





1 Biomedical Institute for Multimorbidity (BIM), Hull York Medical School (HYMS), University of Hull, HU6 7RX Hull, UK
Abstract
Cardiovascular complications claim the lives of up to 70% of patients with diabetes mellitus (DM). The mechanisms increasing cardiovascular risk in DM remain to be fully understood and successfully addressed. Nonetheless, there is increasing evidence in the scientific literature of the participation of platelets in the cardiovascular complications of DM. Multiple reports describe the hyperactivity of platelets in DM and their participation in inflammatory responses. The understanding of the mechanisms underlying the contribution of platelets to cardiovascular pathologies in DM will help the development of targeted therapeutic strategies able to reduce cardiovascular risk in these patients. In this literature review, we summarise our current understanding of the molecular mechanisms leading to the contribution of platelets to cardiovascular risk in DM. Both platelet haemostatic activity leading to thrombus formation and their participation to inflammatory processes are stimulated by the biochemical conditions associated with DM. We also present evidence on how DM affect the efficacy of existing therapeutic treatments for thrombosis and, by converse, how antidiabetic drugs may affect platelet function and the haemostasis/thrombosis balance. Taken together, the growing evidence of the different and unexpected roles of platelets in the progression of DM provides a strong rationale for the design of cardiovascular drugs targeting specifically platelets, their pro-inflammatory activity and their activation mechanisms in this disease. Overall, this article provides an important up-to-date overview of the pathophysiological alterations of platelets in DM, which need to be taken into account for the effective management of cardiovascular health in this disease.
Keywords
- diabetes
- platelet
- thrombosis
- inflammation
- hyperactivity
- cardiovascular
Platelets are anucleate cells representing the second most common blood
component (after erythrocytes). Upon vascular damage, platelets bind to collagen
from the vascular and perivascular tissue (Fig. 1). The binding of von Willebrand
factor (vWF) to collagen promotes the initial deposition of platelets mediated by
the glycoprotein Ib-IX-V (GP-Ib-IX-V) receptor. This event occurs in conditions
of high shear stress and facilitates the direct interaction of the glycoprotein
VI (GPVI) receptor and the integrin alpha2-beta1 of platelets with collagen.
After adhesion to collagen, platelets become activated, leading to their change
of shape from discoid to their activated state with pseudopodia and subsequently,
degranulation of
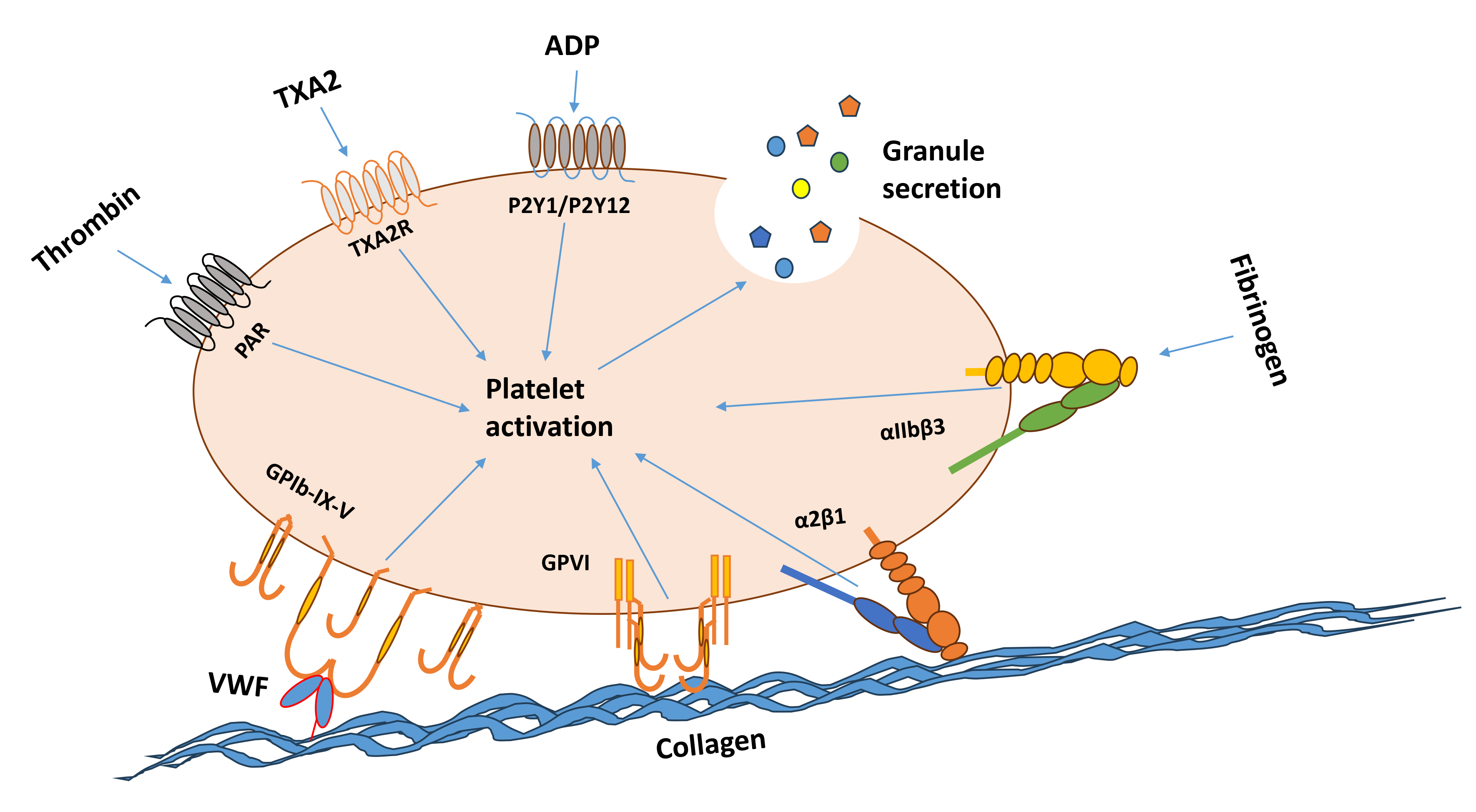 Fig. 1.
Fig. 1.
Schematic representing physiological stimuli of platelet activation. Plasma proteins responsible for platelet adhesion VWF and fibrinogen are shown with their receptors GP-Ib-IX-V receptor and integrin alphaIIb-beta3, respectively. Primary platelet agonists collagen and thrombin are represented with their receptors GPVI/integrin alpha2-beta1 and PARs, respectively. Secondary platelet agonists ADP and thromboxane A2 are displayed with their main receptors P2Y1/P2Y12 and TXA2R, respectively. The release of the content of alpha and dense granules in response to platelet activation is also shown. ADP, adenosine diphosphate; GP-Ib-IX-V, glycoprotein Ib-IX-V; GPVI, glycoprotein VI; PAR, protease-activated receptor; TXA2, thromboxane A2; TXA2R, thromboxane A2 receptor; VWF, von willebrand factor. This diagram was made using Microsoft PowerPoint.
