




 , Yuan Li 1,2,3,†, Chuanghong Lu 1,2,3, Zhongyuan Meng 1,2,3, Ling Bai 1,2,3, Feng Huang 1,2,3,*
, Yuan Li 1,2,3,†, Chuanghong Lu 1,2,3, Zhongyuan Meng 1,2,3, Ling Bai 1,2,3, Feng Huang 1,2,3,* , Zhiyu Zeng 1,2,3,*
, Zhiyu Zeng 1,2,3,*1 Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, 530021 Nanning, Guangxi, China
2 Guangxi Key Laboratory of Precision Medicine in Cardio-cerebrovascular Diseases Control and Prevention, 530021 Nanning, Guangxi, China
3 Guangxi Clinical Research Center for Cardio-cerebrovascular Diseases, 530021 Nanning, Guangxi, China
†These authors contributed equally.
Abstract
Rheumatic heart disease (RHD), which is caused mainly by Group A Streptococcus, leads to fibrotic damage to heart valves. Recently, endothelial‒mesenchymal transition (EndMT), in which activin plays an important role, has been shown to be an important factor in RHD valvular injury. However, the mechanism of activin activity and EndMT in RHD valvular injury is not clear.
Our study was divided into two parts: in vivo and in vitro. We constructed a small interfering RNA (ACVR2A-siRNA) by silencing activin receptor type IIA (ACVR2A) and an adeno-associated virus (AAV-ACVR2A) containing a sequence that silenced ACVR2A. The EndMT cell model was established via human umbilical vein endothelial cells (HUVECs), and the RHD animal model was established via female Lewis rats. ACVR2A-siRNA and AAV-ACVR2A were used in the above experiments.
EndMT occurred in the valvular tissues of RHD rats, and activin and its associated intranuclear transcription factors were also activated during this process, with inflammatory infiltration and fibrotic damage also occurring in the valvular tissues. After inhibition of ACVR2A, EndMT in valvular tissues was also inhibited, and inflammatory infiltration and fibrosis were reduced. Endothelial cell experiments suggested that mesenchymal transition could be stimulated by activin and that inhibition of ACVR2A attenuated mesenchymal transition.
Activin plays an important role in signal transduction during EndMT after activation, and inhibition of ACVR2A may attenuate RHD valvular damage by mediating EndMT. Targeting ACVR2A may be a therapeutic strategy to alleviate RHD valvular injury.
Keywords
- activin
- ACVR2A
- rheumatic heart disease
- endothelial–mesenchymal transition
- valvular injury
Rheumatic heart disease (RHD) is an immune-mediated disease triggered by group A streptococcal (GAS) bacteria [1]. Although the worldwide prevalence of RHD has decreased in low- and middle-income countries, this disease remains a major public health challenge, predominantly affecting younger people, including children, teenagers, and young adults, in these regions [2, 3]. Globally, the number of RHD patients has surpassed a quarter of the total number of cancer patients, and according to relevant data, the number of RHD-related deaths is as high as 250,000 annually [4]. RHD typically results in damage to valves, and almost all cases of heart valvular damage caused by RHD involve the mitral valve [5]. The pathogenic mechanism of RHD and methods for preventing and treating RHD have not been fully elucidated in the last decade of research [6]. Patients with rheumatic mitral valvular dysfunction may undergo surgical repair [7]. However, the adverse effects and complications associated with valvular surgery are far-reaching [8]. Thus, the identification of novel therapeutic approaches and related targets for treating RHD valvular injury is urgently needed.
The mitral valve consists of two layers of endothelial cells closely covering
connective tissue. If the mitral valve is damaged, the endothelial cells become
irritated and inflamed, and when the connective tissue is involved, a fibrotic
scar can form that is directly associated with valvular disease [9].
Endothelial–mesenchymal transition (EndMT), which is linked to endothelial
damage, involves the transformation of endothelial cells from their
characteristic state (expressing vascular endothelial (VE)-cadherin and platelet
endothelial cell adhesion molecule-1 (PECAM-1/CD31)) to a mesenchymal phenotype
(expressing
Activin belongs to the TGF-
Activin is able to bind strongly to activin receptor type II, which includes activin receptor type IIA (ACVR2A) and activin receptor type IIB (ACVR2B) [33]. In recent years, ACVR2A has received much attention in the study of antifibrosis and mesenchymal transition [34, 35]. To gain insight into the relevance and mechanism of action of activin and EndMT in RHD valvular injury, the present study was conducted in vivo and in vitro by silencing ACVR2A. In our in vitro study, we chose human umbilical vein endothelial cells (HUVECs) as our study subjects, and we evaluated the role of activin in the mesenchymal transition of HUVECs via the use of recombinant proteins. Moreover, we constructed a small interfering RNA (ACVR2A-siRNA) that silenced ACVR2A and applied it to a cell model of EndMT to explore its effect. In our in vivo studies, we constructed an adeno-associated virus (AAV-siACVR2A) loaded with silenced ACVR2A sequences, applied it to an animal model of RHD established in female Lewis rats, and explored the role of AAV-siACVR2A by evaluating the valvular organization of the animal model.
