




 , Fatimah Hussain Bu Izran 1
, Fatimah Hussain Bu Izran 11 Department of Anatomy, College of Medicine, Alfaisal University, 11533 Riyadh, Saudi Arabia
Abstract
Bone remains one of the most hospitable—and devastating—destinations for metastatic cancer cells. At the center of this unwelcome alliance is transforming growth factor‑β (TGF‑β), a cytokine stored in the mineralized matrix and unleashed during osteoclastic bone resorption. Once activated, TGF‑β fuels a self‑reinforcing “vicious cycle”: it co‑opts tumor cells to undergo epithelial‑to‑mesenchymal transition, recruits and primes osteoclasts, suppresses osteoblast function, and shapes an immunosuppressive niche that shields malignant clones. The result is a micro‑environment exquisitely tuned for tumor survival, skeletal destruction, and therapy resistance. This review traces the molecular choreography of TGF‑β signaling within the bone tumor microenvironment (TME), detailing its crosstalk with osteogenic, immune, and stromal compartments across breast, prostate, and lung cancer metastases. We synthesize pre‑clinical and clinical efforts to interrupt this pathway, ranging from ligand-neutralizing antibodies and activin receptor-like kinase 5 (ALK5) kinase inhibitors to antisense oligonucleotides and tumor-selective ligand traps—and examine why benefits observed in early trials are tempered by dose‑limiting toxicities and adaptive resistance. Beyond TGF‑β itself, we highlight parallel targets in the TME, including receptor activator of nuclear factor kappa-B ligand (RANKL)‑driven osteoclastogenesis, vascular endothelial growth factor/fibroblast growth factor (VEGF/FGF)‑mediated angiogenesis, and immune checkpoints such as PD‑1, TIM‑3, and LAG‑3, arguing that multi‑pronged combinations guided by real‑time TME profiling offer the most promising path forward. We outline pressing research priorities: mapping the spatiotemporal dynamics of TGF‑β activation, identifying predictive biomarkers for patient stratification, and engineering bone‑targeted delivery systems that preserve normal tissue repair. By decoding and disrupting the TGF‑β‑centered circuitry of bone metastasis, we can move closer to therapies that not only palliate skeletal complications but also prolong life for patients with advanced cancer.
Keywords
- TGFβ signaling
- bone metastasis
- tumor microenvironment
- epithelial–mesenchymal transition
- targeted therapy
Bone metastases represent a significant clinical concern across various cancer types, notably breast, prostate, and lung cancers, due to their high incidence, prevalence, and associated morbidity and mortality. The skeletal system is a common site for metastasis due to its rich blood supply and supportive microenvironment, which facilitates the colonization of tumor cells. Understanding these dynamics informs clinical management strategies and provides insights into patient prognosis, particularly regarding skeletal-related events (SREs) that stem from these metastases.
Bone metastases are reported to occur in a considerable percentage of patients
suffering from primary cancers. Studies indicate that approximately 68% of
metastatic bone diseases arise from three major cancers: breast, prostate, and
lung cancer [1]. Breast cancer is frequently noted as the leading cause of bone
metastases in women, while prostate cancer holds a similar position in male
patients [2]. Prostate cancer metastasizes to the bone in about 70–90% of
patients at the time of advanced disease onset, rendering it one of the most
prevalent forms of skeletal metastases [3, 4]. Lung cancer patients exhibit
notable rates of skeletal involvement, with bone metastases affecting about
30–40% of those diagnosed with advanced stages of the disease [5, 6]. A
recent population‑based SEER analysis of
The morbidity associated with bone metastases primarily manifests through skeletal-related events (SREs), which encompass a spectrum of complications, including pathologic fractures, pain, and hypercalcemia. SREs significantly deteriorate the quality of life and elevate healthcare costs due to increased management requirements such as palliative care, surgical interventions, or radiotherapy [5, 10, 12]. Pathologic fractures are prevalent in this patient population; they occur in 7–35% of patients with lung cancer and account for a significant reason for hospitalization among those with bone metastases from breast and prostate cancers [5, 13]. Hypercalcemia, often considered a paraneoplastic syndrome, emerges in 1–20% of patients and is associated with a poor prognosis due to its debilitating symptoms, including nausea, confusion, and renal dysfunction [5, 12]. Overall, SREs like these not only exacerbate the patient’s clinical condition but also significantly contribute to increased mortality rates, emphasizing the need for effective preventive strategies and therapeutic interventions [14, 15].
The prognosis for patients with bone metastases is generally poor, particularly when considering the burden of SREs. The presence of skeletal involvement has been correlated with reduced overall survival across multiple cancer types, with prostate cancer patients displaying an approximately 70–90% probability of developing bone involvement as the disease progresses [3, 16]. SREs are directly linked to an increase in mortality; data reveal that patients with skeletal metastases experience a markedly escalated risk of dying compared to their counterparts without such complications [14, 15]. Consequently, the early identification of bone metastases and the implementation of appropriate interventions are critical for improving outcomes in affected individuals.
Management of bone metastases, particularly in prostate cancer, may involve bisphosphonates or denosumab, agents that can ameliorate the incidence of SREs while potentially improving quality of life [17, 18]. Recent studies propose novel approaches, including targeted therapies and the utilization of radiopharmaceuticals, which have shown promise in managing advanced prostate and breast cancers alongside their metastatic complications [12, 19]. The integration of palliative care from the point of diagnosis is also crucial to managing symptoms and improving patient-centered outcomes, highlighting the need for a multidisciplinary approach in treating patients with metastatic disease [12, 20, 21].