Since inflammatory responses have been recognised as key components of diabetes mellitus (DM) and its comorbidities, in this section of the review, we present key studies that led to our current understanding of the role of platelets in inflammation. The majority of studies on the role of platelets in inflammation were conducted in preclinical models or clinical samples for diseases different from DM, but the mechanisms that they have highlighted are likely to play a role also in the cardiovascular comorbidities of DM. The validation of platelet-dependent mechanisms of inflammation in DM and the development of targeted therapeutic approaches are promising research fields for the advancement of the healthcare standards in DM. Platelets are considered key players in the immune response and a wide array of inflammatory diseases. To understand the role platelets, play in inflammation, it is important to consider the various ways platelets can promote the establishment of an inflammatory microenvironment. Platelets express surface receptors typically involved in immune response (sections 2.1–2.2) and can release cytokines and other inflammatory mediators (sections 2.3). Moreover, platelets can release pro-inflammatory microvesicles (section 2.4) and interact with leukocytes to modulate their ability to adhere and respond to their surroundings in inflammatory conditions (sections 2.5 and 2.6).
In platelets, TLR expression and activation result in the activation,
aggregation, and release of inflammatory mediators such as cytokines. Although
most platelet TLRs are expressed at low levels, TLR2, 4 & 9 are expressed at
levels sufficiently high to suggest a functional role [2, 3, 4]. Pam3CSK4
(i.e., a synthetic ligand mimicking bacterial lipopeptide) activates platelets
leading to “inside-out”
The CD40L/CD40 binding axis was first described for its effect on platelet
function and thrombus formation in 1998 [11]. They found that when platelets are
stimulated with different agonists (i.e., thrombin, collagen, adrenaline, and
ADP) the surface expression of CD40L is increased via translocation. Thus,
triggering an inflammatory response in cells expressing CD40 such as endothelial
cells. CD40L-stimulated endothelial cells secrete chemokines and express adhesion
molecules leading to recruitment and extravasation of leukocytes. CD40L is
rapidly presented to the platelet surface after platelet stimulation. The
surface-expressed CD40L is subsequently cleaved over a period of minutes to
hours, generating a soluble fragment termed soluble CD40 ligand (sCD40L). The pro-thrombotic role of
platelet CD40L has been demonstrated by André and colleagues [12]. In their
study, these authors found that CD40L genetic silencing reduced the stability of
arterial thrombi and delayed arterial occlusion in vivo, while Infusion
of recombinant soluble CD40L (rsCD40L) restored normal thrombosis. rsCD40L lacking the
lysine-glycine-aspartate (KGD) integrin-recognition sequence did not restore normal thrombosis in
CD40L-/- mice, suggesting that CD40L potentiate platelet aggregation in an
integrin
Platelets also store and release many cytokines and chemokines within their alpha granules (Table 1, Ref. [13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27]). Platelets typically release cytokines when activated and depending on the agonist, the cytokines they release are different.
| Cytokines | Function/s | Reference |
| RANTES/ CCL5 | [13, 14, 15, 16] | |
| CXCL4 | [16, 17, 18] | |
| CXCL7 | [19, 20] | |
| CXCL12 | [18, 21] | |
| CXCL16 | [22, 23] | |
| CXCL7 & NAP-2 | [19, 20] | |
| CXCL3 | [18, 24] | |
| CXCL5 | [25, 26, 27] | |
List of abbreviations in the table: CXCL4, Chemokine (c-x-c motif) ligand 4; CXCL7, Chemokine (c-x-c motif) ligand 7; CXCL12, Chemokine (c-x-c motif) ligand 12; CXCL16, Chemokine (c-x-c motif) ligand 16; CXCL3, Chemokine (c-x-c motif) ligand 3; CXCL5, Chemokine (c-x-c motif) ligand 5; NAP-2, neutrophil-activating peptide-2; RANTES, regulated on activation, normal T-cell expressed and secreted; T2DM, type 2 diabetes mellitus.
Hundelshausen and colleagues [13] demonstrated that regulated on activation, normal T-cells expressed and secreted (RANTES) deposition by platelets is involved in monocyte arrest to inflamed endothelium. More recently, it has been discovered that RANTES binding to inflamed endothelial is important for CD4+ T-cells homing to atherosclerotic regions where they exacerbate atherosclerosis where C-C chemokine receptor type 5 (CCR5) and RANTES interaction is essential [28].
Another important cytokine expressed and released by platelets is Chemokine(c-x-cmotif) ligand 4 (CXCL4) or platelet factor 4 (PF4). Not only is PF4 expressed by platelets but it can also be synthesised by monocytes [29]. PF4 has many functions, including the activation of platelets [30]. PF4 binds and activates the platelet thrombopoietin receptor, cellular myeloproliferative leukaemia protein (c-Mpl), which activates Janus kinase 2 (JAK2), the signal transducer and activator of transcription 3 (STAT3), and STAT5, leading to platelet aggregation. Additionally, it has a role in immunity where it can interact with bacteria and has a role in anti-microbial defence [31]. PF4 is highly expressed in platelets in atherosclerotic carotid arteries [32] and involved in platelet-T-cell interactions [33]. It has been found to synergise with RANTES by forming heterodimers that enhance the activity of RANTES, which leads to increased monocyte recruitment and adhesion in an abdominal aortic aneurysm (AAA) mouse model [16].
CXCL7 is found in platelets where it can be degraded into several derivatives with a role in immunity and inflammation, including neutrophil-activating peptide-2 (NAP-2), which can activate neutrophils [20]. It behaves as a chemoattractant for neutrophils by facilitating their migration through platelet thrombi.
CXCL12 also known as Stromal Cell Derived Factor-1
Platelets also release CXCL16 in response to agonists such as ADP and oxidised low density lipoprotein (ox-LDL). The role of platelet-derived CXCL16 in inflammation is unclear, however, it is thought to be involved in atherosclerosis [22]. Platelets derived from patients with either Acute Coronary Syndrome (ACS) [23] or their first ischemic stroke [35] were found to have significantly increased expression of CXCL16 in comparison to control donors.
Finally, CXCL5 (ENA-78) is a platelet-derived chemokine which is released by platelets upon activation [25]. In peripheral blood from patients with coronary artery disease (CAD) there was a significant increase in the release of CXCL5 in comparison to the control. The authors showed that the release of CXCL5 in response to ox-LDL contributes to inflammatory interactions of platelets and PBMC in CAD.