This study contributes to the understanding of the specific pathogenic mechanisms of RHD valvular injury and provides a useful reference for the prevention and treatment of RHD valvular injury.
Group A Streptococcus (GAS, American Type Culture Collection) was grown in
brain-heart culture medium (024053, Guangdong Huankai Microbial Technology Co.,
Ltd., Guangdong, China) at a steady temperature of 37 °C. After 24
hours, the GAS bacteria were rinsed with normal saline (NS) and then inactivated
by immersion in 4% paraformaldehyde (P1110, Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). After inactivation, the GAS bacteria were
rinsed and placed in NS at a concentration of 4.0
Twenty-four healthy female Lewis rats, aged 10 weeks, were obtained from Beijing
Viton Lihua Laboratory Animal Technology Co., Ltd. (Beijing, China). The rats
weighed between 160 and 180 g. The rats were kept in a laboratory at Guangxi
Medical University’s Animal Experiment Center, which is a specific pathogen-free
(SPF) room. The temperature was maintained at 23
The experimental rats were pretreated with a recombinant adeno-associated virus (AAV) vector (AAV-siACVR2A; Hanheng Biotechnology (Shanghai) Co., Ltd., Shanghai, China) carrying the ACVR2A-siRNA sequence. To analyze the possible effects of the AAV vector on the rats, a negative control group (AAV-control group) was also established in which the rats were pretreated with AAV without the target sequence. The siRNA sequence of the ACVR2A target gene was 5′-GGAAGTTGTTGTGCATAAA-3′. The ACVR2A target gene shRNA sequences were as follows: top strand, 5′-AATTCGGAAGTTGTTGTGCATAAACTCGAGTTTATG CACAACAACTTCCTTTTTTG-3′; and bottom strand, 5′-GATCCAAAAGGAAGTTGTTGTGCATAAACTCGAGT TTATGCACAACAACTTCCG-3′.
The rats were arbitrarily grouped into four groups (6 rats/group): the control
group, RHD group, RHD+AAV-control group (hereinafter referred to as the
AAV-control group) and RHD+AAV-siACVR2A group (hereinafter referred to as the
AAV-siACVR2A group). The RHD experimental model was constructed over 8 weeks via
an approach described in prior investigations [37, 38]. First, a combination of
GAS (4.0
The mitral valve specimens from dead rat hearts were collected in a timely manner, rapidly transferred to liquid nitrogen, and then stored at –80 °C for additional experimental analysis.
After incubation in 4% paraformaldehyde for 12 hours, the tissue blocks were dehydrated through a range of ethanol concentrations. Afterward, the tissue blocks were promptly transferred into molten paraffin to maintain the temperature and subsequently embedded once the paraffin fully enveloped the tissue blocks. Each block was sequentially sliced into 5 µm thick sections for hematoxylin and eosin (H&E) staining and Sirius red staining. The H&E staining process involved immersing the samples in a hematoxylin solution at ambient temperature for 4–10 minutes, followed by 2–3 minutes of immersion in an eosin staining solution containing alcohol. H&E staining kits (G1005) were purchased from Wuhan Servicebio Technology Co., Ltd (Wuhan, China). A BX43 light microscope (BX43, Olympus Corporation, Tokyo, Japan) was used to record the results. The Sirius red staining process involved the use of Sirius red staining solution (MM1036, Shanghai Maokang Biotechnology Co., Ltd., Shanghai, China) at ambient temperature for one hour. Polarized light pictures were then captured with a BX43 confocal microscope (BX43, Olympus Corporation, Tokyo, Japan).
Immunohistochemical analysis was conducted via the use of purified polyclonal
antibodies against rat CD31,
HUVECs (Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd., Shanghai, China)
were grown in endothelial cell medium (ECM, 1001, ScienCell Research
Laboratories, Carlsbad, CA, USA) at 37 °C and 5% CO2. HUVECs were
stimulated with 10 ng/mL recombinant human TGF-
Hanheng Biotechnology (Shanghai) Co., Ltd. (Shanghai, China). provided the siRNAs. HUVECs were transfected with siRNAs via Lipofectamine 3000 (L3000015, Thermo Fisher Scientific, Inc., Waltham, MA, USA). Two microliters of siRNA and 5 µL of Lipofectamine 3000 transfection reagent were added to 250 µL of Opti-MEM (31985070, Thermo Fisher Scientific) solution, mixed separately, and left to stand to form the siRNA-Lipofectamine 3000 complex. The complex was used to treat HUVECs. The ACVR2A-siRNA used were as follows: forward, 5′-GGAAGUUGUUGUGCAUAAATT-3′ and reverse, 5′-UUUAUGCACAACAACUUCCTT-3′. The negative control (NC)-siRNA used were as follows: forward, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse, 5′-ACGUGACACGUUCGGAGAATT-3′.
