








 , Hong Hong 2,†, Lining Zhou 1, Fuying Chu 1, Xiang Chen 1,*
, Hong Hong 2,†, Lining Zhou 1, Fuying Chu 1, Xiang Chen 1,*
1 Department of Laboratory Medicine, The Second Affiliated Hospital of Nantong University and Nantong First People’s Hospital, 226001 Nantong, Jiangsu, China
2 Department of Clinical Laboratory, The Affiliated Hospital of Nanjing University of Chinese Medicine and Nantong Traditional Chinese Medicine Hospital, 226001 Nantong, Jiangsu, China
†These authors contributed equally.
Abstract
As a major contributor to cancer-associated deaths, advanced colorectal cancer (CRC) has a constrained range of effective treatment options. The short isoform of bromodomain-containing protein 4 (BRD4-S) has recently been implicated as a potential oncogenic driver; however, its regulatory mechanisms and functional role in CRC remain incompletely understood.
BRD4-S expression, regulation, and function in CRC were investigated through bioinformatics analyses of the Cancer Genome Atlas (TCGA) datasets, in vitro studies using CRC cell lines (HT29, SW620), and in vivo xenograft models in nude mice. Experimental approaches included quantitative real-time PCR (qRT-PCR), Western blotting, co-immunoprecipitation, RNA immunoprecipitation, immunofluorescence, colony formation, Cell Counting Kit-8 (CCK-8), and scratch assays. Gene enrichment and interaction analyses were performed to identify relevant pathways and molecular partners.
BRD4-S was markedly upregulated in CRC tissues and cell lines, and elevated BRD4-S expression correlated with poorer patient survival. Silencing BRD4-S, but not BRD4-L, significantly impaired CRC cell proliferation, migration, and tumor growth in vivo. Mechanistically, the RNA helicase DEAD-box helicase 27 (DDX27) interacted with Serine and Arginine Rich Splicing Factor 6 (SRSF6) to promote alternative splicing of BRD4 pre-mRNA toward the BRD4-S isoform. Inhibition of SRSF6 phosphorylation suppressed BRD4-S production and blocked activation of the mitogen-activated protein kinase (MAPK)/extracellular regulated protein kinases ERK signaling pathway, identified as a key downstream effector of BRD4-S.
This study defines a novel DDX27–SRSF6–BRD4-S–MAPK/ERK signaling axis that drives CRC progression. These findings underscore the therapeutic potential of targeting BRD4 isoform switching and its regulatory splicing machinery in CRC.
Keywords
- colorectal neoplasms
- alternative splicing
- MAP kinase signaling system
- bromodomain containing proteins
- DEAD-box RNA helicases
According to the Global Burden of Disease Study 2021, colorectal cancer (CRC) was responsible for approximately 1.04 million deaths and affected about 11.68 million people globally, underscoring its significant global health impact as the third most common cancer and the second leading cause of cancer mortality [1]. Despite advances in early detection methods, such as colonoscopy and fecal immunochemical testing (FIT), a considerable proportion of patients are still diagnosed at advanced stages, precluding curative surgical resection [2]. Conventional chemotherapy regimens, including fluorouracil (5-FU) and irinotecan, are limited by their non-selective mechanisms, which cause substantial toxicity in normal tissues and reduce both efficacy and patient tolerance [3]. In recent years, targeted therapies and immunotherapies have emerged as transformative treatment strategies [4]. For example, anti-Epidermal Growth Factor Receptor (EGFR) agents such as cetuximab and panitumumab are standard therapies for Rat Sarcoma (RAS)/v-raf murine sarcoma viral oncogene homolog B1 (BRAF) wild-type metastatic CRC (mCRC), while BRAF inhibitors (e.g., encorafenib) in combination with EGFR antibodies have shown promise in BRAF V600E-mutant cases [5]. Although these targeted therapies have expanded treatment options and improved safety profiles, the proportion of patients eligible to benefit from them remains relatively small [6]. Thus, there is a pressing need for novel therapeutic approaches to effectively suppress tumor progression.
Bromodomain-containing protein 4 (BRD4) is a member of the Bromodomain and Extra-Terminal (BET) protein family, which also includes BRD2, BRD3, and BRDT. In addition to two bromodomains and an extra-terminal (ET) domain, BRD4 possesses a unique C-terminal motif (CTM) that serves to distinguish it from BRD2 and BRD3 [7]. By binding acetylated chromatin via its bromodomains, BRD4 recruits various transcriptional machinery to target genes, thereby directly influencing their transcriptional output. BRD4 is essential for cellular proliferation, cell cycle progression, and DNA repair. Notably, its overexpression in colorectal tumors has been associated with unfavorable clinical outcomes [8].
BRD4 encodes two major isoforms: the long isoform (BRD4-L) and the short isoform (BRD4-S). Compared with BRD4-L, BRD4-S lacks the CTM and the P-TEFb-interacting domain (PID). Owing to these structural differences, it is important to distinguish their functional roles in cancer initiation and progression. Recent studies have shown that BRD4-S exerts oncogenic effects in breast cancer [9], whereas in rhabdomyosarcoma, BRD4-S suppresses metastasis by inhibiting BRD4-L–mediated activation of integrin genes [10]. However, the role of BRD4-S in CRC remains unclear.
The DEAD-box RNA helicase (DDX) protein family in humans comprises 38 distinct members, several of which are highly expressed in tumors and exhibit diverse oncogenic functions. These proteins participate in critical cellular processes, including mRNA splicing, lysosome biogenesis, transcriptional regulation of RNA, and the modulation of RNA replication and degradation [11, 12, 13]. DDX27, a member of this family, has been implicated in multiple cancers by promoting tumor growth and metastasis. In gastric cancer (GC), DDX27 is frequently amplified and overexpressed, correlating with poor patient survival. It enhances the colony-forming ability of GC cells by regulating cell cycle progression independently of apoptosis [14]. In hepatocellular carcinoma (HCC), DDX27 promotes cancer progression through the ERK signaling pathway. It is overexpressed in HCC tissues, where high expression levels are associated with poor prognosis. Mechanistically, DDX27 upregulation induces major vault protein (MVP) expression and enhances ERK1/2 phosphorylation, thereby facilitating metastasis [15]. Although a recent study has reported that DDX27 enhances gene expression in CRC cells [16], the mechanisms by which it contributes to colorectal carcinogenesis and regulates downstream targets remain poorly understood.
Serine/arginine-rich splicing factor 6 (SRSF6) is a critical regulator of both constitutive and alternative splicing and has been implicated in tumorigenesis across multiple cancer types, including colorectal, skin, and lung cancers [17]. SRSF6 promotes exon inclusion by recognizing specific RNA motifs and facilitating spliceosome assembly. Dysregulated SRSF6 expression alters the splicing of various tumor suppressors and oncogenes, leading to the generation of oncogenic isoforms [18]. It is frequently overexpressed in cancers and contributes to tumor progression by modulating the splicing of apoptosis-related genes such as First apoptosis signal receptor (Fas), Bcl-x (B-cell lymphoma-extra large), and Bcl-2 Interacting Mediator of cell death (BIM) [19]. Importantly, SRSF6 has been reported to interact physically and functionally with RNA helicases to regulate splicing outcomes [20]. Given that DDX27 is a member of the DEAD-box RNA helicase family, we hypothesized that DDX27 may cooperate with SRSF6 to control BRD4 isoform switching in CRC.
In this study, we provide novel evidence that BRD4-S is highly expressed in CRC
tissues and cell lines, where it exhibits strong oncogenic activity. Notably,
patients with elevated BRD4-S expression showed significantly reduced survival
rates (p
Cell culture was performed in Dulbecco’s Modified Eagle Medium (DMEM; Cat:11965092, Gibco, Grand Island, NY, USA) enriched with 10% heat-inactivated fetal calf serum, under a humidified atmosphere of 5% CO2 at 37 °C. The human normal colon epithelial cell line (NCM640) and the human CRC cell lines (SW620, HT29, DLD-1) used in this study were all acquired from the American Type Culture Collection (ATCC, Manassas, VA, USA). Transfections of HT29 and SW620 cells were performed using Lipofectamine 2000 (Cat:11668019, Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. All cultures were routinely maintained with mycoplasma scavenger supplementation and regularly monitored for contamination. Cell line authentication was confirmed by short tandem repeat (STR) profiling, and all lines tested negative for mycoplasma.