Transforming growth factor‑
The signaling mechanisms of TGF-
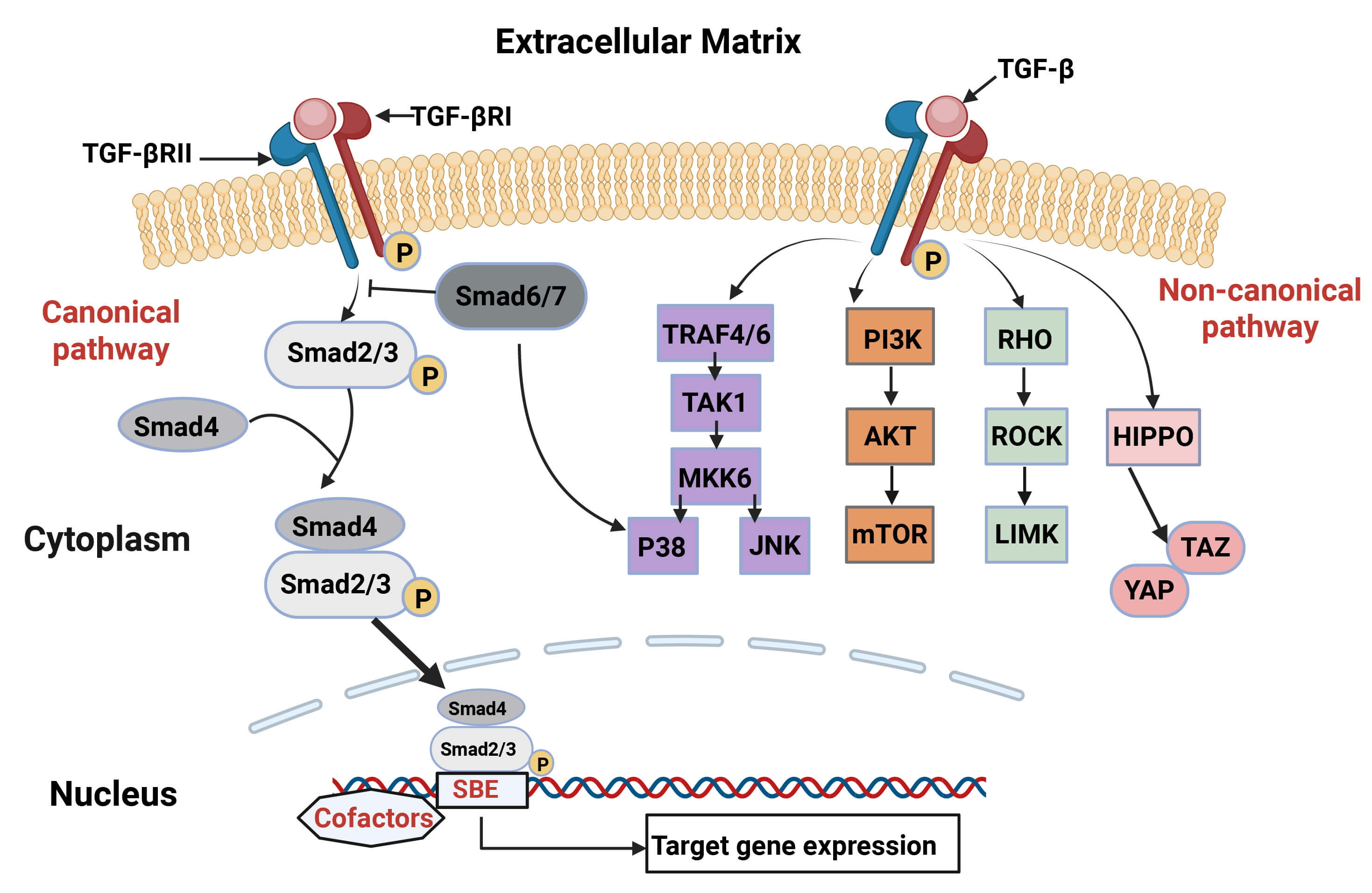 Fig. 1.
Fig. 1.
Canonical and non-canonical transforming growth factor-beta
(TGF-
TGF-
The interplay between these signaling arms enables TGF-
In healthy tissue, TGF-
TGF-
The role of TGF-
TGF-
Given its dual nature as both a suppressor and promoter of tumorigenesis,
targeting the TGF-
Bone metastases are a common phenomenon in advanced cancers, particularly in breast, prostate, and lung cancer. They occur when cancer cells spread to the bone marrow and begin to proliferate, leading to significant complications, including pain, fractures, and systemic symptoms. Understanding the cellular and non-cellular components of the bone metastatic environment is critical for devising therapeutic strategies.
Bone metastasis represents a significant complication in advanced cancer, manifesting a complex interplay of molecular and cellular mechanisms. This process involves interactions between tumor cells and the bone microenvironment, creating a conducive environment for cancer cell survival, colonization, and growth. Specific signaling pathways, cell behavior alterations, and the skeletal microenvironment’s dynamic milieu drive the metastasis of various cancers to the bone.
The mechanisms underlying metastatic spread to the bone can be conceptualized as a cascade involving multiple steps, including local invasion, intravasation into the bloodstream, survival in circulation, extravasation into bone, and, ultimately, establishment in the bone microenvironment. In various cancers, including prostate, breast, and lung cancers, the bone microenvironment is enriched with factors that promote the metastasis process, making it a unique niche for tumor cell colonization [45, 46].
