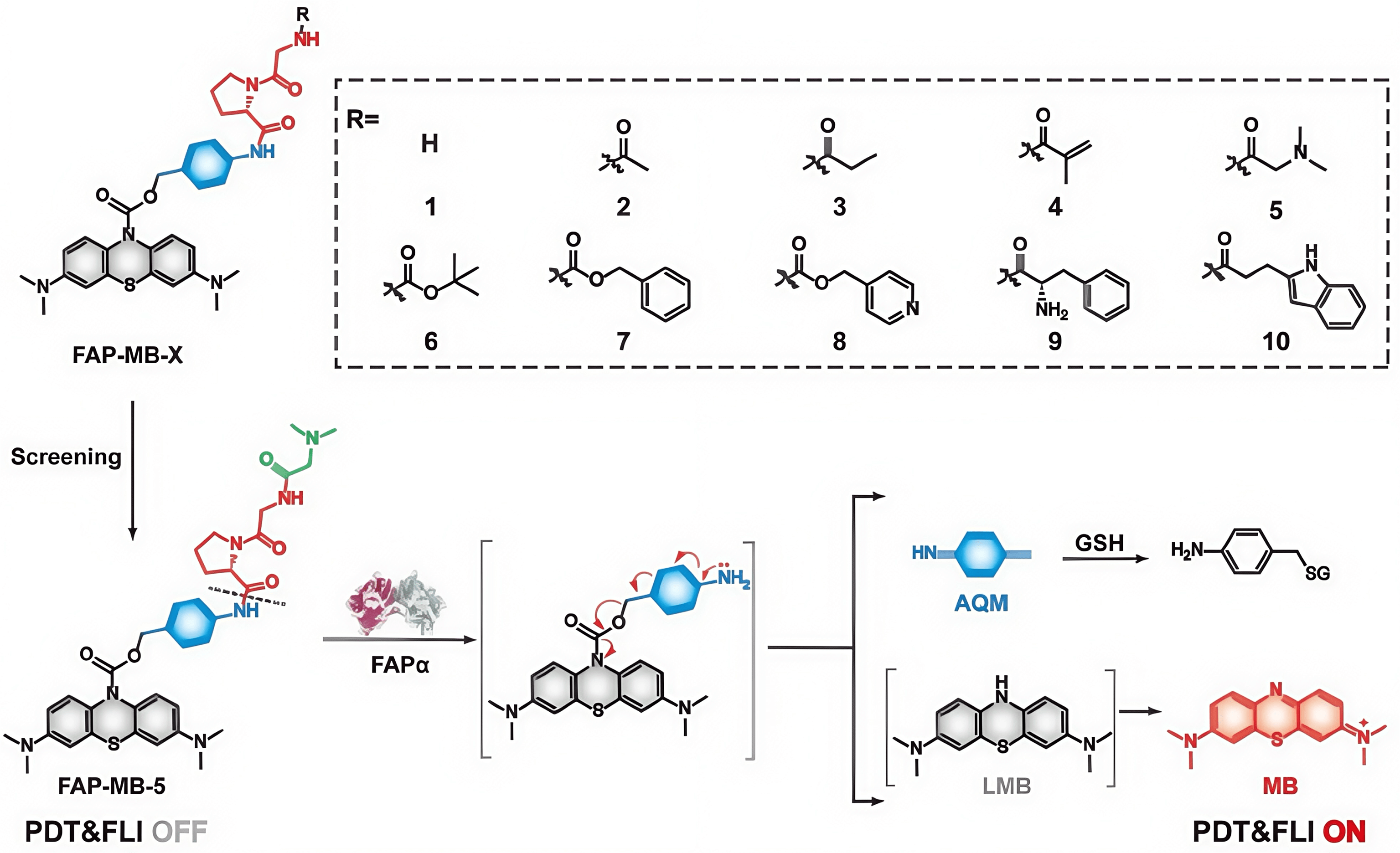 Fig. 4.
Fig. 4.
Schematic of a FAP-activated theranostic photosensitizer class. Structures of FAP activatable methylene blue (MB) derivatives FAP-MB-1–10, and mechanism of action [242]. Derivative FAP-MB-5 has been found to be the most efficiently hydrolyzed by FAP (at dotted line). PDT, photodynamic; FLI, fluorescence imaging; AQM, azaquinone methide; SH, glutathione; LMB, leucomethylene blue. Figure adapted from [242].
A FAP-mediated photodynamic method can be engineered to cause immune stimulation
[243, 244]. ZnF16Pc (zinc hexadecafluorophthalocyanine) is a potent
photosensitizer (
IRDye700DX is a widely used photosensitizer in non-cancerous diseases. A FAP antibody 28H1-700DX has been used to induce FAP-specific cell death in vitro and may become an arthritis therapy [245]. This FAP-directed photodynamic therapy has shown patient-derived fibroblast contraction in a 3D cell culture model [246, 247]. Moreover, 28H1-IRDye700DX has shown promise on rheumatoid arthritis synovial explants and can accumulate in arthritic joints, with very low background signal [248].
Antibodies to FAP were problematic for many years due to the number of commercial antibodies that were not FAP specific [249] and early attempts to target FAP using antibodies failed to deliver clinical benefit. However, specific, potent anti-FAP antibodies are now emerging that have shown that FAP is strongly expressed in a broad range of tumours [91, 250, 251, 252]. Moreover, novel anti-FAP antibodies, including single domain antibodies, are being developed to deliver toxins, radiation, IL2, and CD137 agonism to tumours [143, 144, 145, 250, 251].
Positron emission tomography (PET) imaging, or PET scan, is usually combined with computed tomography (CT) to detect and characterize tumour stages, assessing for tumour metabolism and monitoring treatment response. 18F-FDG PET/CT is widely used in clinical oncology because cancer cells have high glucose uptake and consumption, linked to the Warburg effect [253]. Due to glucose uptake by normal brain, liver, and heart and the unique characteristics of FAP, including its presence in most tumour stroma, FAP-targeted PET tracers, such as FAP inhibitors (FAPIs), are increasingly applied in clinical imaging [254, 255, 256, 257]. FAPI-PET/CT enables highly specific tumour visualization for improving cancer diagnosis and monitoring, especially in sarcoma and in stroma-rich tumours, including pancreatic, gastric, colorectal, biliary, salivary, thyroid, and breast cancers [256, 258, 259, 260, 261].