TRIzol® reagent (15596026CN, Thermo Fisher Scientific,
Inc., Waltham, MA, USA) was used to extract total RNA from heart valves or
cultured HUVECs. The RNA was converted into cDNA via a reagent kit (YFXM0009,
YIFEIXUE Biotech. Co., Ltd., Nanjing, China). RT-qPCR was conducted with
2
| Gene (rat) | 5′–3′ | |
| Forward | GCGTGGCTATTCCTTCGTGACTAC | |
| Reverse | CATCAGGCAGTTCGTAGCTCTTCTC | |
| Col1a1 | Forward | TGTTGGTCCTGCTGGCAAGAATG |
| Reverse | GTCACCTTGTTCGCCTGTCTCAC | |
| Col3a1 | Forward | AGTCGGAGGAATGGGTGGCTATC |
| Reverse | CAGGAGATCCAGGATGTCCAGAGG | |
| Ve-cadherin | Forward | GATGCAGAGGCTCATGATGC |
| Reverse | CTTGCGACTCACGCTTGACT | |
| Acvr2a | Forward | CTTGCTCTTCAGGTGCTATACTTGG |
| Reverse | GTCTGATTGGTTCTGTCTCTTTCCC | |
| Smad2 | Forward | GTCGTCCATCTTGCCATTCACTC |
| Reverse | GTTCTCCACCACCTGCTCCTC | |
| Smad3 | Forward | AGGGCTTTGAGGCTGTCTACC |
| Reverse | TGCTGGTCACTGTCTGTCTCC | |
| Lef-1 | Forward | CACACAACTGGCATCCCTCATC |
| Reverse | GGCTCCTGTTCCTTTCTCTGTTC | |
| Snail1 | Forward | CCGACCGCTCCAACCTACG |
| Reverse | GCAGCCAGACTCTTGGTGTTTG | |
| Twist | Forward | TGAGCAACAGCGAGGAGGAG |
| Reverse | CCGACTGCTGCGTCTCTTG | |
| Zeb1 | Forward | GCACAGCCAAGCACAGAAGAG |
| Reverse | TGGAGAAGGTGGTTCAAGAGACTG | |
| Zeb2 | Forward | GAGATAAGGGAGAGCGTTGTG |
| Reverse | AATTGTGGTCTGGATCGTGG | |
| Forward | GGAGATTACTGCCCTGGCTCCTA | |
| Reverse | GACTCATCGTACTCCTGCTTGCTG | |
| Gene (human) | 5′–3′ | |
| ACVR2A | Forward | GGAACTGGCTTCTCGCTGTACTG |
| Reverse | GCACAACAACTTCCTGCATGTCTTC | |
| SMAD2 | Forward | CGCCGCCAGTTGTGAAGAGAC |
| Reverse | TCCTGCCCATTCTGCTCTCCTC | |
| SMAD3 | Forward | GGAGCGGAGTACAGGAGACAGAC |
| Reverse | CTAAGACACACTGGAACAGCGGATG | |
| LEF-1 | Forward | CACACAACTGGCATCCCTCATCC |
| Reverse | GCTCCTGCTCCTTTCTCTGTTCATG | |
| SNAIL1 | Forward | CCTCGCTGCCAATGCTCATCTG |
| Reverse | CTCTGCCACCCTGGGACTCTC | |
| TWIST | Forward | GACTTCCTCTACCAGGTCCTCCAG |
| Reverse | TCCAGACCGAGAAGGCGTAGC | |
| ZEB1 | Forward | AGTGTTACCAGGGAGGAGCAGTG |
| Reverse | TTTCTTGCCCTTCCTTTCCTGTGTC | |
| ZEB2 | Forward | TGACCTGCCACCTGGAACTCC |
| Reverse | GGCGGTACTTGATGTGCTCCTTC | |
| VE-CADHERIN | Forward | GTACCACCTCACTGCTGTCATTG |
| Reverse | CAGGCACGGACGCATTGAAC | |
| Forward | CTTCGTTACTACTGCTGAGC-GTGAG | |
| Reverse | CCCATCAGGCAACTCGTA-ACTCTTC | |
| VIMENTIN | Forward | ATCGATGTGGATGTTTCCAA |
| Reverse | TTGTACCATTCTTCTGCCTC | |
| GAPDH | Forward | TGACATCAAGAAGGTGGTGAAGCAG |
| Reverse | GTGTCGCTGTTGAAGTCAGAGGAG |
RT-qPCR, real-time quantitative polymerase chain reaction;
PMSF (P0100, Solarbio Science & Technology Co., Ltd., Beijing, China), protein
phosphatase inhibitor (P1260, Solarbio), and RIPA buffer (R0010, Solarbio) were
made into a cell lysate with a ratio of 1:1:100. Valve tissues or cells were
lysed for a period of 1 hour. Protein concentration was determined by BCA assay
and individual samples contained 30 mg of protein. Then electrophoresis on
7.5–12.5% sodium dodecyl sulfate (SDS)/polyacrylamide gel electrophoresis
(PAGE) gels, followed by transfer to polyvinylidene fluoride (PVDF) membranes
(ISEQ00005, EMD Millipore, Darmstadt, Germany). The membranes were blocked with
Protein-Free Rapid Sealing Solution (PS108P, Shanghai Epizyme Biomedical
Technology Co., Ltd., Shanghai, China) for 30 min at room temperature, and
followed by the following specific primary antibodies incubated overnight at 4 °C:
anti-GAPDH (1:10,000, 10494-1-AP, Proteintech), anti-Activin A (1:500, ab89307,
Abcam, Cambridge, UK), anti-ACVR2A (1:1000, ab134082, Abcam), anti-p-Smad2
(1:500, AF3449, Affinity Biosciences, Jiangsu, China), anti-p-Smad3 (1:500,
AF3362, Affinity), anti-Smad2 (1:2000, 12570-1-AP, Proteintech), anti-Smad3
(1:500, AF6362, Affinity, or 1:500, PAC123Hu01, Cloud-Clone Corp), anti-LEF1
(1:1000, ab137872, Abcam), anti-Snail1 (1:500, 13099-1-AP, Proteintech),
anti-TWIST (1:1000, 25465-1-AP, Proteintech), anti-ZEB1 (1:500, 21544-1-AP,
Proteintech), anti-
HUVECs were added to a 6-well plate. Once the HUVECs had adhered and proliferated, a pipette tip was used to gently make a wound in the central area of the cell layer. PBS was added for rinsing, and then, serum-free medium was added for culture. Images of the scratches were acquired with a phase contrast microscope at identical positions for 0 hours and 24 hours. The cell migratory capacity was assessed on the basis of the extent of healing observed in the scratched area.
