

 , Wenxing Li 1,†, Jiaying Diao 2,†, Yang Jiao 2
, Wenxing Li 1,†, Jiaying Diao 2,†, Yang Jiao 21 Department of Cardiology, Guizhou Aerospace Hospital, 563000 Zunyi, Guizhou, China
2 Department of Cardiology, Zunyi First People’s Hospital, 563000 Zunyi, Guizhou, China
†These authors contributed equally.
Abstract
Cardiovascular and cerebrovascular diseases are among the leading causes of death worldwide. Development of these diseases occurs following pathological structural remodeling and functional changes in the vascular wall. Emerging evidence suggests that histone acetyltransferases (HATs) play a role in the pathological processes of the arterial wall. However, there is currently a lack of comprehensive reviews examining the role of HATs in vascular diseases. The aim of this research is therefore to systematically describe the pathological effects of various risk factors on different layers of cells in the arterial vascular wall. The risk factors include abnormal activation of the renin-angiotensin system, hyperglycemia, high-sodium diets, and intermittent hypoxia. The effects regulated by HATs involve the nuclear factor kappa-B (NF-κB)-NOD-like receptor family pyrin domain containing 3 (NLRP3) and AMP-activated protein kinase (AMPK) pathways, and the mitogen-activated protein kinase (MAPK) and vascular endothelial growth factor receptor 2 (VEGFR2) signaling pathways. We also explore the dual role of HATs in vascular protection and injury. Additionally, this study focuses on the prospects of future therapeutic strategies targeting HATs, including innovative approaches such as HAT inhibitors, epigenetic degraders, non-coding RNA interventions, and epigenetic editing technologies. The aim of this review is to provide a basis for the development of selective subtype HAT inhibitors.
Keywords
- histone acetyltransferases
- vascular endothelial cells
- smooth muscle cells
- immune cells
- renin-angiotensin system
- vascular remodeling
- epigenetic therapy
Cardiovascular and cerebrovascular diseases are among the leading causes of
death worldwide. The prevalence of coronary heart disease among adults aged
The arterial vascular wall is primarily composed of three layers: the endothelium, vascular smooth muscle cells (VSMCs), and adventitia. Under the combined effects of long-term environmental factors such as hypertension, hyperglycemia, and genetic factors, each layer of the vascular wall may undergo pathological changes. For example, in the presence of hypertension and hyperglycemia, endothelial cells (ECs) secrete less amounts of important vasodilators, while increasing the expression of adhesion molecules that promote immune cell recruitment and lead to dysfunction of coagulation. VSMCs may shift from a contractile phenotype to a synthetic phenotype, accompanied by cell proliferation and migration, inflammation, and oxidative stress. The adventitia is mainly characterized by infiltration of inflammatory cells and remodeling of the extracellular matrix. Chronic inflammation of the vascular wall is recognized as a fundamental reason for the development of various cardiovascular diseases. Hence, there is an urgent need to find ways to reduce vascular wall inflammation.
Histones are core components of chromatin. Conventional histones consist of two molecules each of H2A, H2B, H3, and H4, forming an octamer that participates in the regulation of DNA expression, folding, and protection. Acetylation is one of the most important modifications of histones. Following the acetylation of histone lysine residues, the chromatin structure becomes more relaxed, facilitating the binding of transcription factors, RNA polymerase, and other transcription-related proteins to DNA, and thereby promoting gene transcription [3]. Histone acetylation is primarily regulated by a balance between histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs can be classified into families such as the MOZ-Ybf2/Sas3-Sas2-TIP60 family (MYST), Gcn5-related N-acetyltransferase family (GNAT), p300/CBP histone acetyltransferase family (p300/CBP), nuclear receptor coactivator family, and general transcription factor family. These enzymes not only acetylate histone lysine residues, but also participate in the recruitment of transcription factors and RNA polymerase [4, 5]. At the same time, HDACs can be divided into four categories, one of which indirectly regulates gene expression by recruiting HATs such as p300 (histone acetyltransferase p300) [6].
Current research indicates that HATs promote pathological processes in various
diseases by having pro-inflammatory roles, regulating excessive gene
transcription, and affecting metabolism. HATs can directly promote the expression
of genes such as nuclear factor kappa-B (NF-
The levels of histone acetylation can influence both systemic and cellular metabolic processes, as well as impact the prognosis of various diseases. For example, several studies have shown that significant increases in histone acetylation levels play a key role in the development and progression of cardiovascular and cerebrovascular diseases. Abnormalities have been observed in the levels and activity of HATs in disease and these may directly drive the pathological processes [11, 12]. Although a large number of studies have explored the role of HATs in cardiovascular and cerebrovascular diseases, the related reviews have not been updated in a timely manner. In particular, there have been few systematic summaries of newly discovered mechanisms in recent years. The goal of this paper is therefore to systematically analyze the histone acetylation characteristics of VSMCs, ECs and immune cells in the arterial walls in cardiovascular and cerebrovascular diseases. This will help to elucidate the disease progression mechanisms driven by abnormal HAT activity and expression, as well as the associated molecular pathways. We also discuss the bidirectional regulatory role of HATs in vascular protection and injury, and analyze the potential clinical value of HATs as therapeutic targets.
Abnormal activity and levels of HATs have been linked to damaging effects in several cardiovascular diseases [7, 13]. In arterial diseases such as hypertension, atherosclerosis (AS) and vasculitis, long-term inflammation and excessive release of reactive oxygen species (ROS) are considered to be the most important steps in disease progression. Recent studies indicate that HATs play a critical regulatory role in these damaging processes.