4.6.3.1 Development of FAP-Targeted Radioimaging in Oncology
FAPI-04 was the first FAP-selective inhibitor developed for FAPI-PET (Fig. 5; Table 4, Ref. [141, 142, 262, 263, 264, 265, 266, 267, 268, 269, 270, 271, 272, 273, 274, 275, 276, 277, 278]). Derived from the University of Antwerp’s UAMC-1110, which was the first quinoline-based FAP selective inhibitor, the University of Heidelberg’s FAPI-04 was selected to have superior affinity for FAP in in vivo pharmacokinetics for tumour retention during PET [279]. Gallium-68 (68Ga) is a short half-life positron-emitting radionuclide that was first used in clinical medicine in the early 1960s, and remains popular as a potent molecule suitable for labeling peptides and small molecules like FAPIs [280, 281]. 68Ga-FAPI-04 FAPI PET/CT is a pan-cancer imaging agent, shown in breast [279, 282, 283], lung [282, 284, 285], prostate [282, 286, 287], colorectal [282, 288, 289], and liver [90, 282, 290, 291] cancers. Superiority of [68Ga] Ga-FAPI-04 over [18F]-FDG-PET has been clearly established, especially for metastases and HCC and CRC tumours [254, 257, 292]. FAPI-PET/CT shows extremely low uptake of 68Ga-FAPI in the liver compared to [18F]-FDG [90, 254, 293].
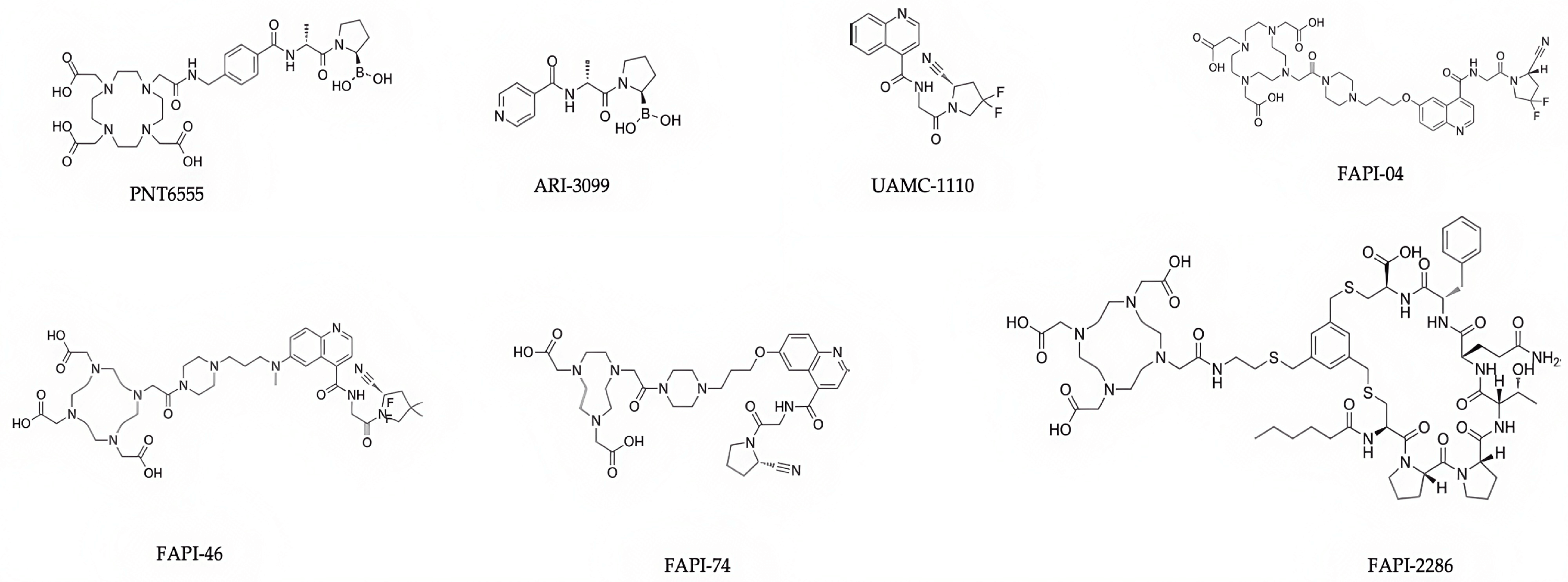
| Name | Chemical Class | Mechanism of Action | Radionuclides | Ongoing Research Field | References |
| PNT6555 | Boronic acid-based small molecule | Potent and selective FAP inhibition | 177Lu (therapy), 68Ga (imaging), 225Ac (therapy), 161Tb (therapy) | Pan-cancer PET, theranostic | [141, 142] |
| PNT3090 | PNT6555 derived | Potent and selective FAP inhibition | 177Lu (therapy), 68Ga (imaging) | Tumour retaining theranostic | [142, 263] |
| ARI-3099 | Boronic acid-based small molecule | Potent, selective FAP inhibition | N/A | Research tool | [264] |
| UAMC-1110 | Quinoline-based small molecule | Highly potent and selective FAP inhibition | N/A | Cancer, fibrotic diseases | [265] |
| FAPI-04 | UAMC-1110-based | FAP targeting, bind CAF | 68Ga (imaging), 18F (imaging) | Pan-cancer PET | [266, 267, 268] |
| FAPI-46 | UAMC-1110-based | FAP targeting to CAF | 68Ga (imaging), 177Lu (therapy), 225Ac (therapy), 90Y (therapy) | Pan-cancer PET, theranostic | [262, 269, 270, 271, 272, 273] |
| FAPI-74 | UAMC-1110-based | FAP targeting for tumour imaging | 18F (imaging), Cold-Kit 68Ga | Various solid tumours (pancreatic, BTC, HCC, gastric, bladder, ovarian, pheochromocytoma, lung, neuroendocrine, mesothelioma, sarcoma) | [273, 274, 275, 276, 277] |
| FAP-2286 | Cyclic peptide | FAP targeting to CAF | 177Lu (therapy), 68Ga (imaging) | Pan-cancer PET, theranostic | [270, 278] |
Table footnote: Ac, actinium; BTC, biliary tract cancer; F, fluorine; Ga, gallium; HCC, hepatocellular carcinoma; Lu, lutetium; Tb, terbium; Y, yttrium.