HUVECs were transfected with ACVR2A-siRNA and treated with activin A. After they reached a sufficient treatment time, they were digested with trypsin. Then, 200 µL of HUVECs were combined with serum-free medium and transferred to the upper chamber of a 24-well dish with an 8 µm well. Additionally, 600 µL of HUVECs mixed with 10% FBS medium was transferred to the bottom chamber. After a 12-hour incubation period, HUVECs were stabilized with 4% paraformaldehyde for 15 minutes and then stained with crystal violet solution (G1064, Solarbio) for 20 minutes. Images of HUVECs located on the underside of the luminal membrane were subsequently captured and quantitatively analyzed via an inverted microscope.
GraphPad Prism 9.5 (GraphPad Software, San Diego, CA, USA) was used for
statistical analysis of the data. The data are shown as the means
H&E staining revealed increased inflammatory infiltration in the mitral valvular tissues of rats in the RHD group. However, this inflammatory infiltration was reduced after the inhibition of ACVR2A (Fig. 1A). Sirius red staining was used to identify the collagen type, with type I collagen (COL1) appearing as tightly packed yellow and red fibers and type III collagen (COL3) appearing as loosely packed green fibers under a polarized light microscope. In nonfibrotic valves, COL1 is the primary collagen type, while the proportion of COL3 steadily increases as fibrosis progresses. As fibrosis progresses, the proportion of COL3 gradually increases, and an increase in the COL3/COL1 (COL3/1) ratio can be used to detect the onset of valvular fibrosis [42]. Our results revealed that the COL3/1 ratio was elevated in the RHD group. However, after inhibition of ACVR2A, the COL3/1 ratio decreased (Fig. 1B). The above results indicated that inflammatory infiltration and fibrotic changes occurred in the mitral valvular tissues of the rats in the RHD group, whereas the inflammatory infiltration and fibrosis of the valvular tissues were reduced after the inhibition of ACVR2A.
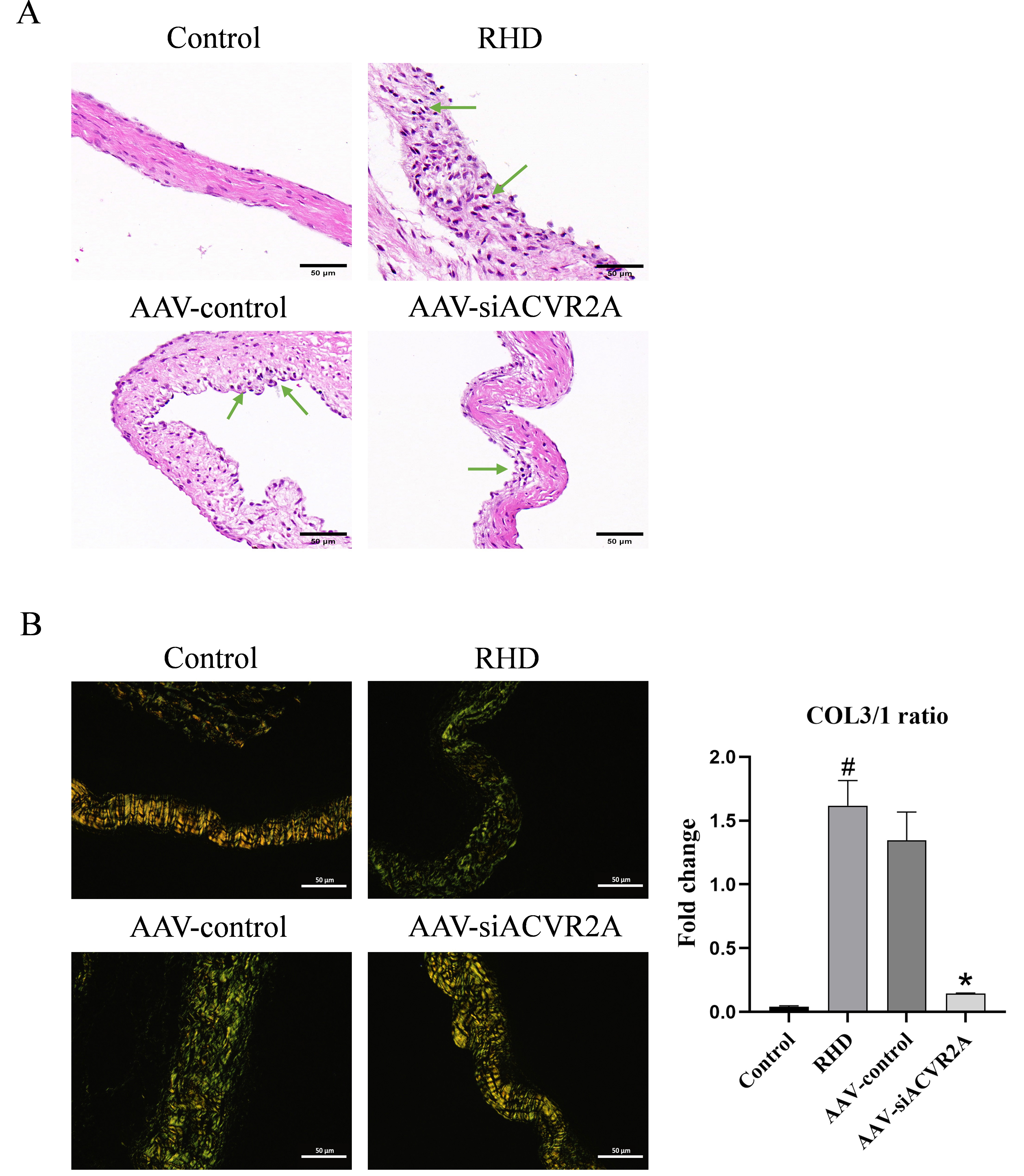 Fig. 1.
Fig. 1.
AAV-siACVR2A attenuates the inflammatory response and fibrosis
in RHD-affected valves. (A) H&E staining showing the severity of inflammatory
cell infiltration in the mitral valvular tissues of the rats in each group.
Magnification,
The immunohistochemistry results revealed that the expression of
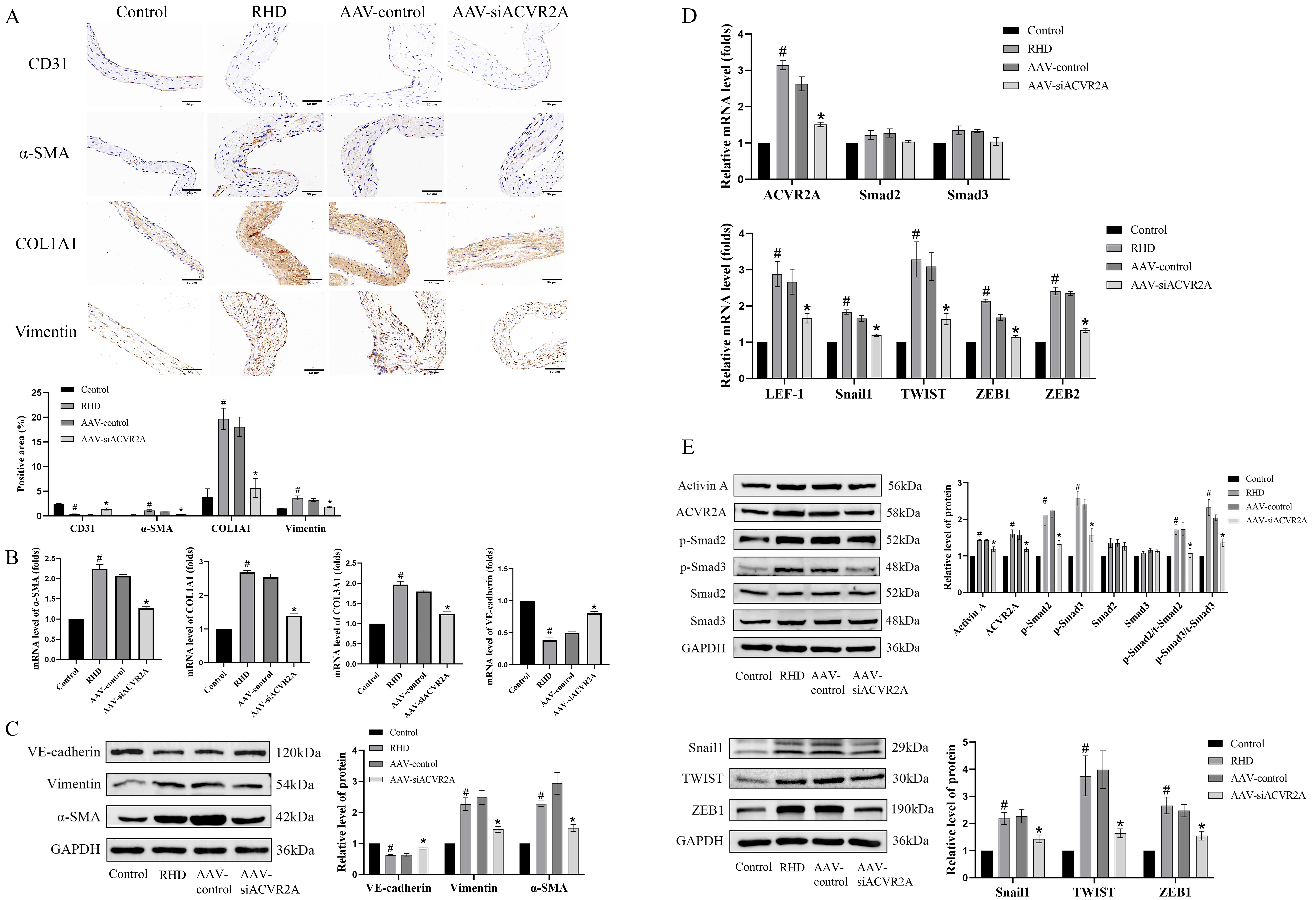 Fig. 2.