NF-
Pathological conditions such as hyperglycemia, hyperlipidemia, and high salt can
directly damage ECs and smooth muscle cells. This causes the release of ROS and
inflammatory factors, thereby activating systemic and local vascular immune cells
and exacerbating tissue damage. During this process, ROS and NF-
ECs form the first barrier of the blood vessel wall. They also regulate
substance exchange, secrete signaling molecules, and maintain the balance between
coagulation and anticoagulation. Early dysfunction of ECs is often the starting
point for the development of various vascular-related diseases. Long-term
exposure to high levels of glucose, lipids, angiotensin, or other harmful
substances can significantly increase the production of inflammatory factors and
ROS within ECs, which can ultimately lead to abnormal EC proliferation, plaque
formation, and even induce the proliferation of VSMCs [19]. It is worth noting
that polyphenolic compounds, such as green tea extract, and substances like
metformin can effectively alleviate endothelial inflammation. Current evidence
suggests that abnormal HATs promote EC injury through regulatory mechanisms
involving NF-
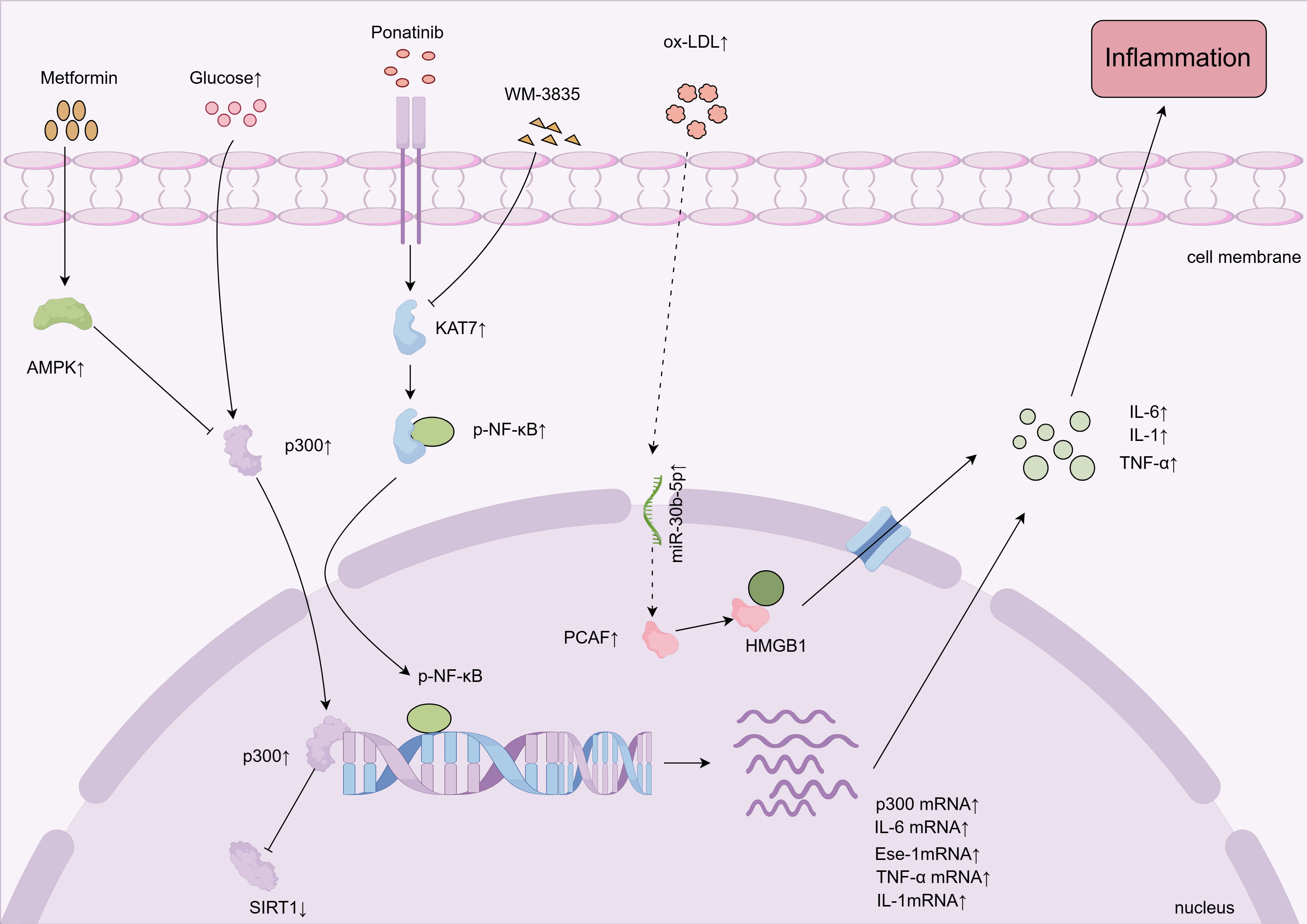
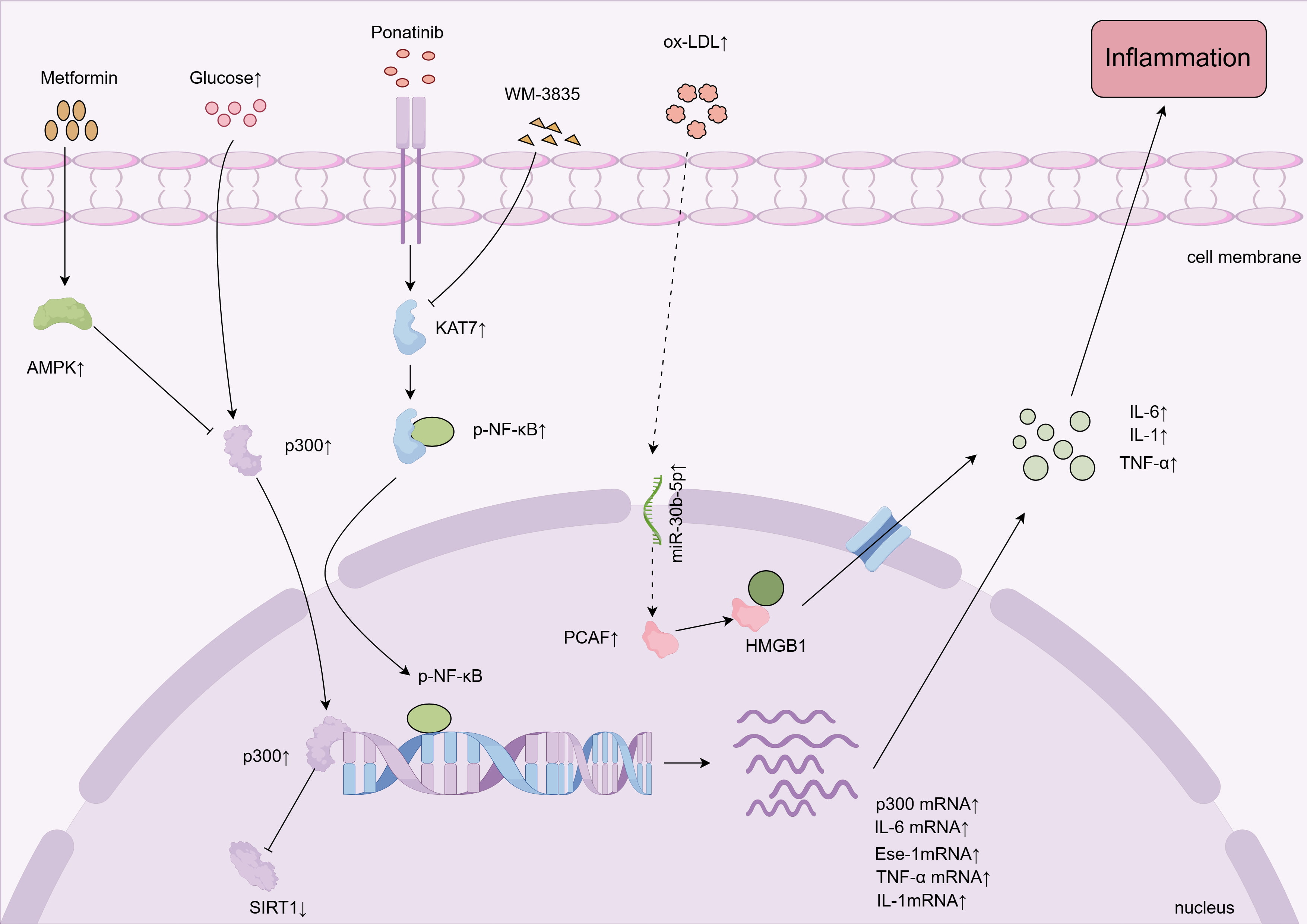 Fig. 1.
Fig. 1.
HATs promote vascular endothelial cell inflammation. High
glucose, ponatinib, and ox-LDL promote vascular endothelial inflammation by
affecting HATs. HATs, histone acetyltransferases; AMPK, AMP-activated protein
kinase; HMGB1, high mobility group box 1; ox-LDL, oxidized low-density
lipoprotein; SIRT1, sirtuin 1; IL-6, interleukin-6; IL-1, interleukin-1;
TNF-
Pathogenic factors such as high glucose, AS and smoking can induce EC
inflammation and senescence by upregulating HATs. Under high glucose stimulation,
the permeability of ECs is significantly increased, accompanied by upregulation
of p300 expression levels [20]. Elevated p300 expression further promotes the
upregulation of epithelial-specific transcription factor 1 in ECs, which then
induces the expression of inflammatory factors such as TNF-
Factors such as aging, AS, palmitic acid, smoking, and angiotensin II (Ang II) can significantly increase the expression levels of p300 and PCAF in vascular ECs, along with upregulating senescence markers such as p53, p21, and p16 [22, 23, 24]. Downregulation of PCAF expression has also been shown to promote the activation of nuclear factor erythroid-2-related factor 2 (Nrf2) in ECs and increase the expression of its downstream anti-aging factors. This ultimately reduces ROS levels in ECs and delays vascular aging [22]. Obesity can also accelerate vascular aging. Research has shown that vascular tissue isolated from the visceral fat arteries of obese individuals exhibits significant ROS-driven endothelial dysfunction, with the expression of acetyltransferase steroid receptor coactivator-1 (SRC-1) being 5.8-fold higher than in normal-weight controls [25]. This finding suggests that SRC-1 may play a key role in obesity-induced endothelial dysfunction, although its specific mechanism requires further investigation.