The earliest characterisation of platelet-derived extracellular vesicles (PDEVs) was in 1946. Later PDEVs were described for their procoagulant activity (i.e., inducing thrombin generation and shortened clotting time) and were understood to be released by platelets during activation [36]. The coagulation capacity of PDEVS is significantly higher than platelets [37]. This increased coagulation capacity is due to the surface of PDEVs being saturated with tissue factor (TF) and three times as much phosphatidylserine (PS) than platelets [38, 39].
PDEVs mainly exert proinflammatory functions through the activation of
endothelial cells [40]. The in vitro cell culture study has shown that
PDEVs activate endothelial cells and monocytes which also led to increased
adhesion between the two cells [41]. An important feature of PDEVs that has been
implicated in inflammation is their ability to deliver proinflammatory cytokines
to target cells. One example is RANTES, which can trigger monocytes to adhere to
endothelial cells and ultimately lead to atherosclerosis [42]. In addition to
RANTES, other Inflammatory compounds that PDEVs contain include, cytokines
associated with proinflammation activity (e.g., IL-1
PLAs are defined as a heterotypic combination of at least one platelet with a
leukocyte. Experimentally, the in vivo study has found that,
these aggregates occur in response to both hemostatic stimuli and inflammatory
stimuli [45]. PLAs may be useful as biomarkers or therapeutic targets for many
diseases. Interestingly, there are many different subtypes of PLAs [46]. During
inflammation, platelets can guide leukocytes to the site of extravasation, they
achieve this by binding to leukocytes, modulating their ability to adhere to the
extracellular matrix. Locally releasing pro-inflammatory molecules, and allowing
tissue infiltration [47]. Importantly, the direct interaction with platelets can
modulate the expression of important functional proteins in leukocytes, as shown
by the upregulation of integrins (
When activated platelets interact with neutrophils to form platelet-neutrophil aggregates (PNAs), which can modulate neutrophil activation and cause neutrophil extracellular trap (NET) production. NETs release critically depends on integrins, P-selectin, and other surface receptors such as intercellular adhesion molecule 2 (ICAM-2) and GPIb on platelets and CD11/CD18 on neutrophils [58, 59]. NETs are defined as lattice extracellular structures made mostly from decondensed chromatin, histones (antimicrobial), defensins and other proteases [60]. They are understood to be a host defence mechanism which can trap and kill hostile pathogens [60, 61]. Although NETs are beneficial for infection control, they can lead to (1) thrombotic complications in response to bacterial or viral infections [62] or (2) chronic inflammatory conditions in the absence of infection, which can lead to vascular complications such as venous thrombosis [63]. The prothrombotic role of NETs has been suggested by several important studies. For example, Brill and colleagues [64], used an experimental deep vein thrombosis (DVT) murine model and found that NETs were able to activate thrombosis. Von Brühl and colleagues [65] also found that in a DVT murine model NETs contributed actively to coagulation as NETs contain tissue factor (TF) and protein disulfide isomerases (activator of blood cell-derived TF). Furthermore, histones within the NET matrix activate platelets via TLR binding [66, 67].
This review focuses on the vascular impairment associated with diabetes mellitus
(DM) (i.e., vascular comorbidities of DM). Thrombosis (i.e., unwanted blood clot
formation and blood vessel occlusion leading to loss of blood flow and tissue
damage) is a critical driver of cardiovascular diseases. Therefore, in this
section, after a brief description of DM, we describe the known cellular and
molecular mechanisms leading to thrombosis in DM. DM incorporates metabolic
disorders which result in chronic hyperglycaemia [68]. Generally, DM can be
broadly split into two categories Type 1 and Type 2. Type 1 DM (or T1DM) is
categorised by impaired insulin secretion which is typically caused by
There is a wide range of health complications associated with DM, including retinopathy, nephropathy, and neuropathy, but cardiovascular diseases [76] are the most life-threatening complications. The risk of vascular inflammation and thrombosis is increased compared to the rest of the population [77]. T2DM is associated with a 2-fold increase in mortality caused by cardiovascular diseases compared to non-diabetic individuals [76]. Recent epidemiological data from the UK show that myocardial infarction, heart failure and ischaemic stroke are significantly increased in patients with T2DM [78]. This is the consequence of a raised tendency of blood to clot in these patients. Both primary and secondary components of haemostasis are dysregulated in T2DM, with reported platelet hyperactivity. This increases substantially the cardiovascular risk for patients with T2DM, for whom canonical antithrombotic drugs show reduced efficacy [79, 80, 81, 82]. Endothelium dysfunction is one of the main reasons for increased platelet activation in T2DM as well as increased blood coagulation and reduced fibrinolysis. An emerging and important role for platelet hyperactivity has also been described [77, 83, 84, 85].
The endothelium is a single cell layer which lines the inner side of vasculature acting as a barrier between the blood and the vessel wall. In addition to its role as a barrier, endothelium has a variety of important functions such as adhesion, tissue repair, angiogenesis, haemostasis, inflammation, platelet and coagulation regulation, fibrinolysis, and blood fluidity.
Hyperglycaemia causes metabolic changes in endothelial cells resulting in
increased vascular damage. In turn, this leads to a reduction of Nitric Oxide
(NO) and prostacyclin release by endothelial cells and, simultaneously, an
increase of reactive oxygen species (ROS) and reactive nitric species (RNS) [77].
One of the main mechanisms of this increased vascular damage is caused by
elevated production of advanced glycation products (AGEs) and an increase in the
expression of AGE receptors (RAGE) [86]. AGEs damage endothelial cells via two
mechanisms: (1) AGEs can directly react with and modify proteins, (2) AGEs can
bind and activate the receptor for advanced glycation products (RAGE), which
causes increases in nuclear factor-k
Another mechanism by which hyperglycaemia causes endothelial dysfunction is the upregulation of the hexosamine pathway (HP). The HP in health is responsible for the production of uridine-5-di-phospho-N-acetylglucosamine (UDP-GlcNAc), a substrate which is used for asparagine (N)-linked glycosylation of secretory and cell surface proteins. This pathway is also needed for O-linked glycosylation, co- and post-translational modifications, all essential for the regulation of protein and enzyme activity [89]. Therefore, the dysregulation of this pathway can result in deregulated glycosylation of proteins which can contribute to diseases such as DM. Paneque and colleagues [89] review the mechanisms of this pathway in extensive detail. Some of the enzymes found in the HP pathway such as glutamine fructose-6-phosphate amidotransferase (GFAT) have been found to have upregulated activity in DM patients, especially those with complications in comparison to healthy controls [90]. There was a significant positive correlation between insulin resistance, hyperglycaemia and oxidative stress markers.