Critical pathways implicated in bone metastasis TGF-
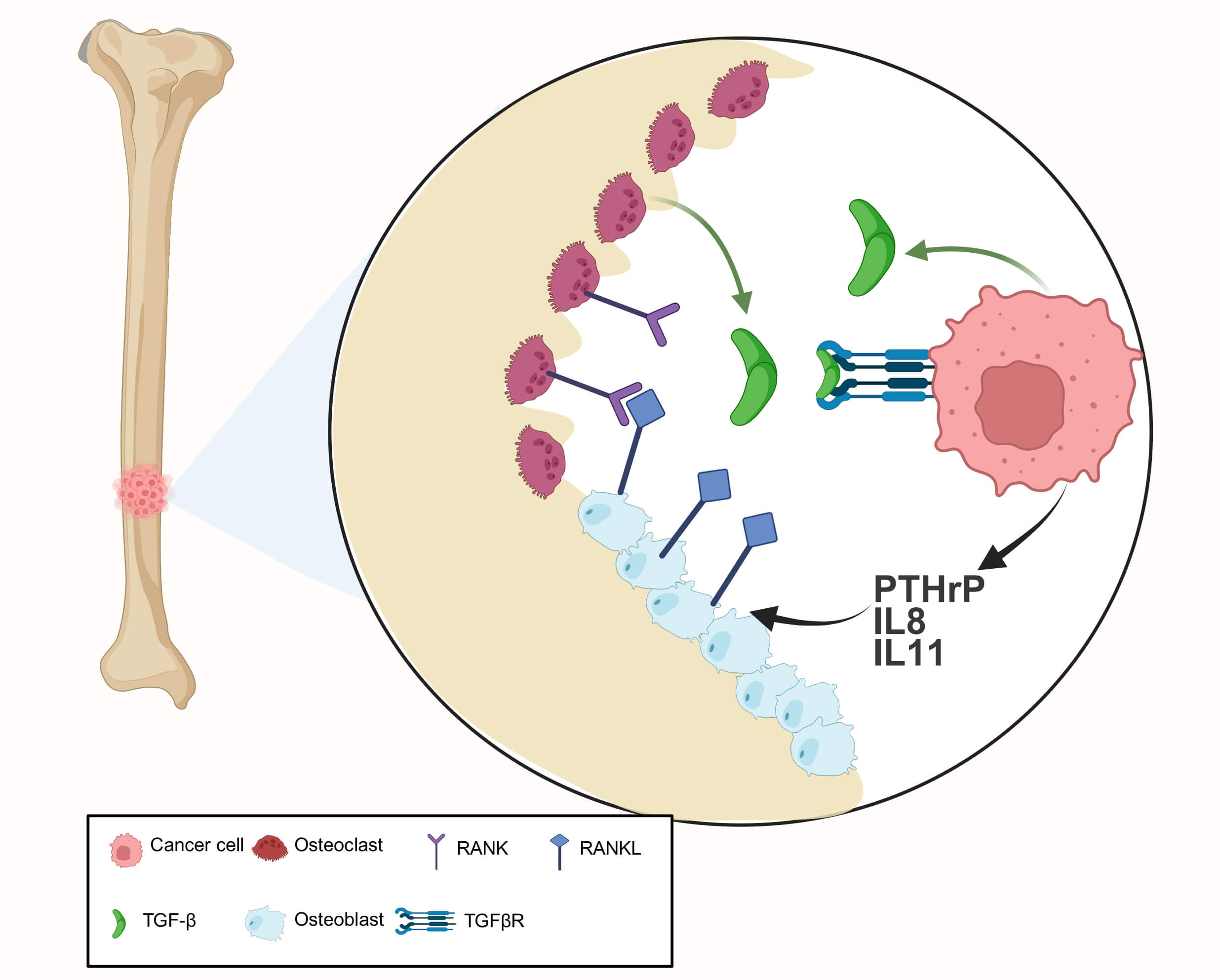 Fig. 2.
Fig. 2.
The TGF‑
The pathobiology of bone metastases is further complicated by the “vicious cycle” of bone metastasis model, which delineates how metastatic cells can induce bone destruction while simultaneously enhancing their own growth [56]. This cycle begins with osteolysis, where cancer cells release factors that stimulate osteoclasts to resorb bone, resulting in the release of growth factors that bolster tumor growth [57]. In prostate cancer, the presence of tumor cells in bone stimulates osteoclast activity leading to increased osteolytic lesions, contributing to severe complications such as SREs [58]. Factors contributing to the bone metastatic process include the expression of specific receptors and adhesion molecules by both cancer cells and bone cells, which govern the dynamics of their interactions. Increased expression of chemokine receptors such as CXCR4 in breast cancer has been linked to enhanced metastatic spread to the bone, suggesting that chemokine signaling is integral to the homing mechanisms of tumor cells [59, 60]. Studying bone metastasis also involves various molecular and cellular technologies to unravel the intricate pathways. High-throughput sequencing has identified dysregulated pathways that govern metastatic behavior, which are being explored to develop novel therapeutics targeting these pathways [61, 62]. In addition, engineered models simulating the bone microenvironment have contributed to our understanding of how cancer cells adapt and thrive, highlighting potential therapeutic interventions to interrupt these processes [63, 64].
Metastatic spread to the bone is a formidable challenge in clinical oncology,
characterized by multifaceted mechanisms involving signaling pathways,
microenvironmental interactions, immune modulation, and cellular adaptations. The
TGF-
Bone metastasis presents significant clinical consequences, including pain, fractures, hypercalcemia, and a notably decreased quality of life for affected patients. These sequelae are compounded by the limitations of current therapeutic approaches, highlighting the complexity of managing metastatic bone disease.
One of the most debilitating aspects of bone metastasis is pain. It affects approximately 68–70% of patients, with the severity ranging from mild to intractable [65]. Bone remodeling becomes disrupted when cancer cells invade the bone marrow, leading to the release of inflammatory mediators and the activation of osteoclasts, which exacerbates pain [66, 67]. Chronic pain significantly detracts from the quality of life, contributing to physical disability and psychological distress [68, 69]. The algorithms defining cancer pain underscore its multifaceted nature, incorporating nociceptive and neuropathic mechanisms [70]. Fractures, especially pathological fractures, represent another major clinical consequence of bone metastasis. Studies indicate that these fractures commonly occur in weight-bearing bones due to the osteolytic activity facilitated by tumor cells [71, 72]. Such fractures lead not only to acute pain but also to increased morbidity by requiring surgical interventions or long-term rehabilitation [66, 69]. In this context, vertebral fractures can lead to spinal cord compression, creating emergency scenarios that necessitate urgent interventions, thereby affecting mobility and independence [69, 73, 74]. Hypercalcemia is frequently associated with bone metastasis and occurs when osteoclastic activity leads to excessive calcium release into the bloodstream [73]. Symptoms can be severe, including nausea, vomiting, and confusion, and can even lead to renal failure or cardiac complications if untreated [69]. This condition presents a complex challenge as it often requires immediate medical attention in addition to standard cancer treatment protocols. The impact of bone metastasis extends to a patient’s overall quality of life. Research reveals that pain resulting from bone lesions significantly correlates with patients’ physical and emotional well-being [66]. The psychological burden includes anxiety and depression due to chronic pain and disability, further complicating treatment outcomes [70, 75]. The multidimensional challenges bone metastases present underscore the necessity for comprehensive management strategies.