HATs promote the pathological effects induced by VEGFA and the Delta-like
ligand-Notch signaling pathway. The expression of VEGFR2 in human umbilical vein
ECs is significantly enhanced under stimulation by high concentrations of
exogenous VEGFA. When p300 is inhibited, the level of histone H3 lysine 27
acetylation (H3K27Ac) modification at the VEGFR2 promoter region decreases,
leading to reduced VEGFR2 expression and a corresponding decrease in angiogenesis
[26]. In glioblastoma cells, HAT1 can acetylate hypoxia-inducible factor
2
HATs are also involved in regulating the Delta-like ligand-Notch signaling
pathway. In VEGFA-treated ECs, the HAT p300 specifically binds to the enhancer
region of Delta-like ligand 4 [28]. Furthermore, in vascular endothelial
inflammation induced by IL-1
Non-coding RNAs (ncRNAs) regulate gene expression at the post-transcriptional level. They also play a critical role in cardiovascular diseases by influencing epigenetic regulators, particularly the activity of HATs. HATs such as p300 and CBP are core enzymes that regulate chromatin accessibility and heart-specific gene expression, and their fine-tuning is closely related to cardiac structural remodeling. Previous studies have found that miRNA-199a is significantly upregulated following myocardial infarction. miRNA-199a targets and inhibits Sirt1, thereby releasing the inhibitory effect of Sirt1 on p300, enhancing p300 acetylation, and activating the Yin-yang 1/soluble Suppression of Tumorigenicity 2 (Yy1/sST2) signaling pathway. This subsequently promotes myocardial hypertrophy and fibrosis. Notably, antimiR-199a can reverse this pathological process by antagonizing the action of miRNA-199a [31]. Animal experiments also showed that miR-330-3p targets the 3′-UTR of CBP in porcine cardiac valve interstitial cells, decreasing its expression. This weakens the acetylation modifications mediated by CBP, leading to upregulation of pro-calcification factors such as bone morphogenetic protein 2 (BMP2) and runt-related transcription factor 2 (RUNX2), which then exacerbate valve fibrosis and calcification [32]. These findings suggest that ncRNAs regulate the expression and activity of HATs, thereby altering histone acetylation levels and acting as a bridge in pathological processes such as myocardial remodeling, fibrosis and valve calcification, and driving the progression of cardiovascular diseases.
ncRNAs also promote inflammatory responses in vascular ECs by regulating the activity of HATs. Studies have shown that ox-LDL induces the upregulation of miR-30b-5p, which subsequently activates the UBE2D2/PCAF pathway. Activated PCAF acetylates high mobility group box 1 protein (HMGB1), leading to the dissociation of acetylated HMGB1 from SIRT1. This dissociation facilitates the migration of HMGB1 from the nucleus to the cytoplasm and its release into the extracellular space, which then induces expression of adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), enhancing endothelial inflammation [33].