FAPI-46 is a modified UAMC-1110 compound [294] superior to FAPI-04 and generally chelated to 68Ga using DOTA [254, 295]. [68Ga]FAPI-46 PET/CT has shown clear superiority to 18F-FDG PET/CT, with, for example, better SUVMAX for staging BTC [262, 295]. However, a [68Ga]FAPI-46 PET/CT limitation is difficulty discriminating between benign hepatic lesions such as focal nodular hyperplasia, hepatic adenoma, and cirrhosis [296].
For head and neck squamous cell carcinoma, 68Ga-FAPI-46 PET/CT has shown
close concordance with 18F-FDG PET/CT for initial staging and
recurrence/metastasis detection [297]. Moreover, such PET/CT achieves comparable
levels of detection of metastases in lymph nodes, liver, and bone [298]. However,
a study of small (
FAPI-74 is probably now the most commonly used FAPI-PET reagent, which can use 1,4,7-triazacyclononane-N, N′,N′-triacetic acid (NOTA) as a chelating agent to enable labeling with 68Ga, or with 18F via a complex with aluminum ([18F]AlF) [300, 301]. Superiority of [18F] AlF-FAPI-74 over 18F-FDG in PET/CT in most primary and metastatic lesions in gastric, pancreatic, and liver cancers has been shown [276, 277, 302]. In addition, [18F] FAPI-74 has a greater synthetic yield and better image resolution than 68Ga-labeled FAPI [274, 275], and is superior to 18F-FDG, based upon SUVmax, TBR, and diagnostic accuracy [303, 304]. However, false-positives can occur, even using both FAPI-PET and FDG-PET, thought to be due to glucose uptake by metabolically active leukocytes and FAP expression by activated fibroblasts in inflamed tissues [305]. Moreover, FAPI-PET false positives have been caused by an external jugular vein thrombus that can prolong blood retention [306].
The cyclic peptide FAPI-2286 (Fig. 5) [270, 307] was developed to address the low tumour retention of FAPI radiotracers that are based on UAMC-1110. Longer tumour retention time is needed for the therapeutic use of a FAPI. Compared to FAPI-46, FAPI-2286 yields greater signal intensity in the kidney, liver, and heart [270, 308]. In preclinical tests, FAPI-2286 shows longer tumour retention than FAPI-46 [309]. Moreover, FAPI-2286 presents a significant advantage in identifying patients with muscle-invasive bladder cancer who may not benefit from radical surgery, as it can discriminate between false-positive and false-negative results that arise from conventional imaging [310]. In summary, FAPI-2286 has displayed superior FAP binding affinity, tumour accumulation, and tumour retention times compared to the UAMC-1110 series of compounds.
Many new FAPIs and antibodies for PET and for radioligand therapy (RLT) are under development [250], with preclinical data published for some [139, 141, 233, 311, 312], but this review is focused on FAP-targeted agents that have published clinical data. Currently, a variety of series of FAP inhibitors and FAP binding peptides are being optimized for clinical use. Many that are published are discussed here. The optimization goals are primarily to maximize tumour uptake and achieve suitable retention times in solid tumours for imaging (brief) and for delivering a therapeutic dose of radiation (prolonged).
FAPI are most commonly bound to 68Ga, but other radionuclide labels are sometimes used, most often 18F, but also 90Y, 89Zr, 11C, 64Cu, 67Cu, 177Lu, and 255Ac [313, 314, 315]. Besides the Gallium-68 (68Ga) and Fluorine-18 (18F), Carbon-11 (11C) can be used in imaging; therefore, some novel inhibitors specifically exploit 11C. The key characteristic of the 11C-RJ1101 and 11C-RJ1102 compounds is their novel 11C-methylated derivatives (Fig. 6, Ref. [315]). Their short half-life could be an important advantage as they persist in vivo no longer than other positrons [315]. FAPI-based therapy generally uses 177Lu [295] or 225Ac [272].
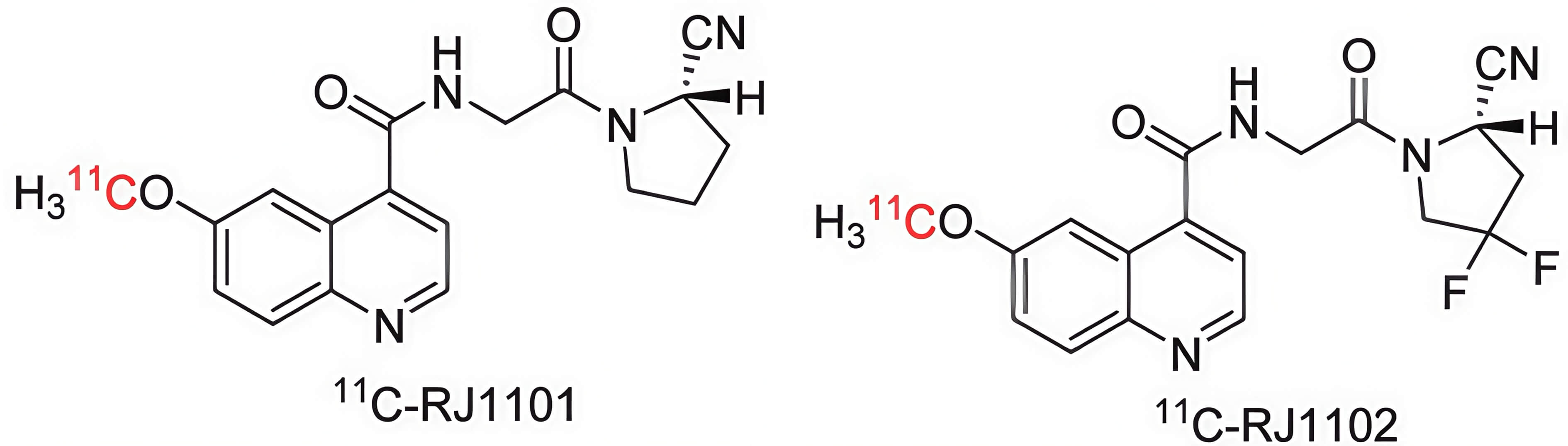 Fig. 6.