Currently, several therapeutic approaches exist for managing complications arising from bone metastases, primarily focused on palliative care and pain management. These include pharmacological interventions such as bisphosphonates and denosumab, targeted at bone density conservation and pain relief [68, 69]. These agents are crucial in moderating SREs and have been shown to significantly reduce the frequency of skeletal complications related to bone metastases [76].
Radiotherapy, particularly palliative radiotherapy, serves a dual purpose: providing pain relief and treating localized tumor burden [74, 77]. Single-fraction radiotherapy has emerged as a favorable approach due to its efficacy in relieving pain with minimal intervention [74, 78]. However, variability in patient responses necessitates an individualized approach to treatment. Although palliative radiotherapy can lead to improved pain management, most studies reveal that a significant proportion of patients may still experience pain flare-ups post-treatment [79, 80].
Despite the advancements in pharmacologic and radiotherapeutic strategies, limitations persist in managing bone metastases. For instance, bisphosphonates do not prevent metastatic spread, nor do they have significant tumoricidal effects [81, 82]. While effective for pain management and quality of life improvements, these therapies can sometimes cause adverse events, including renal impairment and osteonecrosis of the jaw [76]. Prior treatments may also limit Radiotherapy, and the presence of widespread metastatic lesions may impact its efficacy [83, 84].
Alternative innovations, including image-guided therapies and radiopharmaceuticals like strontium-89 and radium-223, promise to alleviate bone pain associated with metastases; however they also have limitations in terms of side effects and response variability [82, 85]. Recently explored modalities, such as high-intensity focused ultrasound and stereotactic body radiotherapy, offer novel approaches for treatment but require further clinical validation before becoming standard practice [86, 87, 88]. The empirical approach toward managing bone metastases often highlights the need for an interdisciplinary team, including oncologists, radiologists, pain management specialists, and palliative care experts. This collaborative strategy is essential as it provides comprehensive patient assessment and a tailored approach to managing complex symptoms associated with metastatic bone disease [66, 76].
While the clinical consequences of bone metastases include severe pain, fractures, hypercalcemia, and decreased quality of life, current treatments offer significant opportunities for intervention but also face notable limitations. Improving understanding of the underlying mechanisms of pain and developing more effective treatments will be crucial in enhancing outcomes and quality of life for patients with metastatic bone disease.
The Tumor Microenvironment (TME) in bone metastases is a complex ecosystem that plays a crucial role in determining the success of metastatic colonization and subsequent tumor behaviors. It includes various components, such as tumor cells, stromal cells, immune cells, and the extracellular matrix, all of which interact actively to influence tumor growth, response to treatment, and overall patient outcomes.
The TME refers to the cellular environment surrounding a tumor, consisting not only of the tumor cells themselves but also of various stromal cells, immune cells, blood vessels, and the ECM [35, 89]. In the context of bone metastases, the TME is specifically specialized to support the survival and proliferation of metastatic cancer cells that have colonized the bone tissues [35, 90]. The unique architecture and cellular composition of the bone microenvironment create a “niche” that is both supportive and conducive for tumor growth, shaped by the interactions between cancer cells and the local stromal components [91, 92].
The cellular microenvironment in bone metastases comprises several key cellular
constituents, including bone-resident cells, hematopoietic and immune cells, and
cancer cells. The mesenchymal stem cells (MSCs) reside within the bone marrow and
contribute to the structure and function of the bone environment. They play a
role in supporting hematopoiesis and in the differentiation of osteoblasts and
osteoclasts. Osteoblasts are bone-forming cells that produce the ECM and
facilitate mineralization. In the context of bone metastasis, osteoblasts can be
influenced by tumor cells through the release of factors such as parathyroid
hormone related protein (PTHrP) and TGF-
In addition to cellular constituents, the non-cellular components also play a
significant role in the bone metastatic microenvironment. These include
cytokines, growth factors, and the ECM. Cytokines are signaling molecules that
modulate the interactions between cancer cells and the host’s bone environment.
TGF-
The matrix proteins provide structural support and actively modulate the
behavior of cancer cells. Cancer cells utilize integrins to anchor themselves to
fibronectin or osteopontin in the ECM. This adhesion is crucial for their
subsequent migration and invasion into surrounding tissues [96, 101]. The presence
of specific matrix proteins, such as TGF-
The primary components of the TME are the metastatic cancer cells themselves, which may exhibit altered phenotypes compared to their origins, often adopting characteristics that favor invasion and survival in the bone environment [107, 108]. For instance, breast cancer cells can undergo an EMT to increase their invasive potential and adapt to the bone stroma [108, 109]. The bone TME comprises various stromal cells, including osteoblasts, osteoclasts, and fibroblasts. Osteoblasts are involved in bone formation, while osteoclasts are responsible for bone resorption [110]. Tumor cells can manipulate these cells to create a favorable environment for metastasis; they may stimulate osteoclasts to enhance bone resorption, leading to osteolytic lesions typically seen in metastatic disease [111, 112]. This interaction results in a vicious cycle characterized by tumor-induced bone remodeling and further tumor growth [113]. The TME in metastatic bone can contain various immune cell types, including TAMs, T cells, and other lymphoid cells. Macrophages within the TME can actively promote tumor growth by releasing pro-inflammatory cytokines, which can enhance tumor cell proliferation and survival [114, 115, 116]. They also create an immunosuppressive environment that allows for tumor evasion from immune surveillance [117, 118]. TAMs, specifically M2-type macrophages, are often enriched in bone metastases and are associated with promoting tumor growth and progression via their secreted factors [118, 119].
The ECM within the bone TME provides structural support and biochemical signals crucial for maintaining tumor cell survival and proliferation [107, 120]. Changes in the ECM composition, driven by factors released from both tumor cells and stromal components, can facilitate the invasion of cancer cells and modify their interactions within the bone [121]. Matrix proteins, such as collagen and fibronectin, can influence the behavior of cancer cells, including their migration and the signaling pathways involved in metastasis.