DM is associated with an alteration of the lipid profile in blood. Increased
low-density lipoprotein (LDL) and decreased high-density lipoprotein (HDL) are
common among T2DM patients [91]. High density lipoproteins (HDL) normally have an
anti-inflammatory effect on endothelial cells and can suppress pro-inflammatory
cytokines (e.g., Tumour necrosis factor-
Oxidised low density-lipoprotein (ox-LDL) accumulates in the endothelial and inner lining of blood vessels where it contributes to endothelial dysfunction as it causes the endothelial to express adhesion molecules (intercellular adhesion molecule-1 and vascular cell adhesion molecule-1) which, ultimately leads to increased migration and adherence of immune cells to arterial walls [94]. Moreover, ox-LDL causes the eNOS pathway to become inactivated which is covered extensively in this review [95]. This causes the endothelial to have reduced protective autophagy and increased apoptosis which contributes to endothelium dysfunction.
The combination of increased LDL and increased oxidative stress leads to high
levels of oxidised-LDL in T2DM, although obesity could be a significant
confounding variable [96]. Interestingly, serum ox-LDL/
Another important mechanism of impairment of endothelial cell function in DM is insulin resistance. Insulin resistance is the development of cellular conditions in which insulin induces a lower biological effect than expected, which leads to defects in insulin-stimulated glucose uptake and glycogen synthesis [98]. Normally, insulin resistance precedes hyperglycaemia and DM [99]. In normal health, insulin acts via two main pathways. The first is the phosphatidylinositol-3-kinase (PI3K) pathway, which ultimately regulates metabolism and different blood cell responses. The second is the mitogen-activated protein kinase (MAPK)-dependent pathway, which is mainly involved in the regulation of gene expression, cell differentiation and cell growth [77]. In endothelial cells, insulin resistance impairs the PI3K pathway. This pathway is normally involved in the production of NO by the endothelial Nitric Oxide Synthase (eNOS), which regulates vasodilation and inhibits platelets. Therefore, a key consequence of insulin resistance is a reduction of NO production by endothelial cells, leading to the development of prothrombotic conditions [100].
Increased plasma levels for different coagulation factors have been reported for DM patients [101]. Fibrinogen (factor I), pro-thrombin (factor II), pre-kallikrein, factor V, factor VII, factor VIII, factor X, and factor XI have been detected at higher-than-normal concentrations in the plasma of T1DM and T2DM patients [102], while kininogen, factor IX, and factor XIII are only elevated in T2D [103]. Tissue factor (TF) is elevated in both T1DM and T2DM [104]. In addition, several anticoagulant proteins have a reduced plasma concentration in both types of DM, including anti-thrombin, protein C, and protein S [105]. Overall, the alteration of plasma levels of coagulation factors promotes excessive coagulation responses (i.e., Hypercoagulation) in DM patients [47]. Fibrinolysis is an essential component for the completion of haemostasis. Fibrinolysis is the process by which the clot/thrombi is degraded to restore normal blood flow. In both T1DM and T2DM, fibrinolysis is reduced, this is known as hypofibrinolysis, which contributes to the pro-thrombotic conditions associated with this disease. One of the key molecules involved in fibrinolysis is tissue plasminogen activator (tPA), this molecule catalyses the production of plasmin, which, initiates fibrinolysis. Ajjan et al. [106] found that in patients with T1DM, the severity of the disease (measured as glycated haemoglobin levels in blood or HbA1c) was significantly correlated with impaired fibrinolysis. The authors found that during hyperglycaemic conditions plasminogen is glycated which inhibits the generation of plasmin, thus, causing a reduction in fibrinolysis. On the other hand, increased fibrin cross-linking caused by hyperglycaemia has been suggested to increase clot strength [107] and reduce the fibrinolytic rate in T2DM patients [108]. Plasminogen activator inhibitor-1 (PAI-1) and thrombin activator fibrinolysis inhibitor (TAFI) are also important inhibitors of the fibrinolysis pathway affected by T2DM. In T2DM, elevated levels of these two inhibitors have been reported. Bryk and colleagues [109] found that PAI-1 and TAFI were significantly correlated with elevated plasma clot lysis time (CLT) and that hyperglycaemia and insulin resistance lead to increase in these two markers amongst others moreover, they were significantly correlated with markers of NETosis (i.e., pathological levels of NET formation) [109]. A Further study using blood from T2DM patients showed that PAI-1 binds to tPA to inhibit its catalytic plasmin production activity and TAFI blocks plasminogen from binding to fibrin, thus, preventing the generation of plasmin also [106]. Taken together, the above studies suggest that reduced fibrinolysis is a likely source of increased thrombotic risk for DM patients.
Platelet hyperactivity is defined as, increased activation, adhesion and aggregation of the platelets in comparison to the normal responses. Platelet hyperactivity has been associated with different abnormalities present in patients with DM, such as hyperglycaemia and dyslipidaemia. A summary of the signalling mechanisms underlying platelet hyperactivity in DM is shown in Fig. 2.
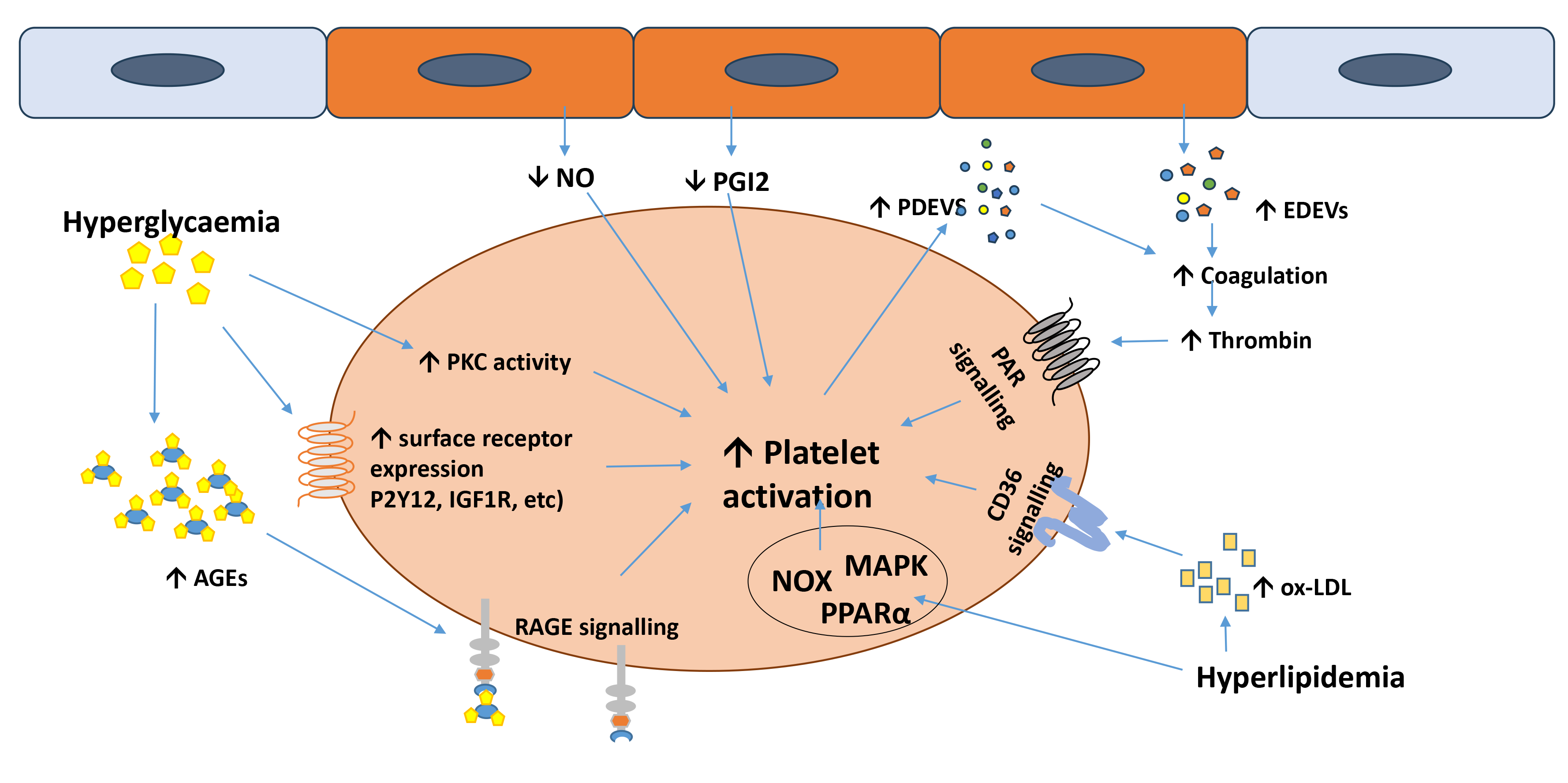 Fig. 2.