Fig. 6.
The radiolabeling structures of 11C-RJ1101 and 11C-RJ1102 [315]. Figure reproduced under CC‑BY license.
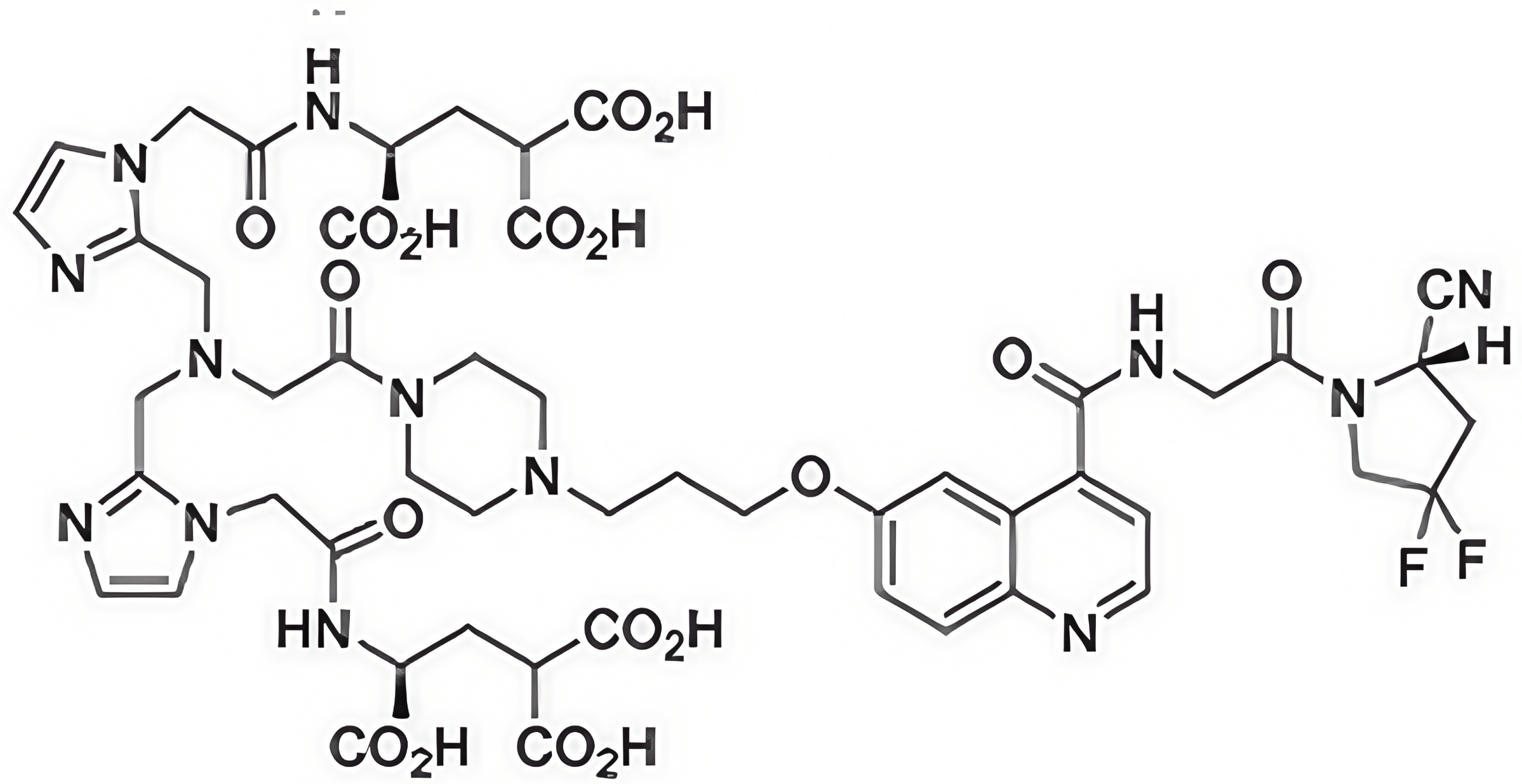
In addition to advances in radionuclides and FAPI for PET/CT, evolving SPECT technology is important for tumour imaging and endoradiotherapy [316]. The most common radionuclide for SPECT images is 99mTc. Advantages of 99mTc SPECT are an affordable price and greater availability of equipment. Therefore, 99mTc conjugated compounds may elevate the popularity of SPECT-based cancer screening [317]. Compared with optimized compounds of the UAMC series, such as FAPI-28, -29, -33, -34, and -43, FAPI-04 has poor intratumoural accumulation because of its high lipophilicity. Intratumoural accumulation has been increased by attaching an asparagine with a neutral carboxamide side chain in FAPI-28 to confer some lipophilicity. When modified for SPECT, 99mTc-FAPI-34 (Fig. 7, Ref. [318]) was a further improvement [316], with this FAPI showing high and reliable uptake in several solid tumours, particularly breast cancer and metastases [319, 320, 321].