The bone TME is rich in cytokines and growth factors that play a crucial role in
stimulating tumor cell growth and survival. Key players include Transforming
Growth Factor beta (TGF-
The TME in bone metastases represents a highly dynamic and interactive environment that profoundly affects tumor behavior and treatment responses. Understanding its complex components and their interactions is essential for developing therapeutic strategies aimed at inhibiting bone metastasis and improving patient
The bone microenvironment plays a pivotal role in supporting tumor colonization and growth, particularly in the context of metastatic disease. The unique cellular composition, including osteoblasts, osteoclasts, osteocytes, immune cells, and various stromal components, creates a supportive niche for tumor cells. The interactions within this microenvironment facilitate tumor growth, modify local bone remodeling processes, and promote the establishment of secondary metastatic lesions.
Tumor cells can stimulate osteoclastogenesis through RANKL (Receptor Activator
of Nuclear Factor Kappa-B Ligand) expression, which binds to RANK receptors on
osteoclast precursors, promoting their differentiation and activation [127]. This
resorption leads to the release of growth factors stored within the bone matrix,
such as TGF-
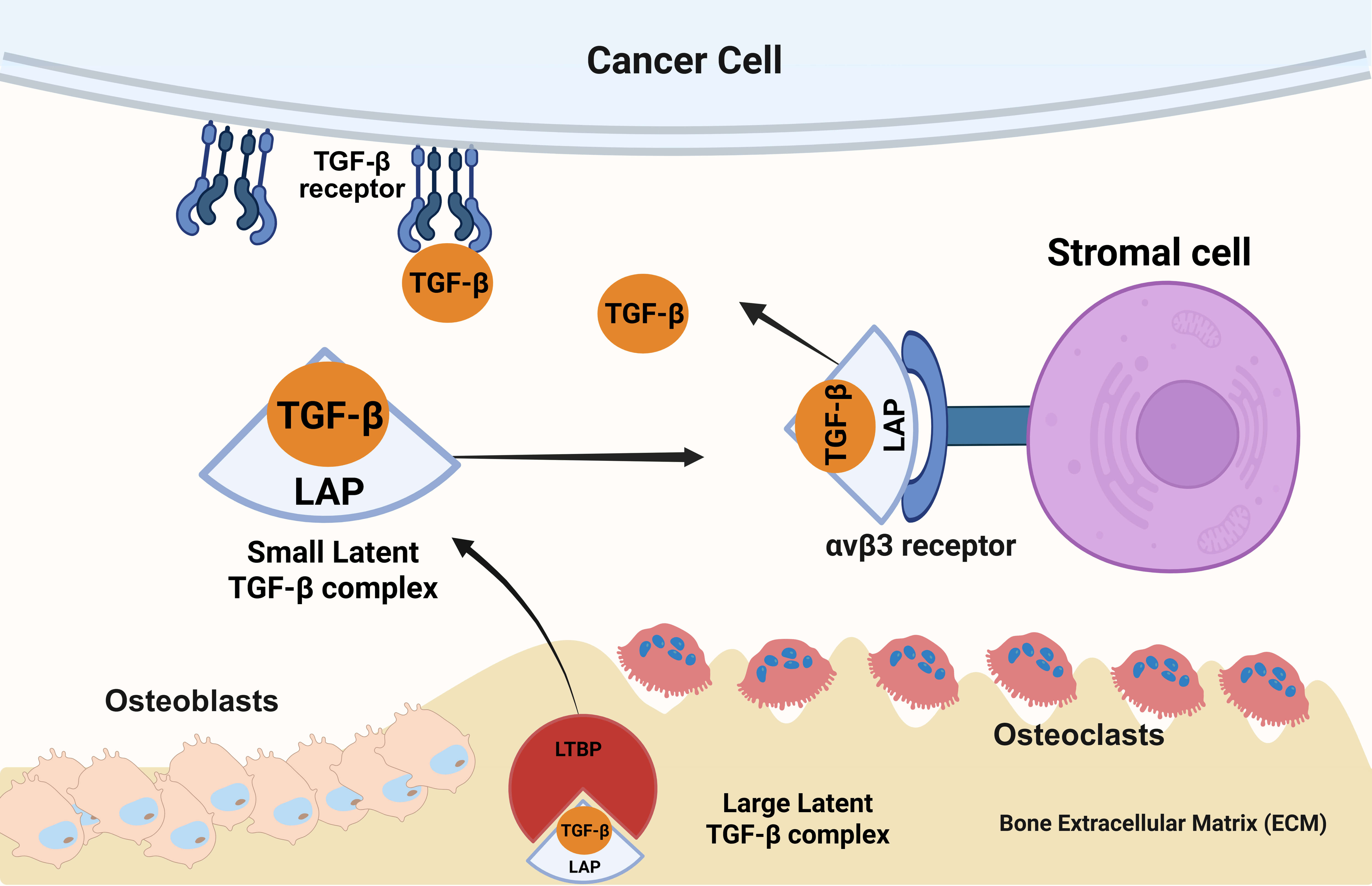 Fig. 3.
Fig. 3.
How inactive TGF‑
The immune landscape in the bone microenvironment is also crucial for tumor colonization and growth. Various immune cells, including TAMs, play a dual role in bone metastases, either promoting or inhibiting tumor growth. TAMs are often polarized towards a pro-tumorigenic M2 phenotype in the presence of cancer cells, which leads to increased inflammation, matrix remodeling, and further recruitment of additional immune cells [110, 130]. This polarization can be enhanced through bidirectional signaling involving cytokines and chemokines produced by both the tumor and stromal cells [130, 131]. Stromal cells, including cancer-associated fibroblasts (CAFs) and mesenchymal stem cells (MSCs), also significantly influence tumor dynamics in the bone microenvironment. MSCs can differentiate into various cell types, including osteoblasts, and release paracrine factors that support cancer cell survival, proliferation, and migration [132, 133]. CAFs contribute to the fibrotic response and create a supportive extracellular matrix, critically altering cell interactions and promoting tumor growth [133, 134]. The interaction between tumor cells and the stromal components can activate various signaling pathways that facilitate proliferation and survival. For instance, the interaction of Jagged1 and Notch signaling in bone cells has been shown to enhance osteolytic metastasis by promoting osteoclast differentiation and activity [135, 136]. Furthermore, factors such as IL-6 are secreted by both tumor and stromal cells, contributing to a pro-inflammatory environment that favors metastasis [48, 110, 137].