Stable knockdown cell lines were generated via lentiviral transduction. In brief, cells at ~90% confluence were co-transfected with a mixture of packaging plasmids (pIP1 and pIP2, 5 µg each), the envelope plasmid pIP/VSV-G (5 µg) from the ViraPower™ Lentiviral Packaging Mix (Cat: K497500, Thermo Fisher Scientific, Waltham, MA, USA), and 5 µg of a lentiviral shRNA construct (non-targeting control pLKO.1, Cat: SHC002; shBRD4-L, TRCN0000021424; shBRD4-S, TRCN0000349782; all from Sigma-Aldrich, St. Louis, MO, USA), using 30 µL of Lipofectamine 3000 (Cat: L30000001, Invitrogen, Carlsbad, CA, USA) as per the manufacturer’s protocol. After 16 hours, the medium was refreshed. The resulting viral supernatants were harvested, concentrated, and used to transduce human CRC cell lines (HT29 and SW620) in DMEM containing 8 µg/mL polybrene (Sigma-Aldrich). Transduced cells were then selected with 1 µg/mL puromycin (Sigma-Aldrich) for 4–5 days prior to expansion and subsequent analysis. The shRNA target sequences were as follows:
shBRD4-L: 5′-CCAACCAAAGTCAGTTCCTTC-3′ (targeting the CTM-coding region of BRD4-L). shBRD4-S: 5′-ATTGGACACGGACTCTTAATA-3′ (targeting the GPA-rich domain of BRD4-S).
To generate the BRD4-S overexpression construct, the full-length coding sequence (CDS) of human BRD4-S was amplified by PCR from cDNA derived from HT29 cells. The PCR product was cloned into the pcDNA3.1(+) vector (Invitrogen) downstream of the CMV promoter, incorporating a C-terminal FLAG tag for detection. The empty pcDNA3.1(+) vector served as the control. All constructs were confirmed by Sanger sequencing.
Male BALB/c (nu/nu) nude mice, aged 6–8 weeks and weighing around 25 grams,
were obtained from the Shanghai Laboratory Animal Center and housed in a
pathogen-free environment under controlled temperature and humidity. HT-29 cells
transduced with either control or BRD4-S shRNA were cultured in McCoy’s 5A medium
at 37 °C with 5% CO2. Cells in the logarithmic growth phase were
harvested, washed, and resuspended in a serum-free medium/Matrigel mixture (1:1)
at a concentration of 1
Total RNA was isolated from target cells with Trizol (Ambion) following the manufacturer’s protocol. Subsequently, 1 µg of the extracted RNA was subjected to reverse transcription using the RevertAid First Strand cDNA Synthesis Kit (Cat: K1622, Thermo Fisher Scientific, Waltham, MA, USA). Quantitative PCR was then performed on the CFX96 Connect™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) using the SYBR Green PCR Kit (Cat: 204145, Qiagen, Hilden, Germany) in 20 µL reactions. The thermal cycling conditions comprised an initial denaturation at 95 °C for 2 minutes, followed by 40 cycles of 95 °C for 10 seconds and 60 °C for 10 seconds. Primers specific for DDX27, GAPDH, BRD4-L, BRD4-S, ZFP64, RBL1, ZNF217, HNF4A, GNL3L, and POFUT1 were designed using Primer3 software (version 1.0; https://primer3.org/). The sequences were as follows: Human DDX27 F (forward): 5′-CTCACTAAAGGCACCGAAG-3′, R (reverse): 5′-GGCAGAGAAGTTGCTTGTGG-3′; Human GAPDH F (forward): 5′-ACGGCAAGTTCAACGGCACAG-3′, R (reverse): 5′-GACGCCAGTAGACTCCACGACA-3′; Human BRD4-L F (forward): 5′-CTCCTCCTAAAAAGACGAAGA-3′, R (reverse): 5′-TTCGGAGTCTTCGCTGTCAGAGGAG-3′; Human BRD4-S F (forward): 5′-TTTCTCTCTCCCTCTACGT-3′, R (reverse): 5′-TTAGGCAGGACCTACGTAG-3′; Human ZFP64 F (forward): 5′-ATGGCTGCAGTTCTGTGTACT-3′, R (reverse): 5′-CTGCAGGATCTGCAGATGT-GGTA-3′; Human RBL1 F (forward): 5′-CGTGATGTCCGTGTACTTTGCAG-3′, R (reverse): 5′-GCTG-CAGTTGC-AAGATGTGCGATG-3′; Human ZNF217 F (forward): 5′-CAGACCTACAGCAACAGCAG-3′, R (reverse): 5′-GCT-GTACTTGGCCTTCATCC-3′; Human HNF4A F (forward): 5′-CGGACTGGGTCTGATGTGCAGTT-3′, R (reverse): 5′-CAGGCATTGCAGTGCCACGTAGAT-3′; Human GNL3L F (forward): 5′-AGCAAGATGGACGAGACCCA-3′, R (reverse): 5′-TCACCAGGTACTTGTAGCCG-3′; Human POFUT1 F (forward): 5′-CGGTTCTGCTGTGGAGATG-TGT-3′, R (reverse): 5′-GCATTGGTGCCAGCTGTGATGTGC-3′; Human STAT5B F (forward): 5′-CAGCCTACCATTGACAGCGT-3′, R (reverse): 5′-TGTCCAGCATCCTTGAACCA-3′.
Cellular proteins were extracted using RIPA buffer supplemented with 1 mM PMSF.
The extracts were then subjected to SDS–PAGE on 8% gels and electrophoretically
transferred onto PVDF membranes (Cat: IPVH00010, Millipore, Billerica, MA, USA).
Membrane blocking was performed with 5% non-fat dry milk in TBST. Primary
antibody incubations were carried out overnight at 4 °C, followed by a
2-hour incubation with corresponding secondary antibodies at room temperature.
Signal detection was achieved with an ECL substrate (Cat: 180-5001, Tanon,
Shanghai, China), and imaging was performed using a ChemiDoc system (Bio-Rad).
The following primary antibodies were used in Western blot analyses: anti-BRD4
(Cat: ab128874, Abcam, Cambridge, UK; 1:1000), anti-DDX27 (Cat: ab177950, Abcam;
1:1000), anti-SRSF6 (Cat: ab38017, Abcam; 1:1000), anti-pSRSF6 (Cat: MABE50, EMD
Millipore, Billerica, MA, USA; 1:1000), anti-
Cell proliferation was evaluated by seeding transfected cells (2500 cells/well) into 96-well clear plates containing complete medium. Proliferation was quantified at designated time points using the Cell Counting Kit-8 (CCK-8; Cat: WH1199, Dojindo Laboratories, Kumamoto, Japan), and the absorbance at 450 nm was measured with a microplate reader.
For immunofluorescence staining, cells were first fixed with 4% paraformaldehyde at low temperature for 20 min and permeabilized with 0.2% Triton X-100 for 10 min. After three Phosphate-Buffered Saline (PBS) washes, non-specific sites were blocked with 1% BSA in PBS for 30 min. The samples were then incubated overnight at 4 °C with primary antibodies against DDX27 (Cat: ab177950, Abcam, 1:200) or SRSF6 (Abcam, ab38017, 1:200), followed by a 1-hour incubation at room temperature with secondary antibodies (Alexa Fluor 488 goat anti-rabbit, Invitrogen, A-11034, 1:40; or Alexa Fluor 594 goat anti-mouse, Invitrogen, A-11012, 1:40). Following additional PBS washes, nuclei were stained with DAPI for 10 min, and the samples were mounted in glycerol for visualization.