Albumin conjugated compounds have been made because albumin can retard excretion
and confer high cell permeability and intratumoural retention time in humans
[322, 323, 324]. 68Ga-FSDD0I (Fig. 8) has a higher binding affinity to
albumin and less hydrophilicity (logP = –1.18
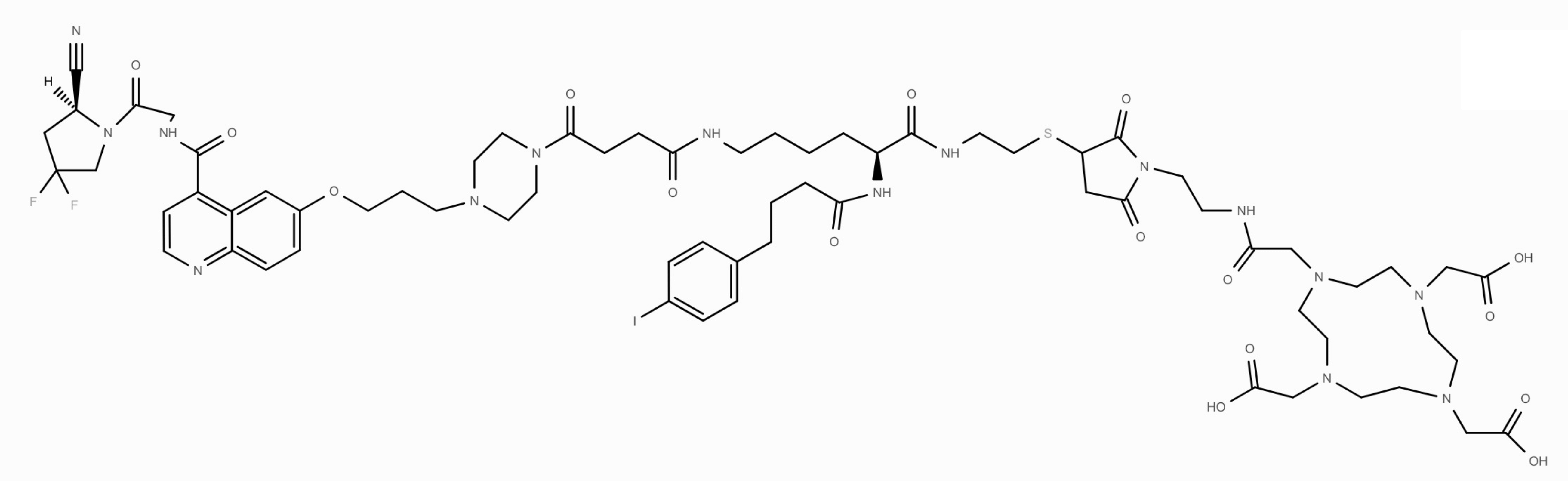 Fig. 8.
Fig. 8.
The structure of FSDD0I. Created using Ketcher v2.25.0.
A number of FAP-targeted agents, including a modification of the cyclic peptide
2286, DOTAGA-FAP-2286-ALB, and derivatives of FAPI-04, TE-FAPI-06 and -07,
include an albumin-binding moiety to increase stability in human serum, with high
FAP binding affinity in vitro and excellent performance in PET and SPECT
imaging [307, 322, 323, 325, 326]. For example, compared with 177Lu-FAPI-04,
TE-FAPI-06 has exhibited high tumour uptake of 7.3
4.6.3.2 Development of FAP-Targeted Radiopharmaceutical Tools in Oncologic Therapy
An interesting example of FAPI-PET/CT potential is its application in tumour classification of consensus molecular subtype (CMS) of colorectal tumours [327]. Most colorectal tumours make FAP [154, 327, 328]. More than that, RNA and protein data implicate FAP as a marker of therapy resistance and poor prognosis and CMS4 [327, 329]. FAPI-PET/CT imaging provides clear discrimination of CMS4 from CMS1-3 CRC [327, 329], showing that FAPI-PET could be very useful in CRC management.
In contrast to FAPI-PET imaging that usually uses 68Ga or 18F-labeled tracers and is designed for tumour localization, staging, and treatment planning, Lutetium-177 (177Lu) is the most common radionuclide in FAPI-targeted RLT because it emits beta particles that can travel from CAFs to cancer cells and thereby kill both cell types. Beta radiation causes localized DNA damage (Table 4). 177Lu has a 6.7-day half-life to allow sustained therapeutic effects, which is useful for systemic treatment of metastatic tumours. Alternative radionuclides for RLT include 225Ac, 67Cu, 90Yitrium, 161Terbium, and 47Scandium [330, 331].
177Lu-FAPI-2286 and 68Ga-FAP-2286 are a promising theranostic (therapy/diagnostic) pair and compared with FAPI-46 [309, 332, 333]. In preclinical PET/CT, 68Ga-FAP-2286 and 68Ga-FAPI-46 are comparable and both can shrink tumours when labelled with 177Lu. However, 177Lu-FAP-2286 is superior to 177Lu-FAPI-46, with longer intratumoural retention time, greater absorbed dose delivered to tumour, and greater tumour inhibition [309]. Limited patient response data have been promising with metastases, including squamous cell carcinoma and adenocarcinoma [278, 332, 333].
Achieving retention of radiation in a tumour, with a high tumour:non-target ratio, has emerged as the greatest challenge for FAPI theranostics. However, the boron-containing FAP selective compound 177Lu-PNT3090 has these characteristics and so is a major advance from the team led by medicinal chemist W.W. Bachovchin [142, 263].
FAP-targeted PET/CT is a potential tool to monitor FAP levels before, during, and following treatment using FAP-targeted and other therapies, as seen using [89Zr] anti-FAP antibody PET/CT when developing FAP CAR-T cells [313]. A simpler method of monitoring the effectiveness of therapies that are designed to eliminate FAP+ cells might be the lowering of circulating levels of soluble FAP (cFAP), as occurs following removal of a cirrhotic liver by liver transplantation [172] (Fig. 4).