The bone microenvironment is a highly orchestrated assembly of specialized cells that provide a supportive niche for tumor colonization and growth. Osteoblasts, osteoclasts, and osteocytes interact dynamically with immune components and stromal cells to create an ecosystem that promotes cancer cell survival and proliferation. These interactions generate a vicious cycle of bone remodeling and metastasis that complicates treatment strategies aimed at managing bone-associated cancers. Thus, understanding the complexities of the bone microenvironment is critical for developing novel therapeutic approaches that target these interactions to hinder tumor growth and improve patient outcomes.
The signaling pathways involved in modulating the TME in bone metastases play critical roles in facilitating the growth and survival of metastatic cancer cells. Several pathways interact with bone-specific cell types and immune components, contributing to the complexities of cancer progression and treatment resistance. Below, we discuss prominent signaling pathways that are implicated in TME modulation specifically related to bone metastases.
TGF-
BMPs are crucial regulators of bone homeostasis and have been implicated in
cancer-induced bone metastasis through their roles in osteogenesis and in
promoting tumor cell interactions with bone cells [140, 142]. BMP signaling
functions are closely linked to TGF-
The Wnt signaling pathway plays a critical role in maintaining bone homeostasis
and influencing tumor biology. In the context of bone metastases, Wnt signaling
induces the differentiation of osteoblasts, enhancing bone formation and
providing a favorable environment for tumor cells [143, 144, 145]. Aberrant
Wnt/
Nuclear factor-kappa B (NF-
The PI3K/Akt signaling pathway is a major regulator of cell proliferation and survival, and it becomes dysregulated in various forms of cancer, including those with bone metastases [149]. In prostate cancer specifically, the pathway has been implicated in the promotion of osteogenesis from mesenchymal stem cells (MSCs), further modifying the TME to favor cancer progression [149, 150]. Akt signaling is vital for osteoclast differentiation and activation, thus contributing to bone resorption and remodeling associated with metastatic lesions [149].
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway is often activated in various cancers and is crucial for mediating responses to inflammatory cytokines in the TME. In the case of bone metastases, such signaling facilitates the transition of macrophages to an M2 phenotype, enhancing the immunosuppressive environment that tumor cells can exploit for growth and survival [122]. The activation of this pathway through cytokine signaling allows cancer cells to evade immune surveillance mechanisms, which is particularly detrimental in the context of bone metastasis [122, 151].
The chemokine signaling pathway, particularly via the CCR2 receptor and its ligand CCL2, plays an important role in the recruitment of immune and stromal cells to the TME. These cells can enhance tumor growth and survival through various mechanisms, including the engulfment of tumor cells and the modulation of inflammation [152]. In the context of bone metastases, chemokine-driven cell recruitment can alter the microenvironment’s composition, creating a more favorable niche for tumor growth.
The tumor microenvironment in bone metastases is modulated by diverse signaling
pathways, including TGF-
The sources of activation for TGF-
TGF-
In the initial stages of tumor development, TGF-
TGF-
Regarding osteoclastogenesis, TGF-
The immunosuppressive effects of TGF-
TGF-
Monoclonal antibodies targeting TGF-
Small-molecule inhibitors, such as ALK5 inhibitors, are also critical in
targeting TGF-
Antisense oligonucleotides and RNA interference, such as Trabedersen and
ISTH0036, represent another innovative approach to inhibit TGF-
TGF-
Targeting TGF-
The role of TGF-
Previous studies demonstrated that TGF-
Efforts to target the TGF-
The targeting of TGF-
TGF-
Several ongoing clinical trials are exploring the targeting of TGF-
TGF-
One promising approach to target the bone microenvironment involves
osteoclast-targeting therapies, such as RANKL inhibitors, with Denosumab being a
notable example. RANKL (Receptor Activator of Nuclear factor Kappa-
In addition to RANKL inhibition, the modulation of osteoblast activity is crucial in bone metastatic contexts. Tumors can educate osteoblasts to adopt traits that benefit tumor growth, such as increased bone formation leading to osteoblastic lesions [193, 194]. The identification of receptors and signaling pathways that facilitate this crosstalk presents another avenue for therapeutic intervention. For instance, the role of parathyroid hormone-related peptide (PTHrP) is significant as it upregulates RANKL in osteoblasts, promoting a feed-forward loop that exacerbates osteoclast activity and further stimulates tumorigenesis in the bone [91, 193]. Targeting PTHrP could reduce osteoclastogenesis and, consequently, the destructive effects of cancer metastases in bone. Stromal cell modulation offers an innovative therapeutic strategy by altering the supportive network that facilitates metastatic growth. The bone stroma itself can influence cancer cell behavior through the release of chemokines and growth factors, enhancing tumor cell migration and proliferation within this niche [195, 196]. For example, the interaction of prostate cancer cells with bone stromal cells has been shown to activate osteoblasts and promote osteoblastic responses, thus changing the bone microenvironment to one that is more conducive to metastasis [101, 197]. Interventions that disrupt these stromal-tumor interactions may lead to reduced metastatic progression.
Therapies targeting the immune components within the bone microenvironment could be advantageous. Investigating the immune landscape, including immune checkpoint inhibitors or agents that modulate myeloid-derived suppressor cells and regulatory T cells, reveals potential strategies to enhance anti-tumor immunity while countering the immune-suppressive effects that bone metastases can elicit [198]. This not only holds promise for reducing tumor growth but also for restoring normal bone remodeling processes that are often disrupted by metastatic lesions.
One major challenge with using Immune Checkpoint Inhibitors (ICIs) in bone metastases stems from the immunosuppressive microenvironment inherent to bone, characterized by regulatory T cells and other factors that can inhibit effective immune responses [199, 200]. Studies indicate that bone metastases often show diminished immune activity compared to primary tumors, complicating treatment outcomes [201]. Nonetheless, emerging strategies suggest promise for ICIs even in this challenging context.