Fig. 2.
Molecular mechanisms underlying platelet hyperactivity in diabetes mellitus (DM).
Hyperglycaemia- and hyperlipidaemia-dependent mechanisms are shown in addition to
endothelial cell-driven mechanisms (i.e., reduced NO and PGI2 release and
endothelial-derived extracellular vesicle release). The role of PKC activation
and surface receptor upregulation is indicated, while the scavenger receptor CD36
and the signalling pathways of NOXs, MAPKs and PPAR
One of the conditions that platelets are exposed to in DM is hyperglycaemia, which consists of a glucose concentration in the blood over the normal range (i.e., higher than 7 mM before meals and over 8.5 mM after meals). A seminal study on human blood from patients with T2DM compared platelet activity (shear-stress induced platelet activation) using P-selectin and lysosomal integral membrane protein (LIMP) surface expression and, urinary 11-dehydro-thromboxane B2 (TxB2) excretion as markers of platelet activation. During acute hyperglycaemic events (induced using the hyperglycaemic clamp approach), platelets displayed increased P-selectin and LIMP surface expression and additionally, the urinary excretion of TxB2 was significantly increased in comparison to normoglycaemic conditions. Thus, demonstrating the induction of platelet hyperactivity by hyperglycaemia [110].
Despite an initial observation of increased glycation of platelet surface
proteins in hyperglycaemic conditions and a reduction in membrane fluidity [111],
these biochemical changes were not associated with an increase in platelet
responsiveness [112]. Nonetheless, the increase in advanced glycation end
products (AGEs) associated with hyperglycaemia can increase platelet
responsiveness [113]. In addition, an upregulation in platelet expression of the
receptor for AGEs (RAGE) in response to hyperglycaemia has been described, which
would increase platelet sensitivity to agonists such as S100, high mobility group box 1 (HMGB1) and amyloid
Interestingly, Keating and colleagues [115] investigated DM and healthy donor platelets activation response to the agonist (ADP) after exposure of platelets from both healthy donors and diabetic patients to in vitro hyperglycaemic conditions for 1 h, which led to significantly elevated platelet reactivity in comparison to platelets incubated in normoglycaemia (as measured by P-selectin surface exposure and fibrinogen binding). The authors found that platelet incubation with high concentrations of mannitol (chemically similar to glucose but metabolically inactive) also augmented platelet activity, suggesting that the osmotic effects of glucose may be responsible for the increase in platelet responsiveness induced by hyperglycaemia in vitro.
Assert and colleagues [116] measured the effect of hyperglycaemia on the
activity of platelet protein kinase C (PKC), which plays a critical role in
platelet activity [117]. Platelets obtained from healthy donors infused with
glucose simulate chronic and acute hyperglycaemia showed significant increases in
the levels of PKC
In addition to the potentiatory effects on signalling pathways described above,
hyperglycaemia has also been shown to induce long-term modulation of receptors
and signalling enzymes that lead to platelet hyperactivity. The expression levels
of the receptor of the negative platelet regulator prostacyclin are decreased in
T2DM, which in turn enhances platelet responsiveness [119]. In parallel, P2Y12, a
key receptor for the secondary agonist adenosine diphosphate (ADP), has been
reported to be significantly upregulated in T2DM platelets [120]. The
upregulation of P2Y12 expression is supported by the activation of oxidative
stress-dependent transcription factor nuclear factor-
Hyperlipidaemia is associated with a prothrombotic phenotype in T2DM and other
metabolic conditions. Hyperlipidaemia is associated with an increase in
