









 , Yingfeng Zhuang 1, Fengying Gao 2,*
, Yingfeng Zhuang 1, Fengying Gao 2,*1 Department of Intensive Care Unit, Fuzhou University Affiliated Provincial Hospital, 350001 Fuzhou, Fujian, China
2 Department of Pulmonary Disease, Shanghai Municipal Hospital of Traditional Chinese Medicine, Shanghai University of Traditional Chinese Medicine, 200071 Shanghai, China
Abstract
Ventilator-induced lung injury (VILI) is a consequence of inflammation and increased alveolar-capillary membrane permeability due to alveolar hyperdistention or elevated intrapulmonary pressure, but the precise mechanisms remain unclear. The aim of the study was to analyze the mechanism by which angiotensin converting enzyme 2 (ACE2) alleviates endoplasmic reticulum stress (ERS) and protects alveolar cells from pyroptosis in VILI by regulating angiotensin (Ang)1-7/Mas.
VILI was induced in mice by mechanical ventilation by regulating the tidal volume. The alveolar cell line, A549, mimics VILI in vitro by cyclic stretch (CS). Ang (1-7) (100 nmol/L) was added to the medium. ERS was induced in cells by stimulating with tunicamycin (TM, 2 μg/mL). ERS was inhibited by tracheal instillation of 4-phenylbutyric acid (4-PBA) (1 mg/kg). ACE2's enzymatic function was activated or inhibited by subcutaneous injection of resorcinolnaphthalein (RES, 20 μg/kg) or MLN-4760 (20 μg/kg). pGLV-EF1a-GFP-ACE2 was instilled into the trachea to increase the protein expression of ACE2. The Ang (1-7) receptor, Mas, was antagonized by injecting A779 subcutaneously (80 μg/kg).
ACE2 protein levels decreased after modeling. Ang (1-7) level was decreased and Ang II was accumulated. ERS was significantly induced in VILI mice, and pyroptosis was observed in cells. When ERS was inhibited, pyroptosis under the VILI condition was significantly inhibited. Ang (1-7) alleviated ERS and pyroptosis under CS. When ERS was continuously activated, the function of Ang (1-7) in inhibiting pyroptosis was blocked. Resorcinolnaphthalein (RES) effectively promoted Ang II conversion, alleviated the Ang (1-7) level in VILI, ameliorated lung injury, and inhibited ERS and cell pyroptosis. Inhibiting ACE2's function in VILI hindered the production of Ang (1-7), promoted the accumulation of Ang II, and exacerbated ERS and pyroptosis, along with lung injury. The Mas antagonist significantly blocked the inhibitory effects of ACE2 on ERS and pyroptosis in VILI.
Reduced ACE2 expression in VILI is involved in ERS and pyroptosis-related injury. ACE2 can alleviate ERS in alveolar cells by catalyzing the production of Ang (1-7), thus inhibiting pyroptosis in VILI.
Keywords
- ventilator-induced lung injury
- angiotensin-converting enzyme 2
- angiotensin (1-7)
- endoplasmic reticulum stress
- pyroptosis
Mechanical ventilation (MV) has widespread utility in managing respiratory failure, anesthesia, surgical procedures, and respiratory support [1, 2, 3]. Notably, its critical role during the recent coronavirus disease 2019 (COVID-19) pandemic highlighted the significant clinical relevance of ventilatory support [4, 5]. Ventilator-induced lung injury (VILI) is a consequence of inflammation and increased alveolar-capillary membrane permeability due to alveolar hyperdistention or elevated intrapulmonary pressure [6]. However, the precise mechanisms underlying VILI remain unclear.
Endoplasmic reticulum stress (ERS) is a prominent feature of injury during ventilator use. Several studies affirm that reactive oxygen species (ROS) production, inflammation, and apoptosis, implicated in ERS, constitute pivotal mechanisms in the pathogenesis of VILI [7, 8, 9]. Pyroptosis, characterized by the cleavage of gasdermin D (GSDMD) by caspase-1, caused attachment of GSDMD-N (N-terminal product) to the cell membrane, thereby disrupting its integrity and permeability [10, 11, 12]. Recent findings have highlighted pyroptosis as a crucial mechanism underlying VILI; for instance, following mechanical ventilation, levels of c-caspase-1 and GSDMD-N in rat lung tissue are markedly elevated [13]. However, the precise mechanism underlying pyroptosis induction in alveolar cells in VILI remains obscure. Interestingly, ERS can also elicit pyroptosis. For example, in diabetic nephropathy, ERS promotes the activation of nucleotide binding oligomerization domain (NOD)-like receptor protein 3 inflammasome (NLRP3) which culminates in pyroptosis [14]. ROS accumulation induced by ERS triggers NLRP3-dependent neuronal pyroptosis [15]. Nevertheless, whether ERS, in the context of VILI, induces pyroptosis remains unclear.