4.6.3.3 FAP Radiopharmaceuticals in Nononcologic Diseases
Significant densities of activated FAP+ fibroblasts occur in liver, cardiac, renal and lung fibrosis and in arthritis and endometriosis, so FAPI PET/CT has been investigated and shows promise for these non-oncologic conditions and diseases (Table 5, Ref. [90, 189, 235, 256, 334, 335, 336, 337, 338, 339, 340, 341, 342, 343, 344, 345, 346, 347, 348, 349]) [140, 296, 312, 334, 349, 350, 351, 352, 353, 354]. Using FAPI-PET or FDG-PET in inflamed tissues can produce false-positive images that are thought to be due to glucose uptake by metabolically active leukocytes and FAP expression by activated fibroblasts in inflamed tissues [305]. Moreover, FAPI-PET false positives have been caused by an external jugular vein thrombus that can prolong blood retention [306].
| Disease | FAPI | Tracer | Reference |
| Liver fibrosis | Various FAPI | 68Ga, 18F | [90, 235, 335, 336, 349] |
| Chronic thromboembolic pulmonary hypertension | FAPI-04 | 68Ga | [337] |
| Acute myocardial infarction | FAPI-04 | 68Ga, 18F | [338, 339, 340] |
| Cardiac injury/fibrosis | FAPI-04, FAPI-46 | 68Ga, 18F | [189, 256, 341] |
| Light-chain cardiac amyloidosis | FAPI-04 | 68Ga | [342] |
| Cardiovascular disease | FAPI-04 | 18F-NOTA-FAPI-04 | [343] |
| Rheumatoid arthritis | FAPI-04 | 68Ga | [334, 344, 345] |
| Pulmonary fibrosis diseases | FAPI-46 | 68Ga | [346] |
| Idiopathic pulmonary fibrosis | FAPI-74 | 18F | [347] |
| Atherosclerotic plaque | MIP-1232 | 125I | [348] |
Table footnote: F, fluorine; Ga, gallium; I, iodine.
Rather than blocking the site of catalysis in FAP enzyme molecules with an inhibitor, prodrugs harness that catalytic activity to release an active form of a drug (the “warhead”) from an inert “prodrug” compound that contains a peptide bond that only FAP can hydrolyze [169, 355]. FAP has a unique catalytic specificity [156, 236]. Therefore, prodrugs contain a pharmacologically active drug (the “warhead”), such as doxorubicin, that is inactivated by covalent linkage to an FAP-specific peptide. Targeting tumours is conferred by the specific and large upregulation of FAP expression by CAFs. If the warhead is a cytotoxin, then activation of the prodrug into an active drug can kill FAP+ cells and nearby cells.
Suitable warheads include drugs that exhibit cytotoxicity at cancer chemotherapy-effective doses, such as doxorubicin [137, 356, 357] and emetine [358]. Emetine is a natural toxin but its cytotoxicity can be blocked by derivatizing its N-2′ position. The FAP-cleavable, DPP4-cleavable peptide Ac-Ala-Ser-Gly-Pro-Ala-Gly-Pro (ASGPAGP) has been added at N-2′ to create an emetine prodrug [358]. Because the FAP-related enzymes DPP4 and prolyl endopeptidase are ubiquitous and thus unsuitable to target in a prodrug, more recent prodrugs are FAP-specific.