Fig. 2.
AAV-siACVR2A inhibited activin-related pathway-mediated endothelial‒mesenchymal transition (EndMT) in
RHD-affected valves. (A) Immunohistochemical staining for CD31,
RT-qPCR analysis revealed that the mRNA levels of ACVR2A, LEF-1, Snail1, TWIST, ZEB1, and ZEB2 were significantly elevated in the RHD group, but these metrics decreased after the inhibition of ACVR2A. The mRNA expression of Smad2 and Smad3 did not significantly differ between the groups (Fig. 2D). Western blotting analysis revealed that the protein expression of activin A, ACVR2A, p-Smad2, p-Smad3, Snail1, TWIST and ZEB1 was elevated in the RHD group, whereas these indices were reduced in the AAV-siACVR2A group. The protein expression of Smad2 and Smad3 did not differ significantly among the groups (Fig. 2E). The above results indicate that during EndMT in RHD valvular tissues, activin is activated, and the expression of ACVR2A is subsequently elevated, which leads to the phosphorylation of Smad2/3 and then the activation of the intranuclear transcription factors involved in EndMT. In contrast, after the inhibition of ACVR2A, activin did not effectively phosphorylate Smad2/3, and related intranuclear transcription factors were not effectively activated, thus inhibiting EndMT in the valvular tissues of RHD rats.
RT-qPCR analysis revealed that the mRNA expression of ACVR2A, LEF-1, Snail1,
TWIST, ZEB1, ZEB2,
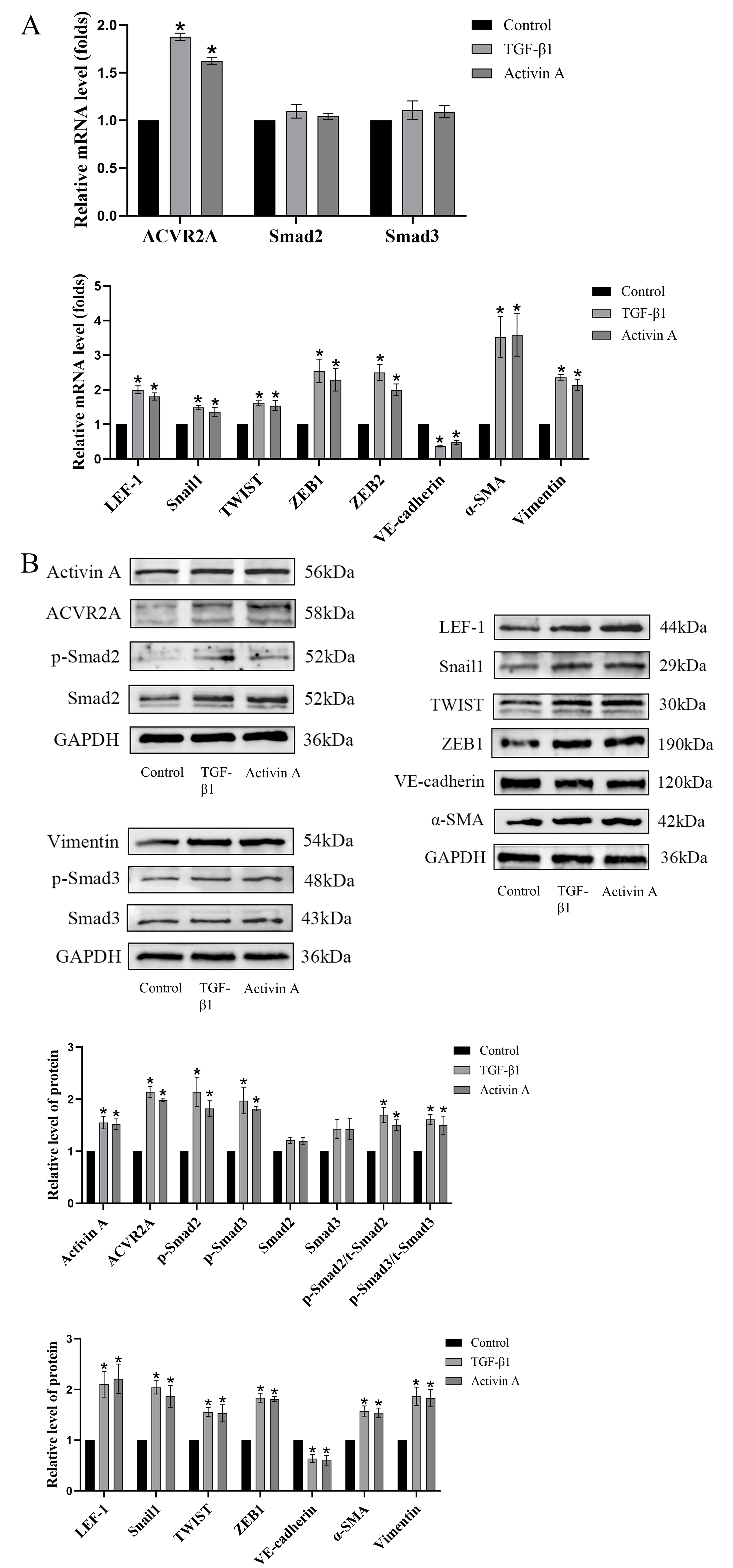 Fig. 3.
Fig. 3.
Activin A promoted EndMT in human umbilical vein endothelial
cells (HUVECs). (A) RT-qPCR was performed
to measure the mRNA expression levels of activin pathway-related factors and
EndMT-related factors in HUVECs during EndMT. (B) Western blot analysis of the
protein expression of activin pathway-related factors and EndMT-related factors
in HUVECs during EndMT. *p
RT-qPCR analysis revealed that the mRNA levels of ACVR2A, LEF-1, Snail1, TWIST,
ZEB1, ZEB2,
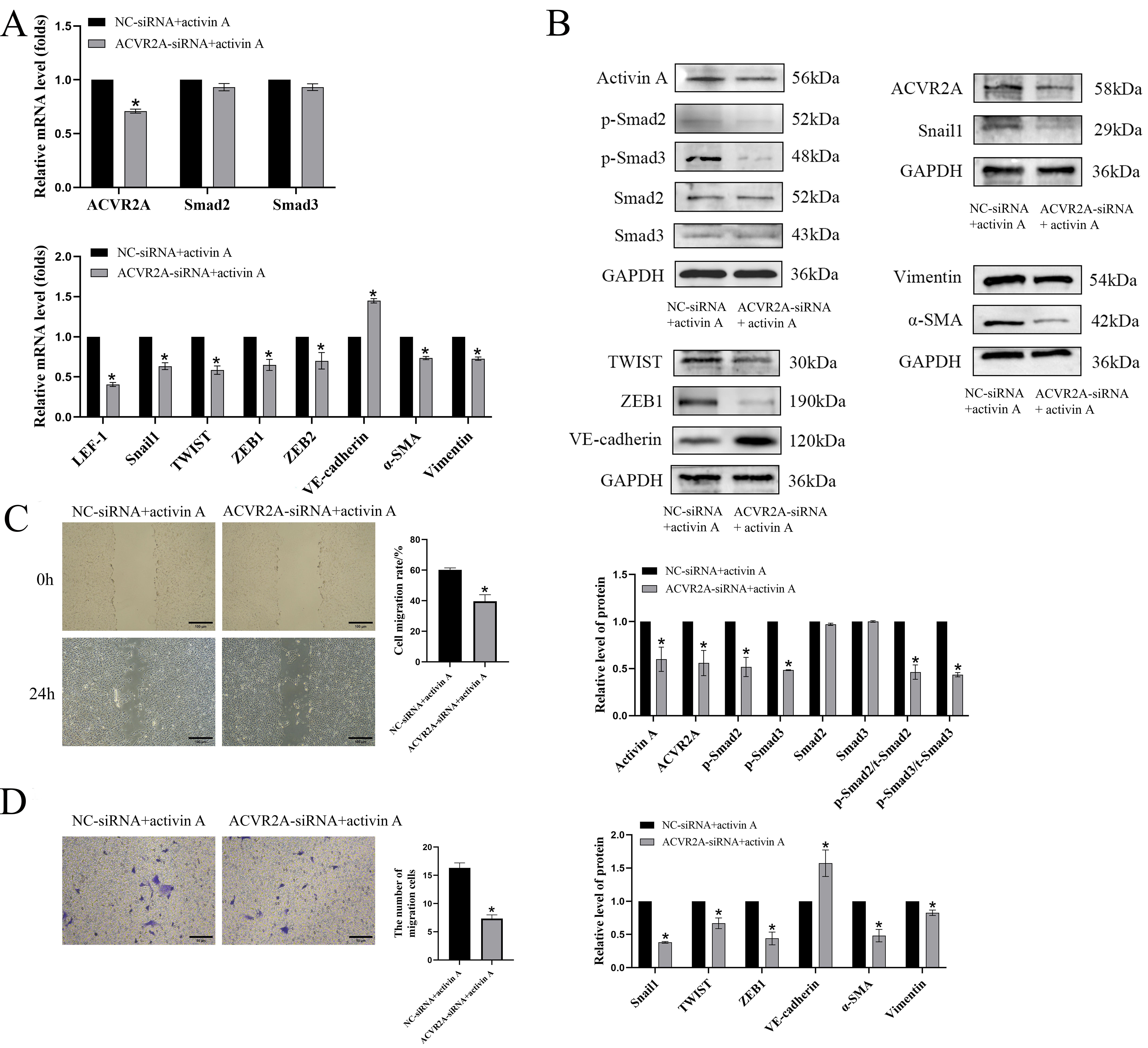 Fig. 4.