circulating oxidised low density lipoproteins (ox-LDL) and choline phospholipids
that serve as high-affinity ligands for the CD36 receptor on platelets. The
activation of CD36 by oxidised lipids has been identified as a source of platelet
hyperactivity in mouse models and patients of T2DM [123]. The signalling pathway
activated by CD36 includes tyrosine kinase- and protein kinase C-dependent
activation of nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) and generation of reactive oxygen species (ROS), ultimately
counteracting the negative regulatory function of the cyclic nucleotides cyclic
adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP). The recent
study from our laboratory highlighted the involvement of both NOX1 and NOX2 in
the signalling of ox-LDL [124] and confirmed the negative modulation of the
cyclic nucleotide pathways by NOXs [125]. In addition, the glycation of LDL to
form glycosylated LDL (GlycLDL) was found to significantly increase intracellular
calcium concentration and heightened aggregation responses to ADP in patients
with T2DMDM [126]. In addition, although the signalling details remain to be
determined, platelet peroxisome proliferator-activated receptor alpha
(PPAR
The development of DM is associated with low-grade chronic inflammation, which
participates in the onset and progression of tissue and organ damage. In this
section of this review, we present a summary of our current understanding of the
pro-inflammatory mechanisms and complications associated with DM and the role
proposed for blood platelets in them. The development of DM is associated with
low-grade chronic inflammation. A case-cohort study conducted by Duncan and
colleagues [128] quantified low-grade systemic inflammation in T2DM, by measuring
the plasma levels of four inflammatory markers (IL-6, CRP, alpha-1-acid
glycoprotein and sialic acid), white cell count and fibrinogen levels, and found
that these inflammation markers significantly in patients with T2DM. Similarly,
chronic low-grade inflammation has been reported in T2DM by other studies, as
evidenced for example by the detection of high IL-6 levels in of T2DM patients
compared to non-DM patients [129]. Markers of low-grade systemic inflammation
present in T2DM are presented in Table 2 (Ref. [128, 130, 131]). Grossmann and
colleagues [130] analysed biomarkers in patients with prediabetes, DM and
normoglycaemia and found that the strongest differences in inflammation biomarker
expression between DM and normoglycaemia were C-reactive protein (CRP),
interleukin-1 receptor antagonist (IL-1RA) and fibrinogen. Interestingly, IL-1RA
is an anti-inflammatory cytokine, which inhibits the pro-inflammatory effect of
IL-1
| Difference of the marker in DM vs normoglycaemia | |
| WBC count | |
| IL-1RA | |
| IL-1 |
|
| IL-1 |
|
| IL-18 | |
| IL-12p70 | |
| IL-17A | |
| IL-6 | |
| IL-8 | |
| IP-10 | |
| CRP | |
| Fibrinogen | |
| GM-CSF | |
| IFN- |
|
| MCP-1 | |
| MIP-1 |
This table combines inflammatory biomarkers found at elevated levels in patients
with T2DM in previous studies [128, 130, 131]. The following abbreviations are
used: CRP, C-reactive protein; IL, interleukin; IL-1RA, interleukin-1 receptor
antagonist; MCP, Monocyte chemoattractant protein; MIP, macrophage inflammatory
protein; WBC, white blood cells; WBC, white blood cells; sICAM-1, soluble
intercellular adhesion molecule-1; IFN-
Platelets have been suggested to participate in the establishment of inflammatory conditions in DM by secreting inflammatory mediators cytokines/chemokines, interacting with immune cells and modulating their responses, and responding to pro-inflammatory stimuli leading to thromboinflammatory complications. The final part of this section and Fig. 3 outline the platelet-dependent mechanisms of inflammation in DM.
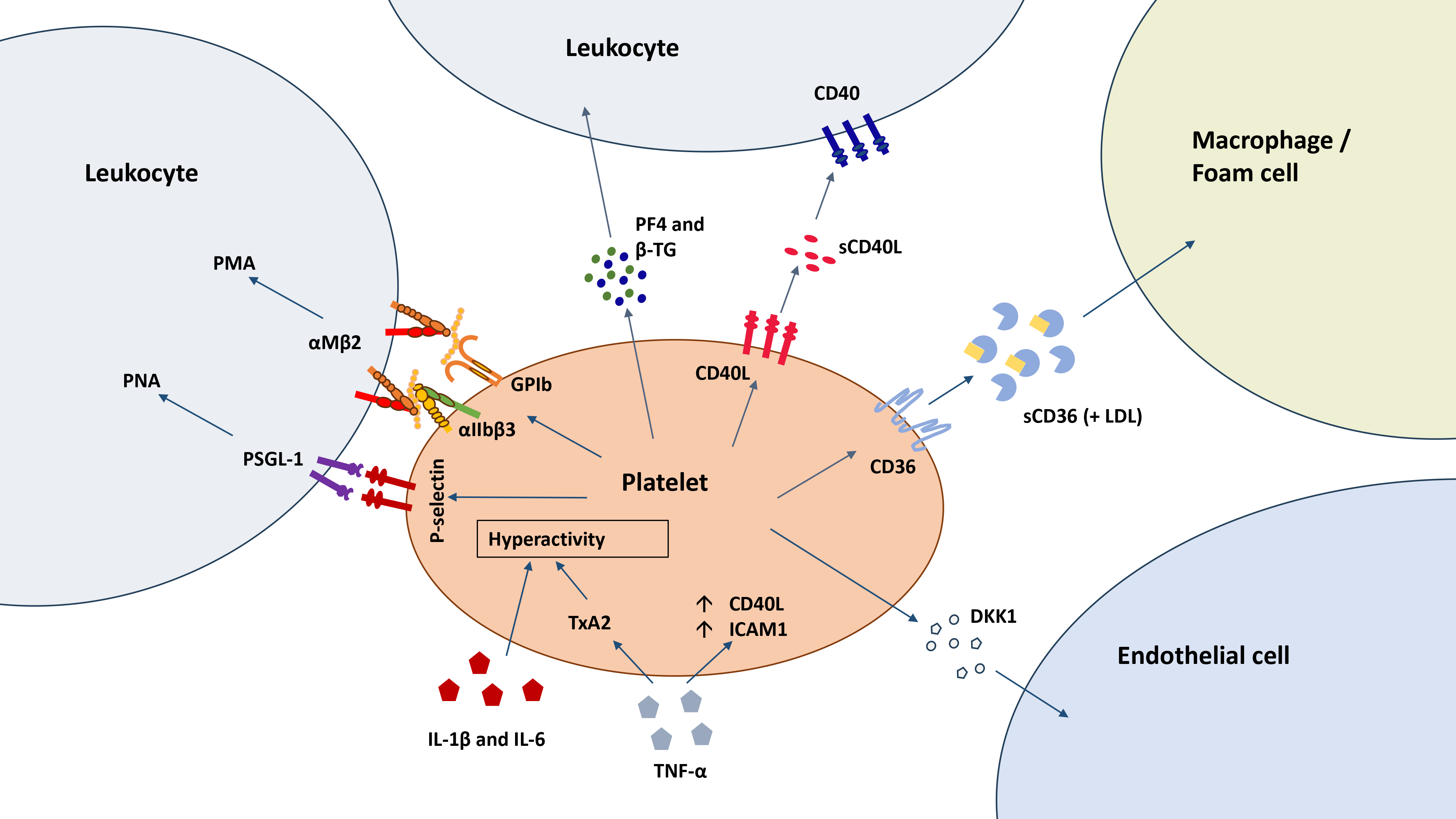 Fig. 3.
Fig. 3.