Epigallocatechin gallate (EGCG) is a polyphenolic compound extracted from green
tea and known for its significant antioxidant and anti-inflammatory properties.
In a polychlorinated biphenyl-induced vascular endothelial injury model, EGCG was
found to significantly reduce the binding of NF-
The inflammation, abnormal proliferation, and migration of VSMCs are known to exacerbate the progression of diseases such as hypertension, AS, and vascular restenosis. Recent studies suggest that excessive modification through histone acetylation can promote the pathological changes observed in VSMCs. Various pathological factors, including intermittent hypoxia, Ang II hyperglycemic environments, and vascular physical injury can exacerbate pathological changes in VSMCs by enhancing the activity of HATs, leading to vascular dysfunction (Fig. 2).
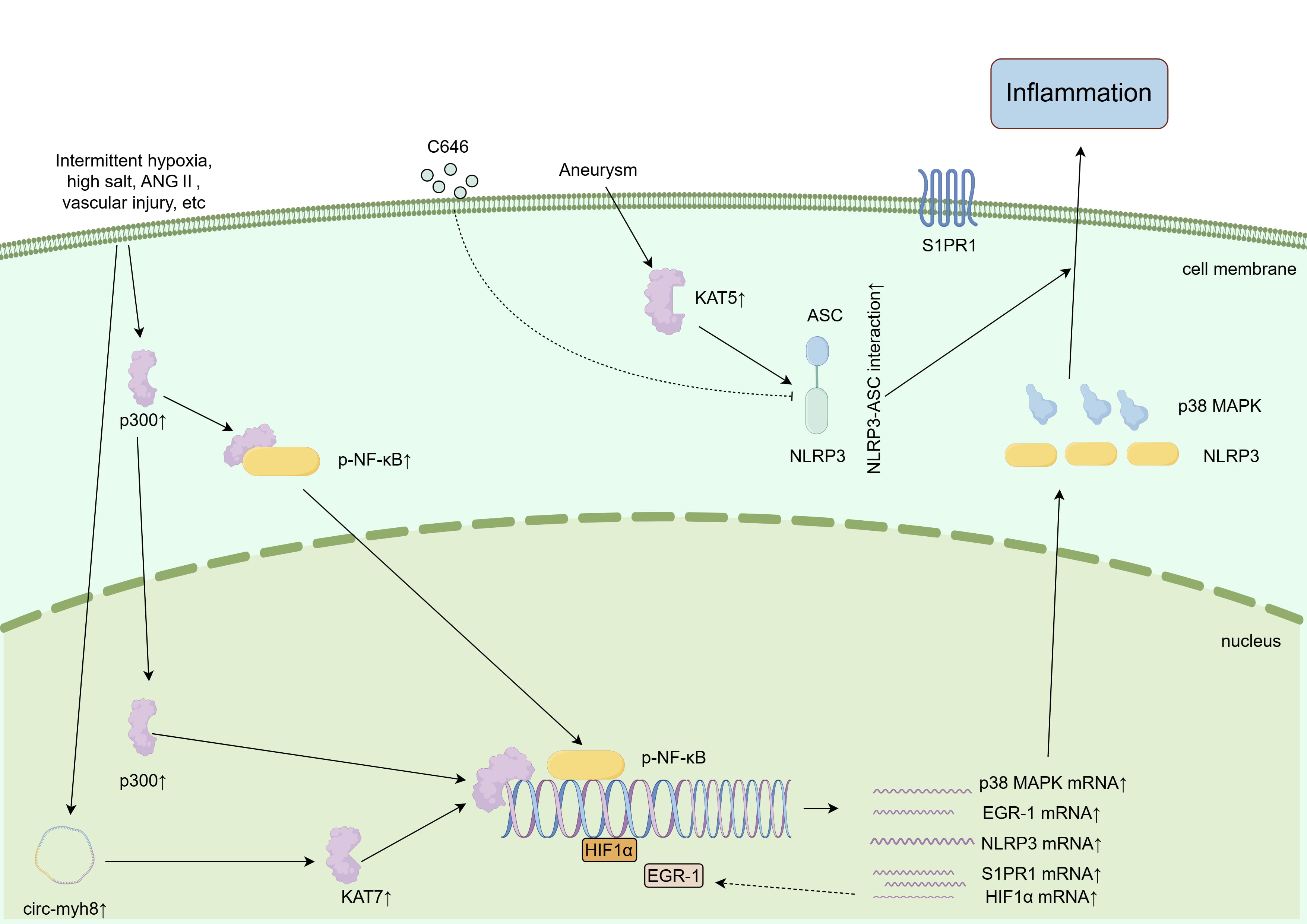
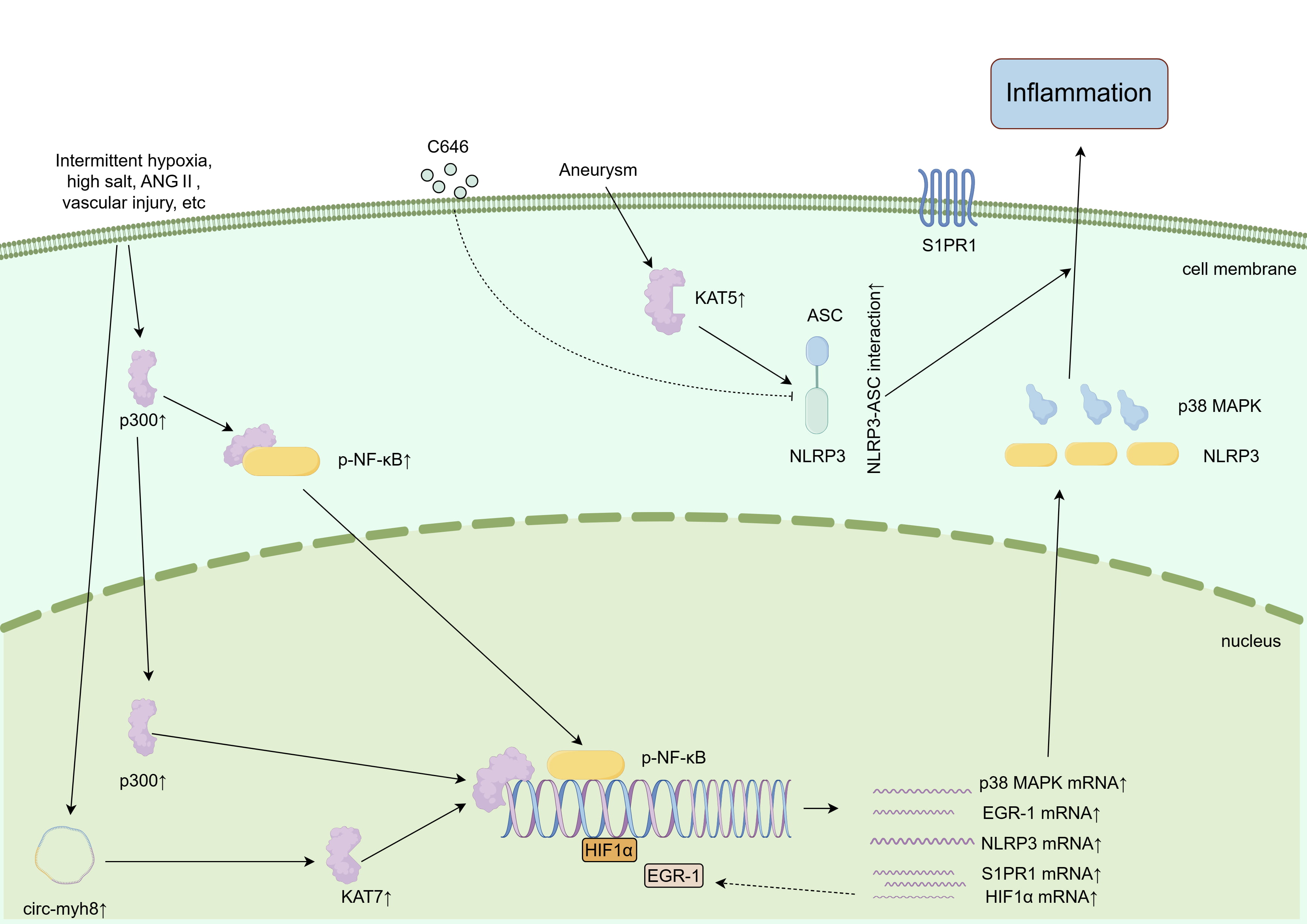 Fig. 2.
Fig. 2.
HATs promote vascular smooth muscle cell inflammation. Multiple pathological factors promote vascular smooth muscle inflammation and injury through HATs. ASC, apoptosis-associated speck-like protein containing a CARD; NLRP3, NOD-like receptor family pyrin domain containing 3; C646, inhibitor of histone acetyltransferase p300. This figure was created using Figdraw (Version 2.0, http://www.figdraw.com), Home for Researchers, China.
Hypoxia-inducible factor-1 (HIF-1) and NLRP3 can promote vascular smooth muscle
injury under pathological conditions. Intermittent hypoxia refers to repeated
cycles of hypoxia and reoxygenation. In experimental animals and patients, this
can lead to symptoms such as activation of the sympathetic nervous system and