The RNA co-immunoprecipitation (RIP) assay was conducted with the Magna RIP Kit
(Millipore, MA, USA) following the manufacturer’s protocol. Briefly, cells were
lysed in ice-cold RIPA buffer containing protease inhibitors. For each
immunoprecipitation, 500 µg of protein lysate was incubated with 40
µL of A/G beads (sc-2003, Santa Cruz Biotechnology, Dallas, TX,
USA). Subsequently, 2 µg of either control IgG (Santa Cruz
Biotechnology) or specific antibody against DDX27/SRSF6 (Abcam) was added, and
the mixture was subjected to overnight incubation at 4 °C. An additional
40 µL of A/G beads were then introduced for a further 6-hour
incubation. The beads were then washed with PBS containing 0.2% NP-40, and the
bound proteins were eluted by boiling in 2
Chromatin immunoprecipitation (ChIP) was carried out with the EZ-ChIP™ Kit (17-295, Millipore, Burlington, MA, USA) per the manufacturer’s protocol. In brief, HT-29 cells were fixed with 1% formaldehyde for 10 min at room temperature, and the cross-linking was terminated by the addition of glycine (0.125 M final concentration). After washing with cold PBS, the cells were harvested and lysed in SDS lysis buffer containing protease inhibitors. The chromatin was then fragmented by sonication using a Bioruptor® Pico (B01060001, Diagenode, Denville, NJ, USA) to achieve fragments ranging from 200 to 500 bp. The sheared lysates were subjected to immunoprecipitation overnight at 4 °C with 2 µg of either anti-BRD4 antibody (Abcam, ab128874) or control rabbit IgG. Protein–DNA complexes were captured with protein A/G magnetic beads and sequentially washed with low-salt, high-salt, LiCl, and TE buffers. Following cross-link reversal (65 °C for 4 h with 200 mM NaCl) and proteinase K digestion, the immunoprecipitated DNA was purified using spin columns. Quantitative PCR analysis was performed on a CFX96 Real-Time PCR System (Bio-Rad) using SYBR Green, with enrichment expressed as the percentage of input and normalized to the IgG control. The following primers were used to amplify promoter regions of ERK target genes:
CCND1 promoter
Forward: 5′-CCTCTCGCTCCGTAACCATC-3′
Reverse: 5′-GCTGGACTTGACCACCTTCC-3′
DUSP6 promoter
Forward: 5′-GAGGCGAGAGAGAGGAAGGA-3′
Reverse: 5′-CCTCCTCCTCCACCTCTTTC-3′
To assess the prognostic significance of BRD4 isoforms in CRC, mRNA expression matrices of CRC tissues and matched adjacent normal tissues were retrieved from The Cancer Genome Atlas (TCGA) database. The mRNA expression values of BRD4-L (long isoform) and BRD4-S (short isoform) were then extracted for comparative analysis. Patients were stratified into high- and low-expression groups according to the median BRD4-S expression value (or predefined clinical cutoffs where applicable). This stratification facilitated systematic evaluation of the association between BRD4-S expression levels and patient survival outcomes.
Transcriptomic data from TCGA were analyzed to identify BRD4-S-related
transcriptional alterations in CRC by comparing tumor and normal tissues.
Differential expression analysis, performed with DESeq2 or edgeR, identified
significant genes using a
To characterize BRD4-L, BRD4-S, and DDX27 -associated genes in CRC, candidate
genes were identified by integrating differentially expressed genes (DEGs) with
BRD4-S targets from the GEPIA2 database. Functional enrichment analysis was
performed using ClusterProfiler (Open-source R package, Bioconductor, Seattle,
WA, USA) or DAVID, with Gene Ontology (GO) terms classified into biological
processes, molecular functions, and cellular components. Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway analysis was also conducted, focusing on
BRD4-S–related pathways such as MAPK/ERK signaling. Significant terms were
defined as those with a false discovery rate (FDR)
Statistical significance was defined as a two-sided p-value
To examine the expression levels of BRD4-L (200 kDa) and BRD4-S (140 kDa) (Fig. 1A) in CRC, we first obtained mRNA expression matrices for CRC tissues
and paired adjacent non-tumor tissues from the Gene Expression Profiling
Interactive Analysis (GEPIA2) database. From these matrices, we extracted the
mRNA expression values of BRD4-L and BRD4-S. Comparative analysis revealed that
overall BRD4 expression was significantly higher in CRC tissues than in adjacent
normal tissues (p
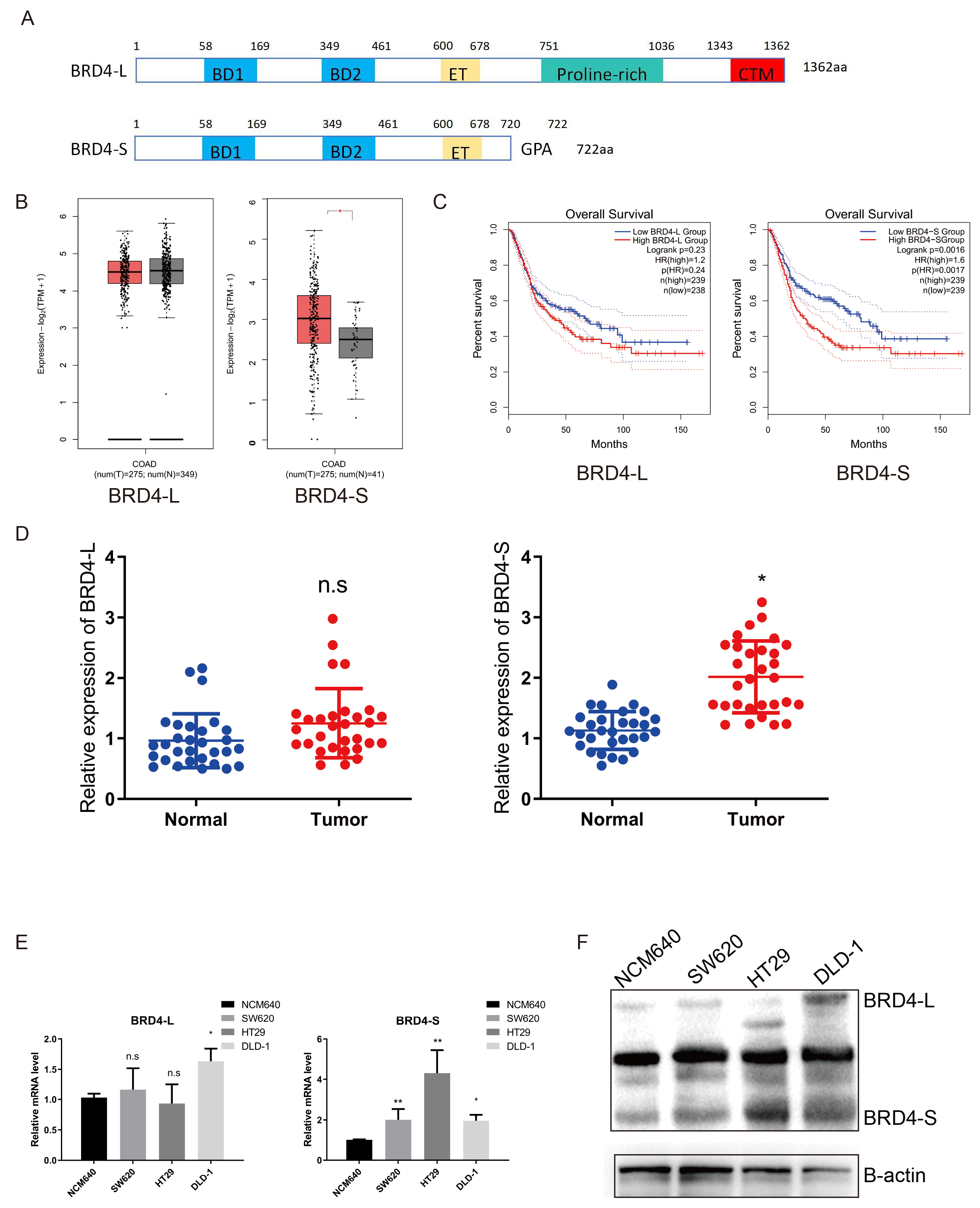 Fig. 1.