Thapsigargin is an early example of a lead compound modified to create a FAP-activated prodrug: A peptide carrier containing a FAP selective cleavage site is coupled to a thapsigargin (TS) analog via a linker [355]. That early study established the FAP-activated prodrug strategy in the literature.
The key power of a prodrug is to greatly increase the intratumoural concentration of a therapeutic with precise tumour targeting to produce a high tumour:uninvolved organ ratio of concentrations. Prodrug development is facilitated by converting an existing, well-understood drug into a prodrug. Ideally, before treatment, a tumour is identified as both FAP+ and sensitive to the chosen drug, such as doxorubicin.
Doxorubicin (Fig. 9, Ref. [137, 359]) is a very effective antitumour drug but is dose-limited by its cytotoxicity. Therefore, concentrating doxorubicin in tumours by exploiting the high prevalence and abundance of FAP in tumours is potentially an ideal synergy. This approach reduces systemic doxorubicin toxicity in mice and humans and produces high tumour to blood ratios of doxorubicin concentration [137, 357]. Doxorubicin prodrugs can have, for example, an N-terminal benzyloxy carbonyl (Z)-blocked peptide [357], Thr-Ser-Gly-Pro as a FAP-specific linker [359], and/or use a photosensitizer [359].
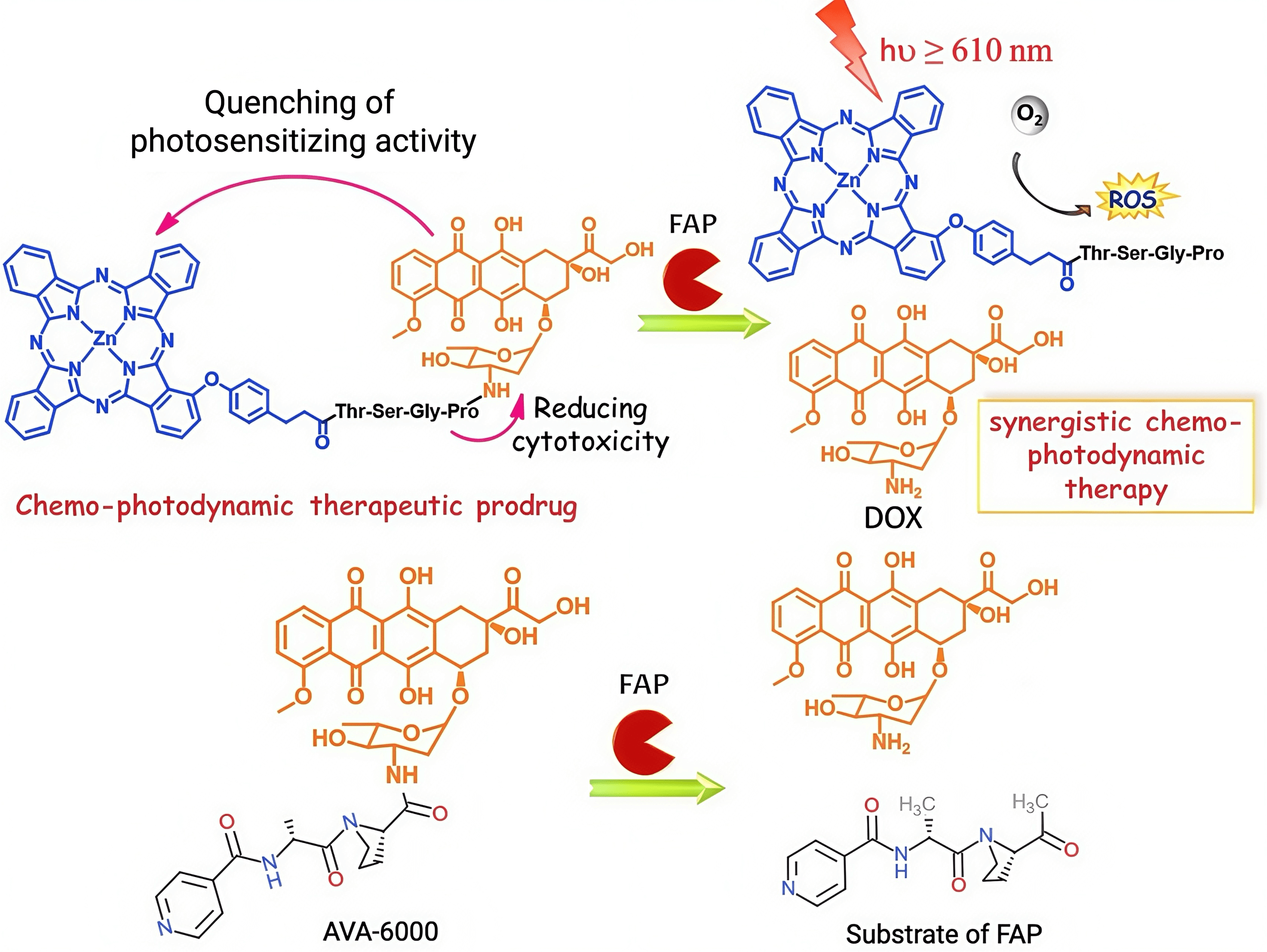 Fig. 9.
Fig. 9.
Schematic illustration of FAP-activated prodrugs that have a
doxorubicin (DOX) warhead. The top schematic illustrates the design and
activation of a FAP-cleavable chemo-photodynamic prodrug [359], a prodrug
constructed by conjugating a photosensitizer (blue) with the chemotherapeutic
agent doxorubicin (DOX, orange) via a peptide linker (Thr-Ser-Gly-Pro) that can
be hydrolyzed by FAP specifically. In its intact form, the prodrug exhibits
quenched photosensitizing activity and reduced cytotoxicity, enhancing systemic
safety. Upon cleavage by FAP, the linker is degraded, releasing active
doxorubicin and restoring photosensitizing activity. Upon irradiation with red
light (
The high specificity of FAP to activated fibroblasts is attracting a variety of approaches (Fig. 10) and the safety in humans of removing FAP+ fibroblasts has been established. Limited efficacy data are emerging. The current major question is which approach has the greatest efficacy.
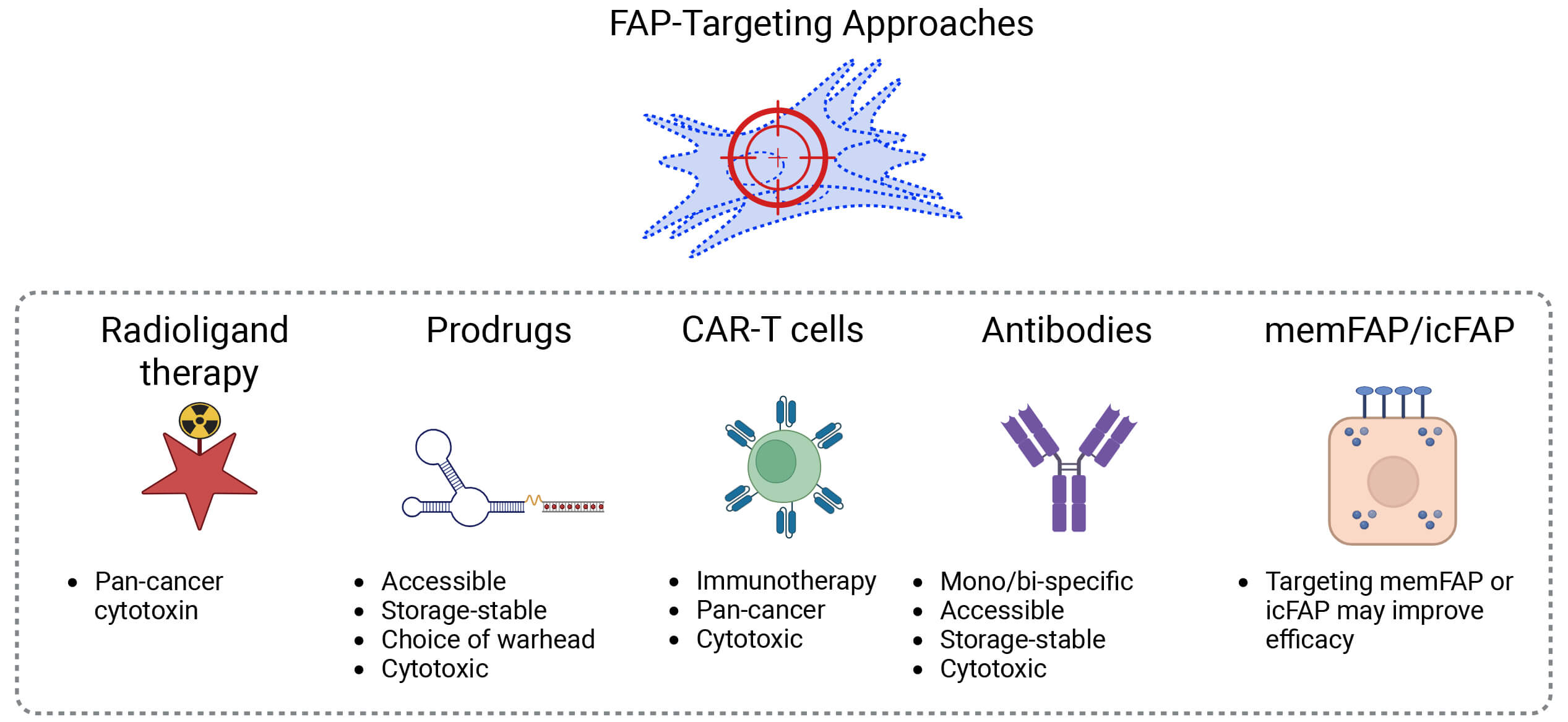 Fig. 10.