Recent clinical case reports have documented complete remission in patients with renal cell carcinoma and melanoma who showed strong responses to ICIs despite having advanced bone metastasis. These results emphasize the potential of ICIs to produce meaningful responses, indicating that even patients with bone metastases may benefit from immunotherapies under appropriate clinical circumstances. Moreover, the presence of immune checkpoint expression (such as PD-1 and PD-L1) within bone metastatic lesions suggests that targeting these pathways could enhance response rates [99, 201].
Combination therapies involving ICIs are showing promise in enhancing therapeutic effectiveness. For example, combining radiation therapy with ICIs has been explored for its potential to induce an “abscopal effect”, where local treatment triggers systemic anti-tumor responses, which could be beneficial for bone metastases [202, 203]. This is particularly relevant as radiation can make the tumor microenvironment more immunogenic, thereby enhancing the effectiveness of subsequent ICI therapy [204]. Combining ICIs with traditional systemic agents, such as tyrosine kinase inhibitors or other immunomodulatory treatments, provides another approach to improve outcomes. These strategies can target both the cancer cells and the supporting stroma in the bone microenvironment [205, 206]. Notably, dual-combination therapies (e.g., anti-PD-1 and anti-CTLA-4) have been shown to improve survival outcomes in advanced melanoma, demonstrating a concept that could be applied to treatments involving bone metastases [207]. While immune checkpoint inhibitors present challenges in bone metastases due to the complex immunosuppressive environment, ongoing research and clinical experience indicate that ICIs can offer therapeutic options for certain patients, particularly when combined with other treatments.
T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) has been identified as a checkpoint that plays a significant role in T-cell exhaustion during chronic infections and cancer [208]. Its expression on T cells can inhibit effective anti-tumor responses, which is particularly relevant in the metastatic niche where the immune system may be suppressed. In the context of bone metastases, where the immune microenvironment can be influenced by factors such as osteoclast activity, targeting TIM-3 may present a strategic opportunity to reinvigorate T-cell responses against metastatic tumor cells [199]. Studies have shown that TIM-3 expression can correlate with tumor progression and poor prognosis in various cancers, linking its elevated levels to reduced overall survival in patients with bone metastases [209].
Lymphocyte-activation gene-3 (LAG-3), another critical immune checkpoint, functions similarly as an inhibitory receptor on T cells and is thought to be co-expressed with PD-1 in the tumor microenvironment [210]. LAG-3’s importance has been illuminated in contexts such as renal cell carcinoma, where increased expression has been associated with metastatic disease [211]. Research indicates that LAG-3 has a unique expression pattern in metastatic tissues compared to primary tumors, influencing patient outcomes and therapeutic responses [210, 212]. The combined expression of LAG-3 with other immune checkpoints like PD-1 has been linked to immune evasion mechanisms, particularly in metastasis, suggesting that simultaneous targeting of these markers may improve immunotherapy efficacy [213]. Recent investigations have also highlighted the significance of the bone microenvironment in modulating immune checkpoint regulator expression. Factors in the bone niche, such as osteoclast and osteoblast signaling, influence the local immune landscape, making the study of specific immune checkpoints like TIM-3 and LAG-3 particularly relevant to metastatic processes [214, 215]. Research into the Siglec-15/sialic acid glyco-immune checkpoint axis has further exposed pathways by which bone metastases can alter immune responses via inhibition of T-cell activity, underscoring the necessity of a comprehensive understanding of these pathways to develop effective targeted therapies [209, 216].
Future studies are warranted to elucidate optimal combinations and timing of these therapies and to improve understanding of the underlying mechanisms that enable immune response in the bone niche.
The treatment of bone metastases benefits significantly from various microenvironment-targeting strategies that extend beyond traditional approaches, specifically by targeting growth factors like vascular endothelial growth factor (VEGF) and fibroblast growth factors (FGFs), along with stromal reprogramming methods. These strategies aim to alter the tumor-promoting signals within the bone microenvironment, which can facilitate cancerous cell growth and spread.
VEGF plays a crucial role in angiogenesis, which is essential for tumor growth and metastasis. In the context of bone metastases, high levels of VEGF can promote osteolysis and metastasis by enhancing vascular permeability and increasing the delivery of nutrients and growth factors to tumors [217, 218]. Multiple studies have examined the efficacy of anti-VEGF therapies like Bevacizumab, noting that while they may reduce angiogenesis and tumor growth, resistance typically arises due to compensatory mechanisms, such as the upregulation of alternative angiogenic pathways, including FGF signaling [219, 220, 221]. Therefore, targeting both VEGF and FGF pathways appears to be a promising strategy. Combinatorial approaches that inhibit both pathways have been shown to improve treatment outcomes by preventing adaptive resistance mechanisms that allow tumors to evade single-agent anti-angiogenic therapies [220, 221].
FGFs, particularly FGF-2, significantly contribute to tumor progression and metastasis by facilitating angiogenesis and influencing the proliferation and differentiation of stromal cells. Inhibition of FGF signaling has demonstrated potential in reducing metastatic behavior in various cancers, including those with bone involvement [222, 223]. For example, FGF-2 participates in the communication between tumor cells and surrounding stroma, promoting an environment conducive to cancer propagation [224, 225]. This has prompted research into multi-targeted therapies that address both VEGF and FGF pathways to mitigate the impact of bone metastases [224, 226].
Reprogramming the tumor stroma offers another innovative strategy to mitigate metastasis and improve treatment efficacy. The tumor microenvironment in bone metastases is not just a passive backdrop; rather, it actively contributes to tumor progression through various cell types, including fibroblasts and macrophages that are often co-opted by tumors to promote metastasis [227]. For instance, inhibiting the activation of fibroblasts can disrupt their production of pro-tumorigenic factors and extracellular matrix components, thereby hindering metastasis-promoting signaling pathways [224, 228].