Fig. 1.
BRD4-S is highly expressed in CRC cells and tissues and related
to poor prognosis. (A) Schematic representation of the domain
structures of the long (BRD4-L) and short (BRD4-S) isoforms of BRD4. (B) Differential expression analysis of BRD4-L and BRD4-S transcripts in normal and
malignant tissues based on the TCGA database. (C) Kaplan–Meier survival curves
comparing overall survival in patients with high BRD4-L versus BRD4-S expression
using TCGA data. (D) qRT-PCR analysis of BRD4-L and BRD4-S expression in
normal colon tissue and CRC tissue. (E,F) qRT-PCR and western blot analyses of
BRD4-L and BRD4-S expression in CRC cell lines, including NCM640, SW620, HT29,
and DLD. Data are presented as mean
We designed isoform-specific shRNA primers targeting the CTM domain unique to
BRD4-L and the GPA domain exclusive to BRD4-S, successfully generating CRC cell
lines with stable knockdown of BRD4-S or BRD4-L (Fig. 2A). To investigate
isoform-specific functions of BRD4 in CRC, we performed CCK-8 and colony
formation assays to evaluate the impact of BRD4-S or BRD4-L silencing on cell
proliferation. Notably, BRD4-S knockdown significantly reduced both the size and
number of colonies (p
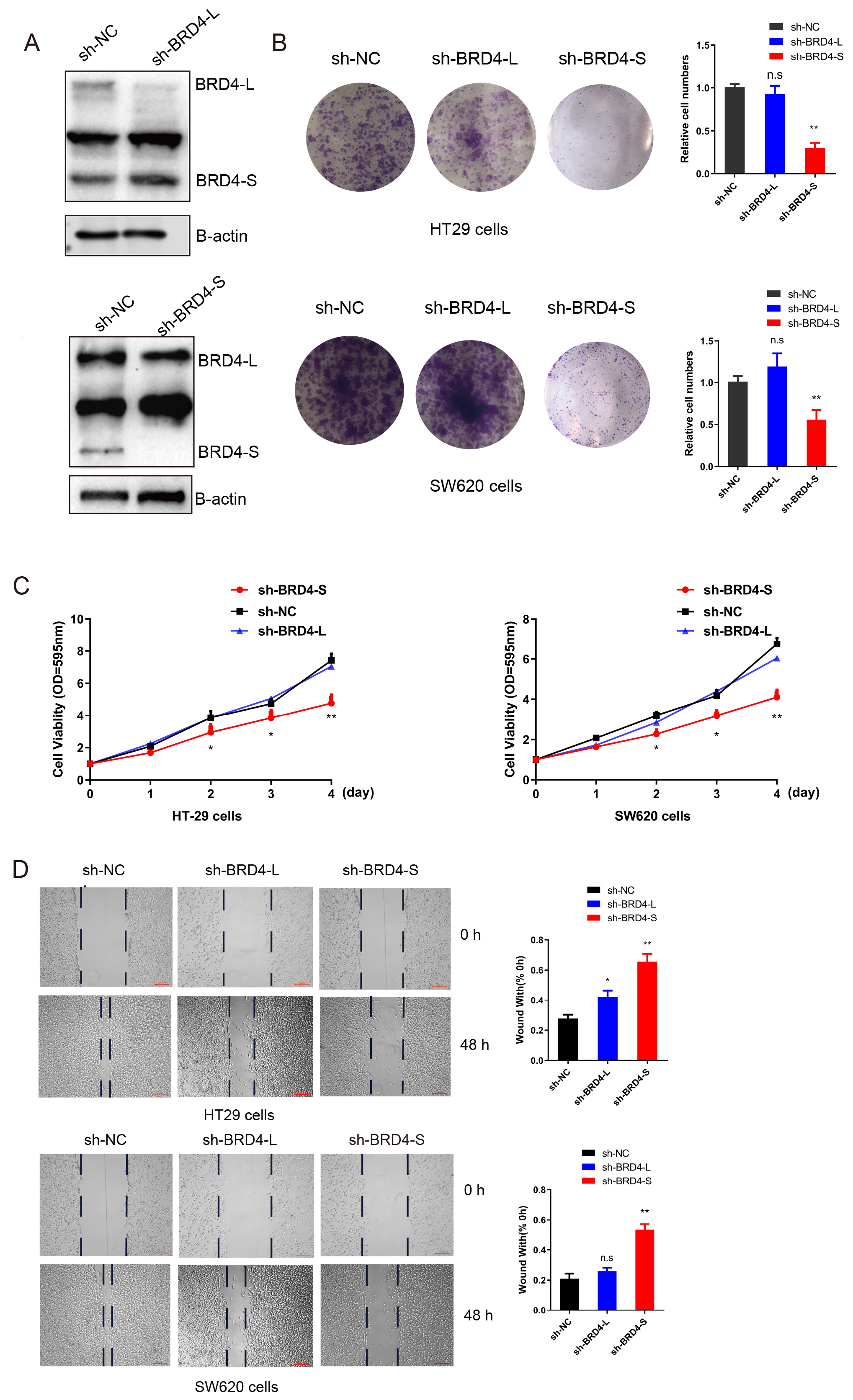 Fig. 2.
Fig. 2.
BRD4-S promotes CRC proliferation and migration in
vitro. (A) BRD4-S and BRD4-L knockdown cell lines were successfully established
in HT29 and SW620 cells. (B) Colony formation assays were performed to evaluate
the sphere-forming ability of BRD4-S and BRD4-L knockdown in HT29 and SW620
cells. (C,D) CCK-8 and scratch assays were conducted to assess the
proliferative capacity of BRD4-S and BRD4-L knockdown cell lines. Data are
presented as mean
To further evaluate the oncogenic role of BRD4-S in vivo, we performed subcutaneous injections of BRD4-L-shRNA cells, BRD4-S-shRNA cells, and control cells into nude mice. In the xenograft model, tumor formation was significantly reduced in the BRD4-S-shRNA group compared with both the control and BRD4-L-shRNA groups (Fig. 3A). Tumor growth curve analysis further demonstrated that BRD4-S knockdown markedly suppressed tumor progression (Fig. 3B). Consistently, both tumor weight and volume were significantly lower in the BRD4-S-shRNA xenografts compared with the other groups (Fig. 3C,D). These results indicate that BRD4-S possesses oncogenic properties that drive CRC tumor growth.