Platelet-dependent mechanisms of inflammation in DM. In
addition to responding to inflammatory stimuli with hyperactivity, platelets
stimulate inflammation by releasing inflammatory mediators (PF4,
In patients with both types of DM, circulating levels of platelet factor IV
(PF4) and beta thromboglobulin (
Similarly, CD40 and CD40L are significantly elevated in DM patients’ blood and
on their platelets, with a strong correlation with HbA1c levels [134]. Lajer and
colleagues [135] confirmed that sCD40L levels are significantly higher in T1DM
than in patients. Interestingly, hyperglycaemia and AGEs can trigger platelets to
express CD40L on their surface and secrete the soluble form [136]. CD40L is
expressed on the surface of platelets (where it promotes homotypic or heterotypic
cellular aggregation by interacting with CD40 on leukocytes or integrin
Another important membrane protein (CD36) upregulated in T2DM platelets is CD36 (or platelet glycoprotein IV). CD36 is a glycosylated transmembrane protein expressed on the surface of macrophages, endothelial cells [138] and platelets [139]. As a consequence of platelet activation, CD36 is cleaved and shed as soluble CD36 (sCD36) [140]. The circulating levels of sCD36 expression are significantly increased in T2DM patients in comparison to non-T2DM donors [141]. This has been suggested to be a consequence of CD36 shedding by platelets, which results from the preactivation/hyperactivation of these cells in T2DM [141]. Although it remains to be clarified whether the sCD36 in the plasma of patients with T2DM is free or associated with extracellular vesicles [142], the ability of this protein to bind and transport LDL to macrophages and other immune cells has been suggested to drive inflammatory and atherothrombotic responses in these patients [143]. Despite a recent study downplaying the potential of sCD36 in the progression of atherosclerosis [144], this hypothesis requires further investigation.
Platelets have also been indicated as the source of increased Dickkopf-1 (DKK-1) in the plasma of T2DM patients [145]. DKK-1 negatively regulates the Wnt signalling pathway and is involved in inflammation, atherogenesis, and glucose regulation [146]. Increased DKK-1 levels, both systemically and within vascular plaques, have been described in patients with atherosclerotic disorders [147]. DKK1 acts as an antagonist of the Wnt signalling pathway, which regulates inflammatory responses in endothelial cells [148]. The physiopathological relevance of this observation and the potential underlying mechanisms remain to be elucidated. Nonetheless, the involvement of Wnt signalling in DM and its comorbidities on different tissues and organs has been documented [149].
Several studies have reported elevated platelet-leukocyte aggregate formation in T2DM (e.g. [150]). PLAs are suspected to play a role in microvascular injury in DM patients. PLAs were significantly more abundant in patients with lesions versus those without, additionally, these levels were concomitantly enhanced by the number of vascular damage events, the more vascular damage patients had the more PLAs were present [151]. This pattern was seen in both T1DM patients and T2DM patients.
The seminal study by Kaplar and colleagues [152] found that there is a significant
difference in the percentage of platelet-monocyte aggregates (PMAs), but not
platelet-lymphocyte aggregates (PlyAs) or platelet-neutrophil aggregates (PNAs)
in patients with T1DM or T2DM in comparison to controls. T1DM patients showed
more marked increases in PMAs compared to T2DM. Independently of the DM type,
patients with proliferative retinopathy and nephropathy showed the highest number
of PMAs. There is currently no agreement regarding the potential cause of high
PMAs in T2DM, although the above-mentioned study highlighted no association
between PMAs and HbA1c values [152], other studies confirming a PMA increase in
T2DM patients showed a strong correlation with glycaemia (measured a HbA1c) in
both diabetic and non-diabetic subjects (e.g. [153]) without association to other
clinical factors, suggesting a central role for glycaemia in the formation of
PMAs. In relation to disease progression, the elevation of PMAs has been
identified as an early marker of T2DM, which precedes and perhaps promotes
inflammation [154]. In this study, the interaction with platelets was associated
with the upregulation of the
Similarly to T2DM, T1DM patients displayed peripheral blood PNAs significantly increased over the normal levels of non-DM donors [157]. The same authors confirmed these observations in a mouse model of T1DM in NOD mice. Platelets associated with PNAs in T1DM displayed markers of activation, as measured by CD62P flow cytometry. Interestingly, this study reports a decline in PNA levels in patients who had T1DM for a year or more compared to patients with recent diagnoses, possibly suggesting a role of PNA and systemic inflammation in the onset rather than the progression of the disease. In parallel, Zahran and colleagues [158] observed a significant increase in PMAs rather than PNAs in T1DM patients, with a linear correlation with HbA1c values. This is in accordance with other studies [159, 160]. In these studies, PMA levels were significantly correlated with levels of HbA1c, total cholesterol, LDL, triglycerides and pro-inflammatory serum markers (CRP and sCD40L).
The hyperactivity of platelets in DM has been abundantly described in the
literature and the contribution of hyperglycaemia and hyperlipidaemia were
addressed in this review (sections 3.3.1 and 3.3.2). In addition, inflammatory
mediators associated with DM have been shown to participate in the state of
hyperactivity of platelets in this disease. The increased levels of circulating
IL-1
Tumor necrosis factor alpha (TNF-
Since the pharmacological management of cardiovascular health is an unresolved challenge for patients with DM and their physicians, in this section of the review we will focus on clinical and preclinical studies making a significant contribution to the current practice and guidelines for the treatment or prevention of cardiovascular diseases in DM.
One of the mainstays of anti-thrombotic treatment is Acetylsalicylic acid (Aspirin). Aspirin works by irreversible inhibition of a cyclooxygenase (COX) specifically COX-1, this action prevents the production of TXA2. Current clinical guidelines for cardiovascular management in DM do not recommend aspirin for primary prevention and is utilised for patients deemed at high or very high risk of cardiovascular events [173, 174]. In DM, in addition to platelet hyperactivity (3.2), the response to aspirin is also impaired [175]. The post-translational modification of the COX-1 enzyme in hyperglycaemic conditions is responsible for the reduced (i.e., acetylation on serine 529 [176]). Another property of platelets in DM that contributes to aspirin resistance is that platelets in DM patients have a higher turnover rate, which increases the amount of active COX-1 enzyme and reduces the efficacy of the inhibitory effect of aspirin [177]. Alternative regimens of aspirin administration are under investigation to overcome this lack of efficacy of aspirin in T2DM. One such alternative strategy is the administration of aspirin twice a day (20 mg) instead or once a day (75 mg) [178].
ADP is a secondary agonist released by platelets that acts in an autocrine manner as a stimulus for platelet activation via the stimulation of the P2Y1 and P2Y12 receptors. The inhibition of P2Y12 receptors has become a successful therapeutic strategy to control platelet responsiveness and reduce thrombotic responses. Thienopyridines are a class of drugs designed to interfere with the P2Y12 receptor on platelets and include commonly used antithrombotic drugs such as Clopidogrel, Ticlopidin, and Prasugrel. Ticagrelor and Cangrelor are also commonly used P2Y12 receptor inhibitors for cardiovascular protection. Most of the time these antagonists are given in combination with aspirin as part of dual antiplatelet therapy (DAPT), which, is explained in more depth in this review [179].