increased blood pressure. Moreover, the expression and activity of p300, HIF-1,
and ROS in the adrenal medulla of rats increase significantly under conditions of
intermittent hypoxia. Inhibition of p300 can effectively lower blood pressure and
reduce the secretion of norepinephrine in rats [37]. In another experiment, the
expression of circ-myh8 was observed to increase significantly in the pulmonary
arteries of hypoxic mice. Circ-myh8 recruits KAT7 to the HIF1
Ang II is considered to be one of the key hormones responsible for the
development of cardiovascular diseases, making it a major target for treatment.
Renin is essential for the synthesis of Ang II. In As4.1 cells, cAMP activates
protein kinase A (PKA), which promotes the recruitment of p300 and H3K27
acetylation, thereby increasing transcription of the renin gene [10]. In another
study of obesity-associated hypertension, the increase in p300 upregulated the
expression of angiotensin-converting enzyme 1 (ACE1) in renal tubular epithelial
cells [40]. HATs significantly enhance Ang II-induced vascular inflammation and
injury. When VSMCs are damaged, histone acetylation levels in the nucleus
increase significantly [41, 42]. Following intervention with Ang II, p300
increases the acetylation of histone H3 lysines (K9/14) in the IL-6 promoter
region, thereby upregulating IL-6 expression. Inhibition of p300/CBP function
through the use of mutant variants suppressed Ang II-induced IL-6 expression [43, 44]. Additionally, under conditions of IL-1
Ang II can regulate histone acetylation levels in the promoter regions of
several key proteins in VSMCs through both direct and indirect mechanisms, thus
affecting cell functions such as inflammation, proliferation, and migration.