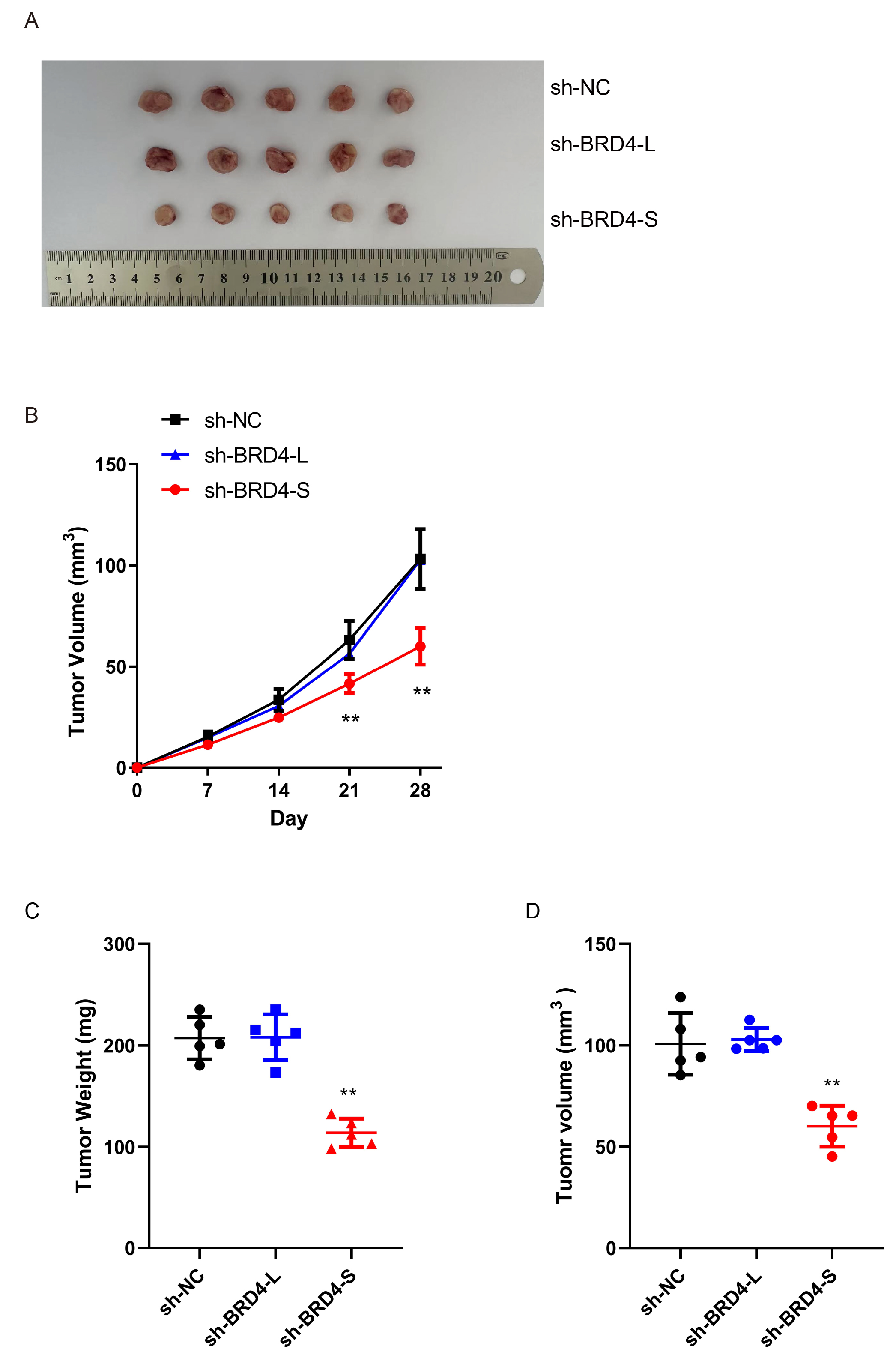 Fig. 3.
Fig. 3.
BRD4-S promotes CRC tumorigenesis in vivo. (A)
Representative images of tumor formation in nude mice at 8 weeks following
ectopic transplantation of HT29 cells with BRD4-S knockdown or empty vector (n =
5). (B) Tumor volume measurements of the two groups (BRD4-S knockdown vs. empty
vector–expressing HT29 cells) at 7, 14, 21, and 28 days after ectopic
transplantation. (C,D) Comparison of tumor weights and volume between BRD4-S
knockdown and empty vector–expressing HT29 cells in nude mice at 4 weeks
post-ectopic transplantation. Data are presented as mean
Using the GEPIA2 database, we systematically screened the top 100 genes
associated with the BRD4-S and BRD4-L isoforms. After excluding
genes co-associated with both isoforms, we identified 63 genes that were
exclusively correlated with BRD4-S (Fig. 4A). Subsequent
differential expression analysis of CRC transcriptomes from the TCGA cohort
revealed 5331 significantly dysregulated genes (
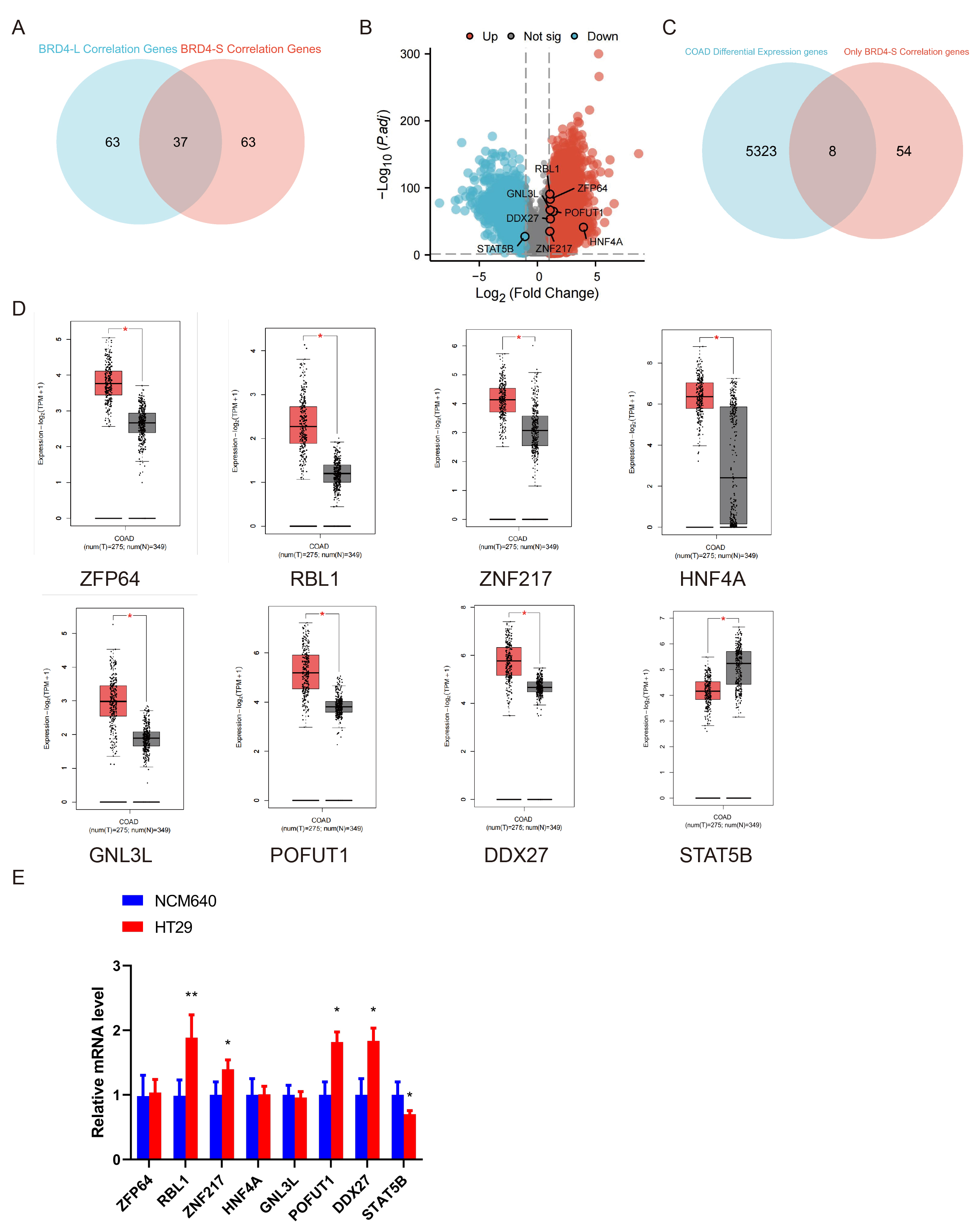 Fig. 4.
Fig. 4.