Fig. 10.
Summary of commentary. Key advantages of each prominent approach to targeting FAP and FAP+ cells. ic, intracellular; mem, membrane. Created in BioRender. Boumelhem, B. (2025) https://BioRender.com/xq3xm9k.
The major hurdle for RLT, which is the persistence of radiation delivery inside the tumour, is being solved. An advantage of RLT is that radiation can kill cells in any tumour type. An advantage of prodrugs is storage and availability because radiation decays rapidly. Moreover, when an array of prodrugs with various warheads becomes available, a prodrug can be selected according to the known susceptibility of the tumour that is to be treated. However, this desirable goal is slowed by the attendant need to develop multiple prodrugs: each delivering a different chemotherapeutic or cytotoxic warhead.
Promising, specific, potent, sometimes bispecific CAR-T cells and antibodies to human FAP are now emerging. Some imaginative approaches have succeeded. Such antibodies can deliver a cytotoxin that has broad or narrow specificity, or can facilitate an immune response or immune cell-mediated killing of CAFs. CAR-T cells remain problematic for solid tumours.
Soluble FAP (cFAP) can conceivably decoy FAP-targeted agents away from tumours, so agents that can target memFAP or intracellular FAP and not react with cFAP (soluble FAP) would be expected to display greater efficacy than indiscriminate FAP-targeted agents. In all FAP-targeted approaches, agents that can achieve slow release in tumours will be advantageous.
The future will reveal exciting new knowledge of which approaches to FAP targeting, including choice of agent, patient, tumour, and treatment timing, are the most effective in humans. The FAP-activated prodrug field as yet contains few warheads: A number of prodrugs will need to be developed, with a variety of warheads, each suited to each tumour genotype. This field has the potential to greatly boost precision oncology.
The question of which other therapy is best combined with a FAP-targeted therapy will need to be explored. The best methods and approaches, and appropriate situations for FAP-targeted clinical imaging, need to be further explored. Assessing treatment success will probably have two aspects: assessing the removal of FAP+ cells and assessing tumour mass, which likely needs separate techniques.
FAP expression by activated fibroblasts extends beyond CAFs, to fibrosis, so the extent to which FAP-targeted CAF removal also alleviates fibrotic disease needs clarification. Taking a step back and targeting triggers of the transition of cells to the mesenchymal phenotype might prevent fibrosis and neoplasia.
CAFs are an attractive target for cancer therapy because they are genetically stable, support tumour growth, and are ubiquitous in solid tumours. The known critical interactions of CAF in the TME make them an exceptional research target for developing therapies for HCC. Many CAFs express FAP, a cell surface protease that is specific to activated mesenchyme and prominent amongst CAF markers. Increasing numbers and varieties of FAP-targeting approaches for therapeutic benefit in cancer and fibrosis are appearing, including FAP inhibitors in RLT, antibody-dependent cytotoxicity or immunotherapy, and FAP-activated prodrugs.
ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; APCs, antigen presenting cells; apCAF, antigen presenting CAF; ATG5, autophagy-related 5; BMSC, bone marrow mesenchymal stem cell; BTCs, biliary tract cancers; C7, complement-7; CAFs, cancer associated fibroblasts; CAR, chimeric antigen receptor; CCL, C–C motif chemokine ligand; CCR, C–C motif chemokine receptor; CMS, consensus molecular subtype; CN-1, type I collagen; CRC, colorectal cancer; CRCLMs, CRC liver metastasis models; CT, computed tomography; CTLA-4, cytotoxic T-lymphocyte–associated protein 4; cDC1, conventional DC type 1; cFAP, circulating FAP; DC, dendritic cell; DDR2, discoidin domain containing receptor 2; DES, desloratatine; ECM, extracellular matrix; EGF, epidermal growth factor; EMT, epithelial-to-mesenchymal transition; EndMT, endothelial-to-mesenchymal transition; EV, extracellular vesicle; FAP, fibroblast activation protein; FAPI, FAP inhibitor; FDG, fludeoxyglucose; FGF-2, fibroblast growth factor 2; FRET, fluorescence resonance energy transfer; FSP1, fibroblast-specific protein 1; GNPs, gold nanoparticles; GRP78, glucose protein 78; HCC, hepatocellular carcinoma; HGF, hepatocyte growth factor; HSC, hepatic stellate cell; HTRA1, HtrA serine peptidase 1; ICC, intrahepatic cholangiocarcinoma; IFN, interferon; IL, interleukin; iCAF, inflammatory CAF; LAG3, lymphocyte activation gene 3; LIANA, ligand–receptor analysis; LncRNA, long non coding RNA; LOX, lysyl oxidase; LPAR, lysophosphatidic acid receptor; LTBP, latent TGF-
Conceptualization, MDG, YAQ, ZVW, JCH, ALF, JMHN, MZ, KAO, BBB, GWM, KL; writing—original draft preparation, YAQ, JCH, ZVW, ALF, JMHN, MZ, KAO, BBB; writing—review and editing, MDG, BBB, GWM, KL; visualization, YAQ, JCH, KAO, ZVW, BBB; supervision, MDG, GWM, KL, BBB; funding acquisition, MDG, GWM, YAQ, JCH, JMHN, MZ. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by postgraduate research scholarships from The University of Sydney to YAQ, JCH, JMHN and MZ, the Joan Krefft bequest to the A.W. Morrow Gastroenterology and Liver Centre, and by anonymous gifts (MDG, GWM).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.