These key proteins include the sphingosine-1-phosphate receptor 1 (S1PR1), EGR-1,
and sodium channels [42, 45, 47, 48]. Ang II upregulates the expression of S1PR1,
for example, which then increases the proliferation of VSMCs. This process is
accompanied by increased histone H3 acetylation levels in the S1PR1 promoter
region [48]. Additionally, Ang II activates its own type 1 receptor, which then
activates an Akt-dependent signaling pathway to specifically phosphorylate Ser570
of peroxisome proliferator-activated receptor
Abnormal microenvironmental conditions, such as high glucose and high salt, can promote pathological changes in VSMCs by regulating PCAF activity. These environmental factors have been shown to induce increased histone acetylation levels in the nucleus, particularly the acetylation of H3K9 [50, 51]. In a high-glucose environment, rat VSMCs and human aortic smooth muscle cells both exhibit increased PCAF activity and expression levels. PCAF then promotes excessive production of ROS within VSMCs, leading to upregulation of synthetic markers (such as collagen type I, osteopontin) and NLRP3 expression, and promoting the abnormal proliferation and migration of VSMCs. Specific PCAF inhibitors can effectively reduce histone acetylation levels and suppress ROS production, thereby mitigating oxidative stress damage to VSMCs [52, 53]. In a high-sodium environment, VSMCs also exhibit significant phenotypic changes, such as decreased expression of contractile markers, and increased expression of synthetic markers and PCAF. Similarly, the inhibition of PCAF activity can promote the transition of VSMCs to a contractile phenotype with reduced NLRP3 expression, thus exerting protective effects [51].
Vascular injury, such as stent interventions or balloon damage, can lead to the proliferation, migration, and inflammation of VSMCs, ultimately resulting in stenosis and thrombosis within the vessel lumen. In a rat carotid artery model with repeated balloon injury, researchers observed a significant increase in histone acetylation levels in the transcriptional regions of inflammation-related genes such as Cebpd, suggesting that HATs may promote the expression of inflammatory genes [41]. In another study, researchers injured the left common carotid artery in Sprague-Dawley rats (SD rats). By inhibiting PCAF expression, a significant reduction in the ratio of intimal to medial area was observed, confirming that PCAF promotes pathological remodeling following vascular injury [54].
Further investigation of signaling pathways revealed the levels of
phosphorylated p38 mitogen-activated protein kinase (p-p38 MAPK) and
phosphorylated serine/threonine protein kinase AKT (p-AKT) were elevated in
lipopolysaccharide (LPS)-induced VSMCs. Upon inhibition of PCAF, the expression
of both p-p38 MAPK and p-AKT decreased significantly, while the proliferation of
VSMCs was inhibited. This suggests that PCAF may affect the proliferation of
VMSCs by regulating the p38 MAPK and AKT signaling pathways [55]. Additionally,
the inhibition of PCAF in PCAF-induced smooth muscle inflammation reduced the
secretion of monocyte chemoattractant protein-1 (CCL2) in the vascular intima,
leading to a 54.8% decrease in the number of macrophages in the intima. In vitro experiments further confirmed that downregulation of PCAF resulted in the
inhibition of LPS-induced VSMC proliferation and migration, as well as nuclear
translocation of NF-
Using a mouse femoral artery injury model, the proliferation and migration abilities of VSMCs were found to be regulated by an interaction between staphylococcal nuclease domain-containing protein 1 (SND1) and serum response factor (SRF). Specifically, SND1, as a co-activator of SRF, recruits PCAF to the promoter region of proliferation and migration-related genes rich in CC(A/T)₆GG (CArG) motifs. By catalyzing histone acetylation modifications, PCAF alters the chromatin structure, thus increasing the binding of SRF to the CArG motif and subsequently activating the transcription of downstream genes. This process ultimately promotes VSMC proliferation and migration, and drives the repair and remodeling process after vascular injury [58].
Non-coding RNAs can exacerbate vascular smooth muscle damage by regulating HATs.
The circular RNA circ-myh8 has been shown to bind to and recruit the HAT KAT7,
which targets acetylation of histone 4 lysine 5 (H4K5) in the promoter region of
HIF1
Histone acetylation and methylation have reciprocal regulatory effects. For example, PCAF acetylates ubiquitin-like with PHD and ring finger domains 1 (UHRF1), a key regulator of DNA methylation maintenance, thereby interfering with its binding to hemi-methylated DNA and disrupting the maintenance of genomic methylation [60]. Additionally, CBP/p300 upregulates H3K27ac levels, which has the effects of promoting ten-eleven translocation 2 (TET2) demethylation, inhibiting DNA Methyltransferases (DNMTs) from binding to the promoter, reducing the methylation of genes like RUNX2, and activating transcription to accelerate vascular calcification [61]. Similarly, methyltransferases can also methylate certain sites on HATs [62]. In diabetic cardiac microvascular disease, the demethylase Alkylation Repair Homolog (AlkB) homolog 5 can reduce the level of m6A methylation in KAT2A mRNA, leading to upregulation of KAT2A. By increasing acetylation levels of the transferrin receptor (Trfc) and heme oxygenase 1 (Hmox1) promoters, KAT2A promotes ferroptosis, thereby exacerbating microvascular disease [63]. Therefore, we hypothesize that the interaction between histone acetylation and DNA methylation may promote arterial damage.
Various immune cells, including T cells, B cells, dendritic cells and macrophages, have been shown to participate in chronic inflammatory processes within the vasculature and are thus considered potential therapeutic targets for various arterial diseases [64]. Recent studies have found that histone acetylation can promote the release of pro-inflammatory cytokines by certain immune cells, thereby enhancing the inflammatory response. For instance, LPS and high glucose can induce acetylation modifications of H3K27, H3K9, and H3K14 in the chromatin of monocyte-macrophages, promoting their conversion to the M1 pro-inflammatory phenotype. This transformation leads to the secretion of more pro-inflammatory cytokines, further exacerbating the inflammatory response. Inhibition of p300 can suppress expression of the pro-inflammatory marker inducible nitric oxide synthase (iNOS) [65, 66]. Additionally, HATs and HDACs also jointly regulate the activity of T cells and B cells [67, 68, 69].