Identification of BRD4-S-specific functionally associated
candidates in CRC. (A) Venn diagram analysis of the top 100 genes most strongly
correlated with BRD4-L and BRD4-S in the GEPIA2 database
identified genes exclusively associated with BRD4-S. (B) Volcano plot
analysis of DEGs in CRC. (C) Venn diagram showing the overlap between DEGs in CRC
and those correlated with BRD4-S expression. (D) mRNA expression of
ZFP64, RBL1, ZNF217, HNF4A, GNL3L,
POFUT1, and DDX27 in paired CRC tissues according to the GEPIA2
database. (E) qRT-PCR analysis of ZFP64, RBL1, ZNF217,
HNF4A, GNL3L, POFUT1, and DDX27 in HT-29
cells. Data are presented as mean
To further investigate the regulatory role of candidate factors in BRD4 splicing, we performed GO and KEGG enrichment analyses on the previously identified genes. This analysis revealed a significant enrichment of DDX27 in spliceosome -related pathways (Fig. 5A). To evaluate the clinical relevance of DDX27 in CRC, we examined its expression in the TCGA CRC cohort. DDX27 mRNA levels were markedly elevated in CRC tissues compared with adjacent normal colon tissues (Fig. 5B), supporting its potential role as an oncogenic factor in CRC. Functional validation through DDX27 knockdown in HT-29 CRC cells (Fig. 5C) demonstrated reduced mRNA and protein levels of the BRD4-S isoform, accompanied by increased BRD4-L protein expression (Fig. 5D,E). Furthermore, correlation analysis using the GEPIA2 database identified SRSF6 as the splice factor most strongly associated with DDX27 (Fig. 5F).
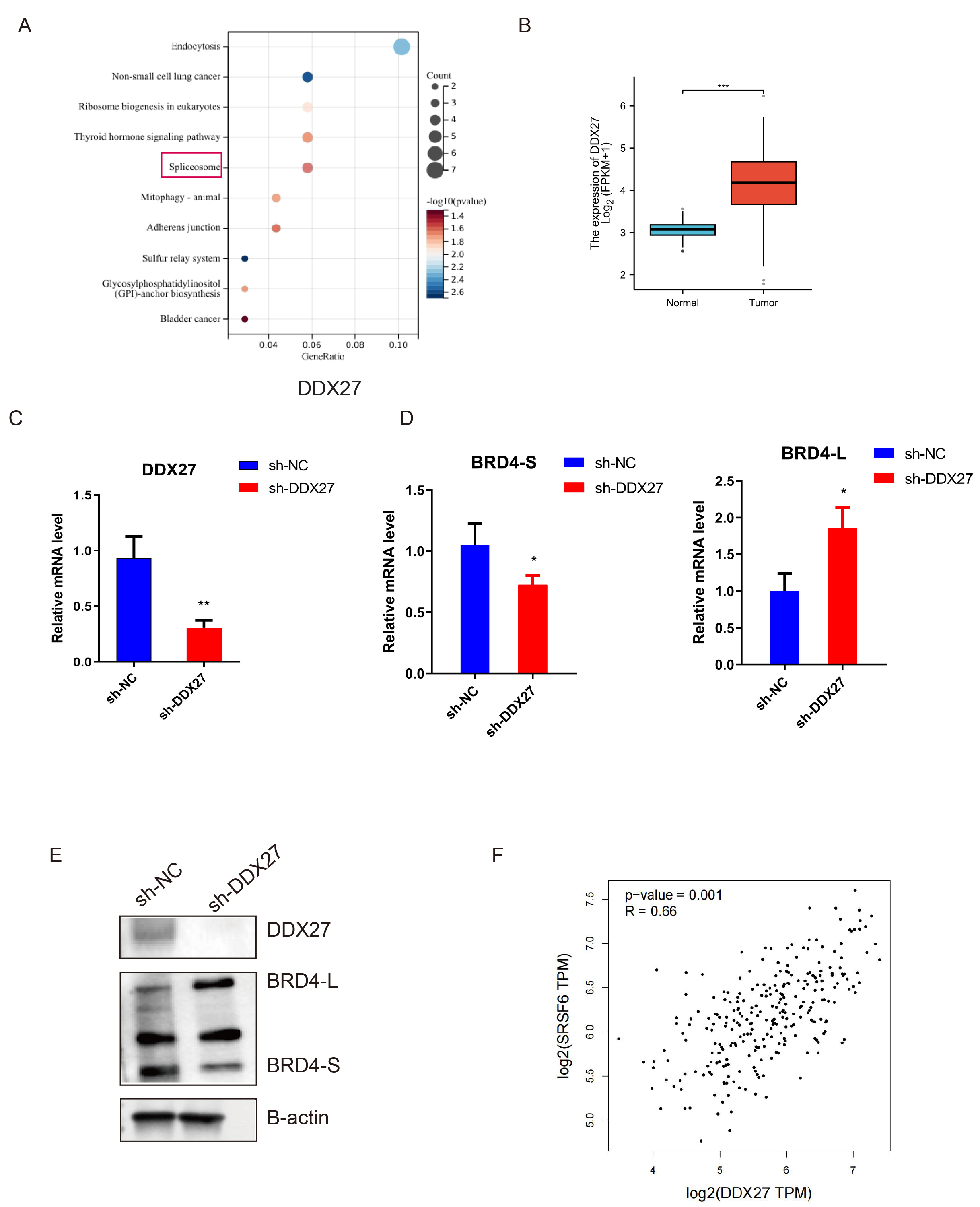 Fig. 5.
Fig. 5.
DDX27-mediated BRD4 splicing regulation modulates isoform
switching through SRSF6 interaction in HT-29 cells. (A) GO and KEGG enrichment
analysis of DEGs in samples with high DDX27 expression. The red box indicates the
association between DDX27 and the spliceosomal pathway investigated in subsequent studies. (B) Analysis of DDX27
mRNA Expression in Paired Colorectal Cancer Tissues from TCGA Cohort. (C)
DDX27 knockdown cell lines were successfully established in HT29 and SW620 cells.
(D,E) qRT‒PCR and Western blotting analyses of BRD4-L and BRD4-S expression in
DDX27 knockdown HT-29 cells. (F) Correlation analysis between the
splicing factor SRSF6 and DDX27. Data are presented as mean
Our study identified DDX27 as a regulator of BRD4-S expression in experimental models. To elucidate the molecular mechanisms underlying this regulation, we examined its interactions with splicing factors. RNA immunoprecipitation (RIP) assays confirmed the association of DDX27 with BRD4 pre-mRNA in HT29 cell extracts. The RIP assays also demonstrated that SRSF6 interacts with BRD4 pre-mRNA (Fig. 6A). To further investigate the specificity of the DDX27–SRSF6 interaction, co-immunoprecipitation (Co-IP) and immunofluorescence analyses were performed in HT29 cells using anti-DDX27 and anti-SRSF6 antibodies. These experiments suggested that DDX27 and SRSF6 likely form a complex (Fig. 6B,C). Moreover, knockdown of DDX27 resulted in a marked reduction of phosphorylated SRSF6 levels (Fig. 6D), indicating that DDX27 promotes SRSF6 phosphorylation. To assess whether SRSF6 phosphorylation is required for DDX27-mediated splicing activity, HT29 cells were treated with the SRSF6 phosphorylation inhibitor SPHINX31 at concentrations of 0, 1, and 2 µM for 24 hours. Inhibition of SRSF6 phosphorylation effectively suppressed the alternative splicing of BRD4 toward the BRD4-S isoform (Fig. 6E).