Macrophages, particularly tumor-associated macrophages (TAMs), play a dual role in cancer; while they can exert anti-tumoral effects, they often become polarized towards a pro-tumorigenic phenotype, facilitating metastasis and tissue remodeling [227]. Strategies that repolarize these macrophages to a more anti-tumoral phenotype, potentially via immune checkpoint inhibitors in conjunction with stroma-targeting therapies, represent a promising direction in combating bone metastases [205, 220, 229].
Modifying the extracellular matrix (ECM) can alter the interactions between tumor cells and their microenvironment. Changes in ECM composition and stiffness can directly influence cancer cell behavior, including proliferation, invasion, and migration. By targeting the signaling pathways that regulate these properties within the ECM, researchers can potentially destabilize the supportive niche required for tumor growth and dissemination [219, 230, 231].
Targeting Multiple Pathways: Cancer cells in the bone microenvironment engage in crosstalk with different cell types, including osteoclasts, osteoblasts, fibroblasts, and immune cells, creating a dynamic and supportive niche for metastasis [232, 233, 234]. Effective treatment necessitates strategies that simultaneously disrupt these interactions. For example, targeting the RANK-RANKL pathway involved in osteoclastogenesis alongside inhibiting growth factors like VEGF or FGF can counteract the mechanisms that promote bone degradation and cancer cell proliferation [109, 233]. Recent research suggests that dual inhibition can effectively break the crosstalk between cancer cells and bone cells, which is pivotal for tumor progression [234].
Modulation of the Tumor Microenvironment: The bone TME is characterized by immunosuppressive elements, such as macrophages and myeloid-derived suppressor cells (MDSCs), which foster tumor growth and hinder effective immune responses [91, 198, 235]. Combination therapies that entail immune checkpoint inhibitors alongside TME reprogramming, aimed at altering the profile of infiltrating immune cells, can potentially create a more favorable environment for therapeutic response [111, 236]. For instance, the introduction of therapies that convert tumor-associated macrophages (TAMs) from a pro-tumor phenotype to an anti-tumor phenotype may improve the overall immune response against bone metastases [111, 236].
Overcoming Resistance Mechanisms: Resistance to therapy is a significant hurdle in treating bone metastases. Cancer cells can adapt to targeted therapies by activating alternative signaling pathways [222, 224, 237]. By employing combination therapies that simultaneously inhibit multiple growth factors or signaling pathways, it may be possible to prevent or delay the onset of resistance. For example, studies have shown that targeting both FGFs and VEGF can reduce compensatory angiogenesis that aids in tumor survival and resistance [222, 223, 224].
Personalized medicine anchored in TME profiling is paving the way for more tailored therapeutic strategies against bone metastases. Assessing the unique characteristics of the TME can inform treatment decisions and improve outcomes. Personalized treatment approaches can utilize molecular profiles from patient tumor samples to identify active signaling pathways and immune cell infiltrates within the TME [238, 239]. For example, profiling for the expression of RANKL, PTHrP, or various cytokines can provide insights into the specific requirements of a patient’s cancer, allowing for targeted interventions that are best suited to disrupt critical interactions in the bone environment [109, 190]. With advancements in screening technologies, identifying biomarkers that predict response to specific therapies is becoming increasingly feasible [120, 239]. Such biomarkers may include the expression levels of integrins or immune checkpoint proteins within the TME, which can indicate how responsive a patient’s cancer may be to immunotherapies or targeted therapies [111, 240]. Adaptive Treatment Strategies: The TME can change dynamically over the course of treatment, especially with the introduction of new therapies. Conducting regular assessments of the TME can allow oncologists to adjust treatment protocols based on the current status of the tumor and the surrounding microenvironment, thus optimizing therapeutic strategies [241, 242].
We believe that combination therapies targeting multiple components of the TME, alongside personalized medicine approaches grounded in TME profiling, are necessary strategies for effectively managing bone metastases (Table 1). These approaches not only hold the potential to improve therapeutic efficacy but also to address the complexity and adaptability of the tumor microenvironment.
| # | Therapeutic class/example agents | Principal target/mechanism | Development status | Key limitations/considerations |
| 1 | TGF‑ |
Sequester active TGF‑ |
Phase I–II NCT01401062 | Dose‑limiting skin & cardiac toxicities; systemic wound‑healing concerns |
| 2 | ALK5 (TGFBR1) kinase inhibitors (vactosertib, galunisertib) | Block receptor phosphorylation and downstream SMAD2/3 | Phase I–II NCT02160106, NCT02423343 | Narrow therapeutic window; rebound via parallel pathways |
| 3 | Antisense oligos/RNAi (TGF‑ |
Deplete TGF‑ |
Early clinical AP12009-P001, NCT03436563 | Delivery to bone lesions; off‑target immunomodulation |
| 4 | Tumor-selective ligand traps/bone‑targeted nanoparticles | Localized capture of TGF‑ |
Pre‑clinical NCT03834662 | Formulation, targeting specificity, scalability |
| 5 | RANKL inhibitor (denosumab) | Suppresses osteoclast differentiation & activity | Approved NCT01824342 | Does not inhibit tumor growth directly; hypocalcemia risk |
| 6 | Bisphosphonates (zoledronic acid, low‑dose regimens) | Induce osteoclast apoptosis; reduce SREs | Approved NCT00320710 | Renal toxicity, ONJ; no effect on visceral disease |
| 7 | Radiopharmaceuticals (radium‑223, strontium‑89) | Approved/Phase II NCT00699751 | Myelosuppression; limited to osteoblastic lesions | |
| 8 | Immune‑checkpoint blockade (anti‑PD‑1 |
Re‑activate exhausted T cells in bone TME | Approved (solid tumors) NCT02834013 | Immunosuppressive niche dampens responses; bone‑flare phenomena |
| 9 | Next‑gen checkpoints (TIM‑3, LAG‑3 mAbs) | Overcome alternative T‑cell exhaustion pathways | Phase I NCT05367401 | Biomarker selection; combinatorial toxicity |
TGF‑
Conceptualization, KSM, and FHB; writing—original draft preparation, KSM, and FHB; writing—review and editing, KSM. Visualization, KSM, and FHB supervision, KSM; project administration, KSM. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.