The vascular pathological tissues and peripheral blood of patients with AS and abdominal aortic aneurysm show elevated chromatin acetylation levels in the monocyte-macrophage system and in T cells [70, 71, 72]. Moreover, the protein levels of p300, KAT1, H3K27ac, and NADPH oxidase 5 (Nox5) were also elevated in atherosclerotic tissues. Immunohistochemistry and immunofluorescence staining experiments indicated these proteins co-localized in CD45⁺/CD68⁺ immune cells and lipid deposition regions within atherosclerotic plaques. In vitro experiments showed the levels of KAT1, H3K27ac, H3K9ac and Nox5 increased significantly in cultured human macrophages under LPS stimulation, with both p300 and KAT1 recruited to active transcriptional regions of the Nox5 gene promoter. In addition, HAT inhibitors reduced Nox5 mRNA and protein expression in LPS-stimulated macrophages, thereby decreasing ROS production and inflammatory responses [70].
The interaction between HATs and certain non-coding RNAs not only regulates the prognosis of some cardiac diseases, but also promotes macrophage activation during coronary AS, thereby exacerbating the progression of the disease. In this pathological process, oxidized low-density lipoprotein (ox-LDL) can induce the upregulation of miR-21-5p (a non-coding RNA), which then activates P300. Activated P300 subsequently catalyzes the acetylation of HMGB1, causing it to migrate from the nucleus to the cytoplasm and be secreted into the extracellular microenvironment. Here, it induces macrophages to polarize into the M1 phenotype, thus driving the development of AS [73]. Related studies have also shown that miR-486 binds directly to the 3’ untranslated region of KAT1 mRNA, inhibiting its post-transcriptional expression. This reduces its regulation of the downstream target ATP-binding cassette transporter A1 (ABCA1), impeding cholesterol efflux in macrophages and promoting cholesterol accumulation, which ultimately worsens AS [74].
PCAF also participates in the recruitment of inflammatory cells. Intervention with PCAF siRNA, or the PCAF inhibitor garcinol, can significantly reduce the secretion of CCL2 in the vascular intima, leading to a 54.8% reduction in the number of macrophages in the intima [56]. Studies have also found that PCAF knockout attenuates the VSMC-induced increase in leukocytes and inflammatory response [75]. Lastly, the atherogenic phospholipid phosphatidylcholine promotes AS by upregulating the activity of HATs and increasing the H3K14ac modification of chromatin, thereby upregulating the expression of tat-interactive protein (TIP) genes in human aortic ECs. This activation triggers trained immunity pathways, initiating sustained chronic inflammation [76]. The above findings suggest the HAT system exacerbates vascular injury by regulating the activation of immune cells and the production of inflammatory factors.
In chronic damage conditions such as hypertension, hyperglycemia and hypoxia, the HAT levels in the vascular wall increase, and histone acetylation in the nucleus is significantly elevated. At the same time, abnormal expression of HATs exacerbates inflammation, senescence, and damage in the vascular wall, forming a positive feedback loop. This bidirectional interaction between inflammation and HAT activity is an important mechanism for the persistence and amplification of the inflammatory response.
The NF-
The MAPK pathway also promotes vascular inflammatory responses, immune cell regeneration, release of inflammatory mediators, and vascular remodeling. Following LPS induction of VSMCs, the expression of p-p38 MAPK and p-AKT increases. Downregulation of PCAF significantly inhibits the phosphorylation of both proteins, thus reducing VSMC proliferation. Mechanistically, VEGFR2, a key receptor in the MAPK pathway, is transcriptionally regulated by p300. Additionally, researchers have found that KAT7 increases the transcription of MRAS (a small GTPase from the RAS superfamily) by acetylating histone H3 at Lys14, which then activates the MAPK/ERK pathway and promotes colorectal cancer progression [79].
AMPK is a key energy metabolism-regulating kinase that is generally considered
to play a protective role in vascular wall inflammation. The AMPK/SIRT1/mTOR
pathway is the main pathway through which AMPK exerts its anti-inflammatory
effects. AMPK can also inhibit inflammation by suppressing HATs. Furthermore,
AMPK can activate SIRT1 and directly phosphorylate p300, thereby downregulating
p300 activity [80]. In diseases like hypertension and AS, the levels of HATs and
NF-
HATs primarily amplify inflammation and promote damage in vascular wall lesions.
However, under certain conditions, HATs can also play a protective role in the
vasculature. For example, KAT3A promotes the expression of miR-322 by stabilizing
the HIF-1
HATs regulate inflammation-related pathways, promote oxidative stress responses, and accelerate vascular cell aging (e.g., by affecting the p53/p21 signaling axis), thereby contributing to the formation of atherosclerotic plaques, vascular remodeling, and other pathological processes. Given the multifaceted regulatory roles of HATs in the core pathological mechanisms of arterial diseases, the targeting of HATs has become a promising therapeutic strategy in the cardiovascular field, drawing widespread interest from researchers.
Several commonly used clinical drugs, including metformin, curcumin, melatonin, and green tea extracts, have shown significant potential for HAT inhibition. Among them, metformin has shown clear supporting evidence of cardiovascular protective effects. In vitro experiments have confirmed this is primarily mediated by activating the AMPK signaling pathway, which in turn inhibits p300 acetyltransferase activity [89]. Clinical trials showed that oral curcumin significantly lowers plasma BNP levels in patients with hypertensive heart disease [90]. Similarly, the cardiovascular protective effects of natural bioactive substances such as curcumin, melatonin, tea polyphenols, garcinol, and urushiol are closely related to their molecular mechanisms involving inhibition of p300 and PCAF [22, 34, 53, 91]. Table 1 (Ref. [7, 12, 34, 37, 39, 56, 70, 92, 93, 94, 95, 96, 97, 98, 99]) summarizes the most important findings to date of research on HAT inhibitors in relation to cardiovascular diseases. Intraperitoneal injection of CTK7A, a p300 inhibitor, effectively lowers blood pressure in rats and reduces norepinephrine secretion [37]. Pentamidine, an inhibitor of acetyltransferase Kat5, mitigates damage from myocardial infarction [92]. WM-3835, a KAT7 inhibitor, significantly improves hypertension and endothelial damage induced by punatayin in SD rats [7]. The p300 inhibitor A-485 and the imidazole ketone erastin induced ferroptosis in human aortic smooth muscle cells [93]. C646, a specific p300 inhibitor, and CPTH2, a KAT2A inhibitor, reduced macrophage activation to varying degrees following LPS-induced activation [70]. In terms of clinical translation, significant progress has been made in the development of targeted drugs for histone acetylation modifications. Notably, the histone deacetylase inhibitor (HDACi) CS1 has entered phase II clinical trials for PAH [100], while several other HDACis have been approved for the clinical treatment of malignant tumors.