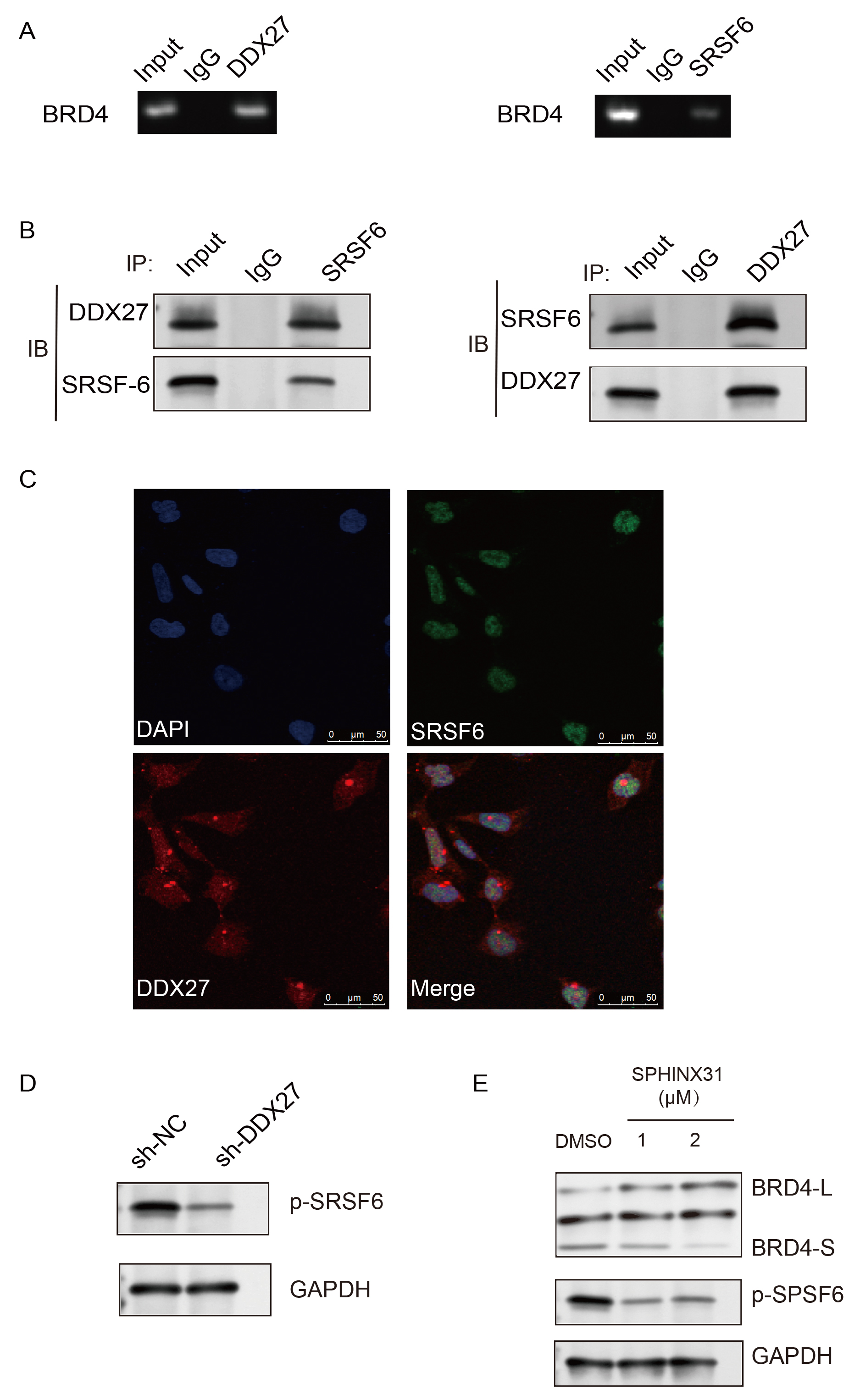 Fig. 6.
Fig. 6.
DDX27 Mediates BRD4-S alternative splicing through phosphorylation dependent interaction with SRSF6. (A) Immunoprecipitation from HT-29 cells using DDX27 and SRSF6 antibodies followed by RT-PCR or qPCR with BRD4-specific pre-mRNA primers. BRD4 pre-mRNA levels were quantified as fold enrichment relative to the IgG control. (B) Co-immunoprecipitation (Co-IP) of HT-29 cells with SRSF6, DDX27, or IgG antibodies, followed by western blot detection of SRSF6 and DDX27 in both precipitates and cell lysates. (C) Immunofluorescence analysis showing co-localization of SRSF6 (green) and DDX27 (red) with DAPI-stained nuclei (blue). Scale bar = 50 µm. (D) Western blot analysis of SRSF6 phosphorylation in HT-29 cells following DDX27 knockdown. (E) Western blot analysis of BRD4-S, BRD4-L, and p-SRSF6 levels in HT-29 cells treated with the SRSF6 phosphorylation inhibitor SPHINX31.
To elucidate the downstream pathways regulated by BRD4-S in CRC, we performed GO-KEGG enrichment analyses on both BRD4-S and BRD4-L. To avoid confounding effects from overlapping signals, pathways commonly enriched in both isoforms were excluded. The analysis revealed a significant enrichment of BRD4-S in the MAPK signaling pathway (Fig. 7A). To validate this finding, we examined MAPK pathway alterations in HT-29 cells following BRD4-S knockdown. Western blot analysis showed a reduction in ERK1/2 phosphorylation, whereas phosphorylation of JUN and P38, as well as total protein levels of ERK1/2, JUN, and P38, remained unchanged (Fig. 7B). Similarly, treatment of HT-29 cells with the SRSF6 phosphorylation inhibitor SPHINX31 suppressed ERK1/2 phosphorylation (Fig. 7C). To further establish a causal link between BRD4-S and MAPK/ERK pathway activation, we overexpressed BRD4-S in HT-29 cells and treated them with the clinically approved MEK inhibitor trametinib. Notably, while trametinib effectively reduced baseline ERK1/2 phosphorylation in control cells, BRD4-S overexpression not only enhanced p-ERK1/2 levels but also substantially reversed trametinib-mediated suppression of ERK phosphorylation (Fig. 7D). Finally, to assess whether BRD4-S directly regulates the transcription of ERK target genes, we performed chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR) in HT-29 cells using an anti-BRD4 antibody. The results demonstrated significant enrichment of BRD4-S at the promoter regions of CCND1 and DUSP6 compared with the IgG control (Fig. 7E). These findings suggest that BRD4-S is directly recruited to the chromatin of key ERK target genes, thereby facilitating their transcription downstream of ERK activation.
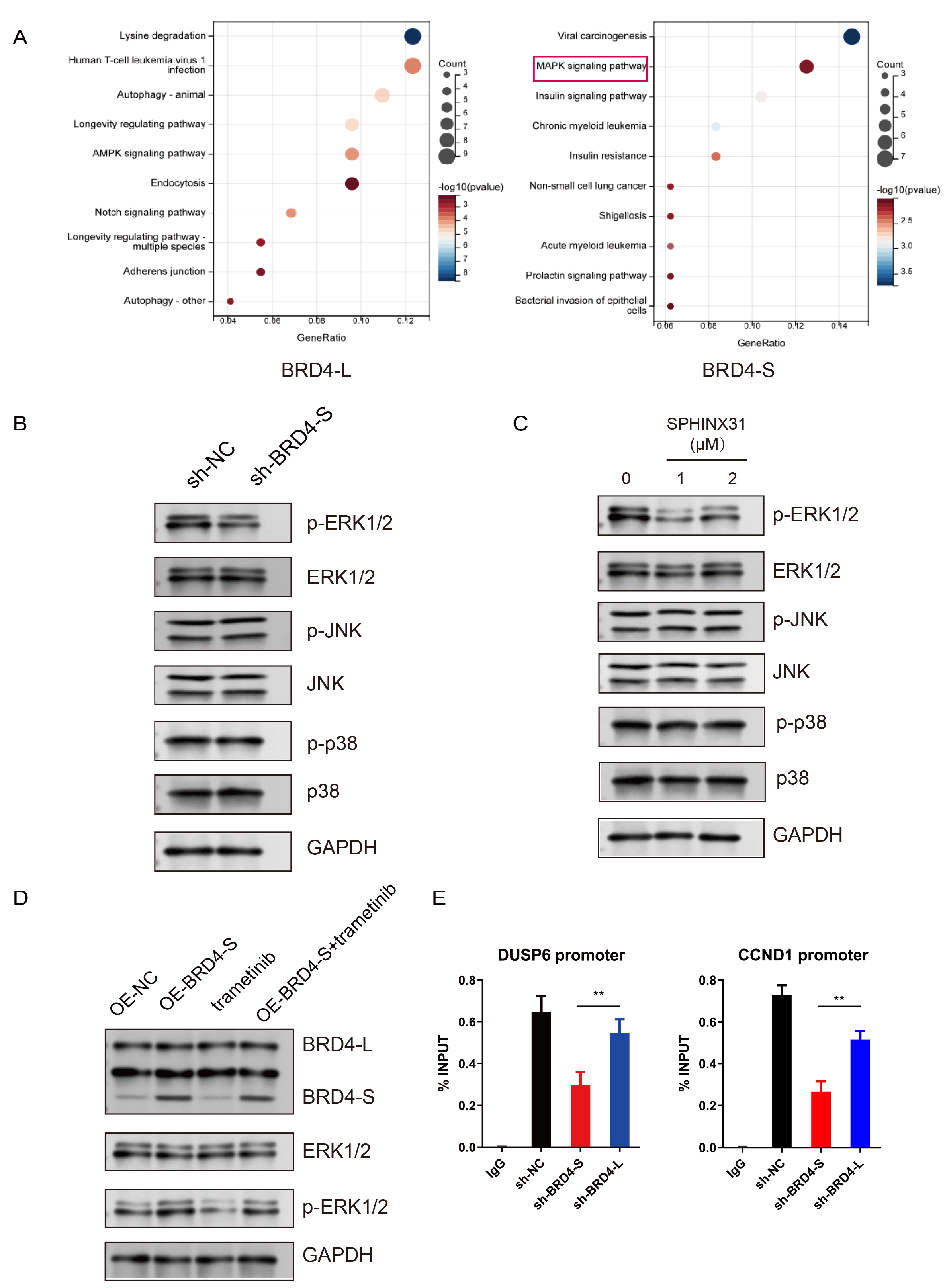 Fig. 7.