Fig. 4.
ACVR2A-siRNA inhibited EndMT in HUVECs. (A) RT-qPCR analysis of
the mRNA expression levels of activin pathway-related factors and EndMT-related
factors in HUVECs after ACVR2A-siRNA treatment. (B) Western blot analysis of the
protein expression of activin pathway-related factors and EndMT-related factors
in HUVECs after ACVR2A-siRNA treatment. (C) Cell scratch assay using HUVECs.
Magnification,
EndMT is a complex biological process by which endothelial cells lose their endothelial phenotype and acquire a mesenchymal phenotype [10, 11]. EndMT has been demonstrated to be involved in the development of the atrioventricular valve and is associated with the development of various inherited and acquired diseases, such as malignant, cardiovascular, inflammatory and fibrotic diseases. In the cardiovascular field, atherosclerosis, pulmonary hypertension, valvular heart disease and cardiac fibrosis are all associated with abnormal EndMT [43]. Notably, RHD is a disease that involves fibrotic changes in heart valves, and the role of EndMT in RHD valvular damage has recently been studied [32]. In our study, EndMT occurred in the valves of the RHD model rats. It is associated with mitral fibrosis.
The TGF-
In our study, we revealed inflammatory infiltration and fibrotic changes in the valvular tissues of rats in the RHD disease model, and during the above pathology, the valvular tissues underwent EndMT, which was accompanied by the activation of activin and associated pathway factors. However, after inhibition of ACVR2A, activin pathway factor signaling was also suppressed, and mesenchymal indices were reduced, suggesting that EndMT in valvular tissues was suppressed and that inflammatory infiltration and fibrotic changes in valvular tissues were simultaneously reduced. In our in vitro study, activin A stimulated the mesenchymal transformation of endothelial cells, which was consistent with previous findings [49]. After inhibition of ACVR2A, mesenchymal transformation of endothelial cells was also inhibited. These results indicated that activin was activated by binding to ACVR2A, activated relevant intranuclear transcription factors via the Smad2/3 phosphorylation pathway, and caused mesenchymal transformation of endothelial cells, which, in turn, could lead to inflammatory infiltration and fibrotic changes in the valvular tissues of RHD rats. After inhibition of ACVR2A, there was a reduction in damage to valvular tissues. Because there is a relative paucity of treatments for RHD valvular injury, we believe that targeting ACVR2A may be a promising approach for the treatment of RHD valvular injury.
This study has several limitations. Firstly, we did not extract primary endothelial cells from the mitral valvular tissues of the RHD rat model, and further extraction of the primary valvular endothelium is needed for validation. Secondly, our conclusions were based on the RHD rat model, which does not accurately describe the pathomechanisms of human RHD valvulopathy accurately, and more subsequent combinatorial studies are needed to confirm these findings.
In conclusion, the present study demonstrated that inhibition of ACVR2A could mediate EndMT to attenuate valvular injury in RHD rats, further elucidating the mechanism of action of activin and EndMT in RHD valvular injury.
The raw data supporting the conclusions of this article will be made available
by the correspondence author
ZYZ, FH and CHL conceived and designed the study. ZRL and YL participated in the experimental design. ZRL and YL conducted the experiments. ZYM and LB analyzed the data. ZRL and YL drafted the manuscript. All authors contributed to editorial changes in the manuscript. All authors approved the final version. All authors agree to be accountable for all aspects of the work to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The protocol involving animals and patients has been approved by the Medical Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (Approval Number: 202209145).
The authors would like to express their gratitude to American Journal Experts (https://www.aje.cn) for the expert linguistic services provided.
This work was supported by the National Natural Science Foundation of China (Grant No. 81960082), the Guangxi Key Laboratory of Precision Medicine in Cardio-cerebrovascular Diseases Control and Prevention (Grant No. 22-035-18), the Guangxi Clinical Research Center for Cardio-cerebrovascular Diseases (Grant No. AD17129014) and the Guangxi Medical High-level Backbone Talents “139” Program (Grant No. G201901006).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.