There is growing evidence that Clopidogrel has limited effectiveness in DM patients, specifically T2DM. Several studies have demonstrated that the inhibitory effect of Clopidogrel in T2DM is reduced. Geisler and colleagues [180] found that patients with Coronary Artery Syndrome (CAD) and T2DM undergoing percutaneous coronary intervention (PCI) had significantly higher post-treatment with Clopidogrel aggregation in response to ADP and collagen compared to patients with CAD only. This is in agreement with Angiolillo and colleagues [181] who also found that T2DM patients with CAD had sub-optimal responses to Clopidogrel in comparison to non-DM patients. In this study, despite an increased dose of (150 mg daily) 60% of the patients showed sub-optimal response to Clopidogrel, demonstrating the need for personalised DAPT strategies to account for patient variability. A reduction of the plasma concentration of the Clopidogrel active metabolite has been indicated as the reason for the decreased efficacy of this drug in T2DM patients [181]. In addition, as reported in previous sections of this review [121], the upregulation of P2Y12 receptors has been indicated as the cause for increased platelet activation and reduction of the efficacy of P2Y12 receptor inhibitors in T2DM patients [182]. Prasugrel is an alternative P2Y12 receptor inhibitor for which contrasting reports on the efficacy in T2DM patients have been published. While some studies report no reduction of efficacy in T2DM patients compared to non-DM (e.g. [183]), other studies show that Prasugrel does not significantly reduce death from cardiovascular causes in comparison to Clopidogrel in T2DM patients (e.g. [184]).
Among P2Y12 antagonists, Ticagrelor showed a stronger inhibitory effect on platelet function compared to Prasugrel, both in non-DM and T2DM patients [185]. Therefore, although some reduction in its efficacy was observed in T2DM patients, Ticagrelor appears to be the P2Y12 inhibitor of choice for these patients, as it has also been shown to achieve a higher amount of inhibition of platelet aggregation in comparison to Clopidogrel [186]. Similarly, Laine and colleagues [187] found that Ticagrelor was consistently effective at reducing platelet reactivity (Vasodilator-stimulated phosphoprotein - VASP index) and was superior to Prasugrel in high-risk T2DM patients with acute coronary syndrome (ACS).
Anti-diabetic agents which aim to control blood glucose in patients have been found to have potential antithrombotic effects. This may be due to the indirect effect on platelets due to glucose or to a direct inhibitory effect on platelets.
Metformin acts as an insulin sensitiser (i.e., it increases cell responses to insulin) and by stimulating muscle tissues to consume glucose. Different clinical studies have demonstrated that metformin influences platelets in DM patients. One such study investigated the effect of metformin in newly diagnosed T2DM platelet activation (which was measured as 11-dehydro-thromboxane B2 urinary excretion) [188]. They found that metformin treatment significantly decreased platelet activation. Interestingly, metformin treatment also resulted in significantly reduced 8-iso-prostaglandin F2α (8-iso-PGF2α) urinary excretion, thus, less oxidative stress. Another earlier study found that platelet superoxide anion production (measured by a lucigenin assay with washed platelets derived from patients) found that superoxide anion level in patients treated was significantly lower in patients treated with metformin than untreated and treated with glibenclamide [189].
Another anti-diabetic drug class are sulphonylureas which control glucose levels
in DM by stimulating insulin secretion from pancreatic
TZDs are used as insulin-sensitisers (i.e., they increase cell responses to
insulin) and they act upon proliferator-activated receptor gamma
(PPAR
Glucagon-like peptide-1 (GLP-1) is secreted by cells in the gastrointestinal tract and the pancreas; it causes insulin secretion and glucagon suppression in a glucose-dependent manner when it binds to and interacts with the GLP-1 receptor (GLP-1R). This receptor is mainly expressed in vascular smooth muscle cells [201]. However, recently platelets have also been shown to express GLP-1R [202]. The incubation of human and mouse platelets with the GLP-1 agonist exenatide led to a significant inhibition of thrombin-, ADP- and collagen-induced aggregation [203]. The same study also found that exenatide treatment in a mouse model of diabetes significantly inhibited thrombus growth. It is hypothesised that the anti-thrombotic effects observed were due to the enhancement of endothelial nitric oxide synthase (eNOS) activity observed. Similarly, the GLP-1 agonist liraglutide inhibited ADP- and thrombin-induced platelet aggregation [202]. A similar study on liraglutide (and other GLP-1 agonists) showed that the inhibitory effects on platelets are due to an increase in NO bioavailability [204]. Additionally, the authors found that PI3K and MAPK/ERK-2 pathways were less activated in the presence of GLP-1 agonists. Kristensen and colleagues [205] found that death from cardiovascular causes and all-cause mortality were significantly lower in GLP-1-treated patients. However, further investigation is needed to validate these data and clarify the molecular mechanisms underlying the effect of GLP-1 on platelet activity.
We are only starting to understand the contribution of platelets to inflammation and their contribution to the inflammatory state associated with DM is even less understood. Nonetheless, the information available in the literature begins to draw a picture in which platelets are important contributors to the progression of DM and its complications affecting different tissues and organs. In view of the importance of cardiovascular complications in the health management of patients with DM, our understating of the molecular mechanisms underlying the dysregulation of platelets in this disease will be essential to design new and effective drugs. For this reason, this literature review firstly briefly describes the known contributions of platelets to the physiological response to vascular damage (section 1) and to inflammatory response (section 2). Then, the focus moves to the molecular mechanisms proposed for platelet hyperactivity and related thrombotic risk in DM (section 3) and the inflammatory response in DM in which platelets have been proposed to play a key role (section 4). Finally, we present clinical evidence on how DM affects the efficacy of antithrombotic drugs and how antidiabetic drugs can modulate the balance between physiological haemostasis and pathological thrombosis (section 5). In view of the high cardiovascular risk for DM patients, a better understanding of the mechanisms leading to vascular degeneration is critical for healthcare. Amongst, different mechanisms of cardiovascular disease onset and progression, the contribution of platelet hyperactivity to vascular inflammation is receiving increasing attention. Growing experimental evidence suggests that the ability of platelets to modulate leukocytes either via direct interaction (e.g., PLA formation and NET stimulation) or paracrine modulation is crucial for vascular degeneration in patients with DM. The molecular mechanisms underlying these phenomena are only partially understood, which has not permitted yet the design of selective drugs. This is made strikingly clear by the epidemiology data showing how much higher cardiovascular risk is in DM and how less effective existing drugs are. Despite the importance of receptors such as P-selectin (responsible for PLA formation and involved in NET stimulation) and CD36 (responsible for the exacerbation of platelet responses in hyperlipidemic conditions), effective drugs targeting them have not yet been developed. This has limited our ability to tackle thromboinflammation, which is an important component of the cardiovascular risk in patients with DM. This area of biomedical research and drug discovery is likely to lead to important advances in the management of cardiovascular health in patients with DM in the future. With this review, we present the state of the art in this research field and hope to facilitate the fruition of existing literature by basic and clinical scientists considering the cardiovascular management of patients with DM for their research.
JG and GP designed and wrote the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
Given his role as the Editorial Board member, Giordano Pula had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Ioanna-Katerina Aggeli. The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.