Fig. 7.
BRD4-S drives CRC progression via MAPK/ERK signaling activation. (A) GO and KEGG enrichment analyses of highly expressed DEGs in BRD4-L and
BRD4-S samples from the TCGA database. (B) Western blot analysis of
ERK1/2, p-ERK1/2, p38, p-p38, c-Jun, and p-c-Jun in HT29 cells with BRD4-S
knockdown. (C) Western blot analysis of ERK1/2, p-ERK1/2, p38, p-p38, c-Jun, and
p-c-Jun in HT29 cells treated with the SRSF6 phosphorylation inhibitor SPHINX31
(0, 1, 2 µM). (D) Western blot analysis of ERK1/2 and p-ERK1/2 in
NC–overexpressing and BRD4-S–overexpressing HT29 cells treated with or without
the MEK inhibitor trametinib (5 µM). (E) ChIP-qPCR analysis showing
enrichment of BRD4 at the promoter regions of ERK target genes CCND1 and DUSP6 in
HT-29 cells. Data are presented as mean
The rising incidence and mortality of CRC highlight the urgent need for novel therapeutic strategies and biomarkers. CRC is a heterogeneous malignancy driven by complex genetic and epigenetic alterations, with aberrant transcriptional regulation playing a central role [22]. Dysregulation of BRD4 has been implicated in tumorigenesis, metastasis, and therapeutic resistance, underscoring its potential as a therapeutic target in CRC management [23]. Moreover, elevated BRD4 expression has been linked to poor prognosis in CRC patients, emphasizing its importance in disease progression and treatment response [24]. In this study, we report for the first time the distinct regulatory roles of BRD4 isoforms in the initiation and development of CRC.
In healthy populations, BRD4-L and BRD4-S maintain a dynamic equilibrium. BRD4-L has been shown to inhibit tumor progression and suppress metastasis in specific tissues and organs affected by breast cancer [9]. By contrast, both BRD4-L and BRD4-S are markedly elevated in patients with high-grade serous ovarian carcinoma (HGSOC) [10], while in rhabdomyosarcoma, BRD4-S displays tumor-suppressive properties [25]. In tumor contexts, BRD4-L primarily acts as a transcriptional co-activator, promoting RNA transcription through RNA Polymerase II pause-release mechanisms [26, 27]. Through the mechanism of phase separation, BRD4-S drives the transcription of proliferative genes, thereby potentiating oncogenic activity in cancer cells [28, 29]. Thus, whether BRD4-L and BRD4-S function as antagonistic or synergistic regulators across different tumor types remains to be determined.
Our study demonstrates that BRD4-S plays a critical role in the progression of CRC, whereas BRD4-L exerts a less pronounced influence on tumor development. In HGSOC, although BRD4-L overexpression showed no significant effect on tumor growth or migratory capacity compared with controls, subsequent studies reported that elevated expression of both BRD4-L and BRD4-S was associated with increased patient sensitivity to combination therapy with paclitaxel and cisplatin [30, 31]. In the present work, we investigated the role of BRD4 isoforms in CRC through an integrated approach combining bioinformatics analyses, cellular assays, and molecular techniques. By delineating the specific contributions of BRD4 isoforms to CRC cell proliferation, migration, and invasion, we sought to clarify the mechanisms driving tumor progression. Our findings provide evidence for the differential regulation of target genes by BRD4 isoforms and highlight their potential impact on patient outcomes. Although BRD4-S exhibited a more pronounced oncogenic role, further studies are needed to explore alternative contributions of BRD4-L to CRC progression. Collectively, this work is expected to inform the development of targeted therapeutic strategies that exploit BRD4 dysregulation in CRC, ultimately contributing to improved clinical management.
To further investigate the regulators of BRD4-S, we conducted a comprehensive analysis of the TCGA database, which identified DDX27 as a factor uniquely correlated with BRD4-S and upregulated in CRC. DDX27, a member of the DEAD-box RNA helicase family, has been implicated in diverse cellular processes, particularly those associated with tumorigenesis and progression [32]. In GC, elevated DDX27 expression has been linked to poor patient prognosis, likely through its role in regulating the alternative splicing of metastasis-related genes [33]. Our experimental validation confirmed that DDX27 plays a critical role in regulating BRD4 alternative splicing. GO and KEGG pathway analyses revealed significant enrichment of DDX27 in spliceosome-related pathways, while further experiments demonstrated that DDX27 interacts with SRSF6 and facilitates its phosphorylation to promote BRD4 splicing. Collectively, these findings suggest that DDX27 may represent a potential therapeutic target in cancers characterized by BRD4-S dysregulation.
In many cancers, aberrant activation of the mitogen-activated protein kinase (MAPK) pathway is a well-established contributor that sustains both tumor initiation and advancement. The MAPK pathway plays a central role in regulating cell proliferation, differentiation, and survival, and its activation has been strongly associated with tumor invasiveness and metastatic potential in malignancies such as lung, breast, and CRCs [34, 35]. In our study, BRD4-S was significantly enriched within the MAPK signaling pathway. Knockdown of BRD4-S selectively inhibited the aberrant activation of downstream ERK1/2, while phosphorylation of P38 and JUN remained unaffected. These findings are consistent with our mechanistic predictions.
Our study has several limitations. The observed downregulation of the BRD4 short isoform (BRD4-S) and the associated reduction in MAPK pathway activity following DDX27 knockdown were validated exclusively in CRC cell lines. Therefore, the clinical relevance and therapeutic potential of these findings require further confirmation in more physiologically relevant models.
Collectively, our findings demonstrate that DDX27 promotes CRC progression by activating the MAPK signaling pathway through modulation of BRD4-S. These results provide novel mechanistic insights into colorectal carcinogenesis and establish a translational foundation for the development of targeted therapeutic strategies. We recommend that future research prioritize BRD4-S as a potential therapeutic target and further investigate its pathobiological roles across diverse cancer types to enhance clinical translation.
The datasets generated and analyzed during this study are available from the corresponding author upon reasonable request.
Conception and design: CW, XC and LZ. Methodology development and validation: HH and FC. Manuscript writing, review, and editing: CW and FC. Study supervision: XC. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Animal Ethics: All animal experiments in this study were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee and were approved by the Experimental Animal Ethics Committee of The Second Affiliated Hospital of Nantong University (Approval Number: P20250303-039). Human Ethics: The study was carried out in accordance with the guidelines of the Declaration of Helsinki. The study was approved by the Ethics Committee of The Second Affiliated Hospital of Nantong University (Approval Number: 2025KT080, March 17, 2025). Informed consent was obtained from all participants.
Not applicable.
This study was supported by the Wu Jieping Medical Foundation Clinical Research Special Grant Program (320.6750.2024-03-70).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL46158.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.