| HAT inhibitor | HATs | Target cell | Inhibitory/Promotional pathological effects |
| A-485 | p300/CBP | Human aortic SMCs | Ferroptosis [93] |
| Curcumin | p300/CBP | Rat VSMCs | Smooth muscle cell inflammation [39] |
| WM-3835 | KAT7 | Rat mesenteric artery ECs | EC senescence and inflammation [7] |
| EGCG | p300/CBP | Human ECs | EC inflammation [34] |
| CPTH2 | KAT2A | Human macrophages | Oxidative stress [70] |
| C646 | p300/CBP | Human macrophages | Oxidative stress [70] |
| Garcinol | PCAF | White blood cells | Intimal leukocyte recruitment [56] |
| Anserine | p300/CBP | Mouse cardiomyocytes | Myocardial hypertrophy [94] |
| Andrographolide | p300/CBP | Mouse valve interstitial cells | Aortic valve calcification [95] |
| Anacardic acid | p300/CBP | Mouse cardiomyocytes | Myocardial hypertrophy [96] |
| Metformin | p300/CBP | Rat cardiomyocytes | Myocardial hypertrophy [12] |
| Curcumin | p300/CBP | Human atrial fibroblasts | Atrial fibroblast senescence [97] |
| Anacardic acid | PCAF | Mouse cardiomyocytes | Myocardial hypertrophy [98] |
| L002 | p300/CBP | Cardiac fibroblasts | Myocardial hypertrophy [99] |
| Pentamidine | KAT5 | Mouse cardiomyocytes | Myocardial infarction area [92] |
| CTK7A | p300/CBP | pheochromocytoma-12 cells | Oxidative stress [37] |
Despite the current intense research into the clinical application of HAT inhibitors, several major issues have limited the progress in this field. For example, HATs are key enzymes that catalyze histone acetylation (‘writing’). However, the ‘reading’ mechanism, which relies on bromodomain proteins recognizing acetylation modifications, and the details of its coordination with the ‘writing’ process remain unclear [101]. This makes it difficult to precisely intervene in the activity of HATs, or to target specific gene loci during transcription. Single-cell epigenomics techniques (such as scChIP-seq, CoBATCH) have enabled high-throughput analysis at the single-cell level by optimizing cell isolation and library preparation processes, thereby refining epigenetic research. Secondly, HAT inhibitors lack subtype selectivity, and their broad target range results in many side effects [102]. Compared to traditional HAT inhibitors, epigenetic degraders offer higher selectivity and low-dose catalysis, making them a promising alternative to HAT inhibitors [102].
In addition to HAT inhibitors and epigenetic degraders, non-coding RNAs and
epigenetic editing technologies are also key areas of research. Non-coding RNAs
target KATs through direct binding or competitive binding. They play a critical
role in the development of cardiovascular diseases, thus providing new directions
for disease diagnosis and treatment. For example, miR-185-5p binds to the
3′UTR of KAT7 to suppress its expression, while lncRNA ADAMTS9-AS1 competes
with miR-185-5p to upregulate KAT7, inhibiting myocardial cell hypertrophy [103].
Circ-myh8 recruits KAT7 to the HIF1
Although drug developments that specifically target HATs are still in the preclinical stage, an accumulating body of basic research evidence suggests that HATs could become a novel molecular target for cardiovascular disease intervention. This view is gaining increasing recognition and attention among researchers.
Currently, HATs are primarily considered to promote vascular pathological changes. This review mostly discusses the pro-inflammatory mechanisms of HATs, including their enhanced activity, upregulated expression, and increased acetylation levels of chromatin histones, all of which contribute to vascular wall damage. We systematically explore several common pathological factors, including hyperglycemia, high salt, intermittent hypoxia, and activation of the renin-angiotensin system, and delve into their potential molecular mechanisms, such as the Nfkb-NLRP3 pathway, MAPK signaling pathway, AMPK pathway, and VEGFR2 signaling pathway. Additionally, this review discusses the dual role of HATs in vascular homeostasis, both as a protective agent for blood vessels, as well as a participant in the vascular injury process. We also explore the potential clinical applications of HATs, with a focus on innovative and targeted intervention strategies for HATs.
In recent years, multiple studies have demonstrated that HATs promote the expression of inflammatory mediators, such as IL-6 and IL-1, by influencing the promoter regions of these factors. Notably, as histone acetylation levels increase, there is a tendency for upregulation of various inflammation-related genes, cell proliferation proteins, and chemokines, suggesting there may be a complex network of interactions between these proteins and HATs that warrants further investigation. Although the functional regulation of HATs in the vascular system is starting to be revealed, significant gaps remain in our understanding of the epigenetic mechanisms underlying HATs. Further elucidation of the epigenetic regulatory networks mediated by HATs will help to identify new drug targets and provide theoretical support for screening specific HAT inhibitors. This should eventually lead to new directions for the targeted prevention and treatment of vascular-related diseases.
In conclusion, HATs are significantly implicated in vascular pathological changes. Their unique regulatory mechanisms and therapeutic potential open new pathways for the treatment of vascular diseases, while expanding knowledge in this field.
WL, JD, YJ and QJ conceived the study topic and the manuscript design. WL, JD, and YJ contributed to figure preparation and making the table. All authors performed the literature search and manuscript writing and made substantial contributions to the interpretation of the literature. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
2024 Annual Municipal and Hospital Joint Science and Technology Cooperation Fund Project, Project No: Zunyi City Science and Technology Cooperation HZ (2024) 110.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.