Angiotensin converting enzyme 2 (ACE2) is a critical enzyme catalyzing the
conversion of angiotensin (Ang) II to Ang (1-7). Ang II plays a pivotal role in
regulating vasoconstriction and the renin-angiotensin system [16]. For instance,
it can regulate the electrolyte and fluid balance by aldosterone [17]. Ang (1-7)
exerts an antagonistic effect against the activity of Ang II by binding to its
receptor, Mas receptor. The involvement of Ang II in the onset of VILI is
well-established [18]. An animal study has documented the upregulation of Ang II
levels in VILI. Ang (1-7) or the ACE inhibitor captopril increased Ang (1-7)
levels, captopril decreased Ang II levels, and both protected mice from VILI
[19]. Ang II has been implicated in ERS and pyroptosis induction in lung injury
following seawater inhalation. ACE2 mitigates ERS and apoptosis by facilitating
the conversion to Ang (1-7) [20]. Ang II participates in cardiac hypertrophy by
provoking cardiomyocyte pyroptosis [21]. Mice with ACE2 knockout exhibit
heightened levels of ERS, while ACE2 overexpression diminishes ERS in the liver
through the IKK
This study primarily aimed at exploring the role of ERS in the induction of pyroptosis and assessing whether ACE2 could inhibit ERS and pyroptosis in VILI by inducing the conversion of Ang II to Ang (1-7). The findings are expected to provide a more comprehensive understanding of mechanisms underlying VILI mediated by the ACE2/Ang II axis, holding promise for improving the prognosis of patients with VILI.
The experimental protocol adhered to the national standard laboratory animal guidelines for ethical review of animal welfare (GB/T 35892-2018) and was approved by the Fuzhou University Affiliated Provincial Hospital (the original name: Shengli Clinical Medical College of Fujian Medical University) Animal Experiment Committee. Specifically, 130 C57BL/6 male mice, 18 weeks old, were procured from Shanghai Xipur-Bikai Experimental Animal Co., Ltd and maintained in a pathogen-free environment. We only used male adult mice to avoid possible interference due to the physiological cycle and female hormones in the activity of the renin-angiotensin system. Ten mice were included in each group (N = 10). In experiment I, 20 mice in total were randomly assigned to the Control and VILI groups; in experiment II, 30 mice were randomly assigned to Control, VILI, and VILI+4-phenylbutyric acid (4-PBA) groups; in experiment III, 40 mice were randomly assigned to Control, VILI, VILI+ Resorcinolnaphthalein (RES), and VILI+MLN-4760 groups; in experiment IV, 40 mice were randomly assigned to Control, VILI, VILI+ACE2, and VILI+ACE2+A779 groups. Measurements were repeated thrice for each animal.
Injectable anesthetics, ketamine (100 mg/kg, H20023609, Zhejiang Xianle Pharmaceutical Co., Ltd., Xianju, Zhejiang, China), and xylazine (10 mg/kg, B25730, Yuanye biotechnology, Shanghai, China), were administered intraperitoneally (i.p.) for anesthetizing the animals. One-quarter of the initial dose was added every 30 min to maintain anesthesia. A No. 20 catheter (Cusabio Life Science, Wuhan, Hubei, China) was sterilized and inserted into the trachea. Mechanical ventilation (MV) for 4 h was conducted using a ventilator (R500, RWD Life Science Co., Ltd., Shenzhen, Guangdong, China) with a high tidal volume of 20 mL/kg at a rate of 80 breaths per minute and a breathing ratio of 1:1 without exerting positive end-expiratory pressure [23]. Mice in the MV group underwent ventilation for 4 h, whereas mice in the control group were intubated and spontaneously anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). After the 4-h MV was completed, mice were allowed to breath spontaneously. Vital signs of the animals were monitored by evaluating the color of the mucous membrane in the mouth, limb skin color, temperature, and heart rates. At 2 hour after the 4-h MV, the bronchoalveolar lavage fluid (BALF) and lung tissue were collected from the mice.
Bronchoalveolar lavage fluid (BALF) was collected 2 h after the mice were resuscitated. A small incision was made in the midline of the neck to expose the trachea to collect BALF after the mice were fully anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg). A sterile tracheal cannula was inserted and secured with a ligature. Phosphate-buffered saline (PBS) was injected into the tracheal cannula such that it entered the lungs. Controlled instillation was performed by gently pushing PBS in and out four times to dislodge and collect lung samples. The lavage fluid was aspirated into the syringe, and this instillation-aspiration process was repeated until 1.5 mL BALF was collected from each animal.
After euthanizing the animals by cervical dislocation, lung tissues were
collected for subsequent experiments, including hematoxylin-eosin (H&E)
staining, measuring the wet/dry (W/D) weight of lungs, immunohistochemical staining,
and western blotting. The model establishment of lung injury was confirmed by
significant changes in H&E staining, lung edema (lung wet/dry ratio), and
concentrations of interleukin-1 beta (IL-1
To inhibit ERS in vivo, 4-PBA (1 mg/kg, HY-A0281, MedChemExpress, Monmouth Junction, NJ, USA) was instilled through the trachea 6 h before MV. To reduce the dosage and its effect on the whole body, 4-PBA was administrated through the trachea instead of injecting i.p. In the pilot experiments, we tested NLRP3 expression at the indicated intervals and found that, after 6 h, the expression of NLRP3 was markedly decreased (Supplementary Fig. 1). ACE2’s enzymatic function was either activated or inhibited by subcutaneous injection of resorcinolnaphthalein (RES, 20 µg/kg, HY-122445, MedChemExpress, Monmouth Junction, NJ, USA) or MLN-4760 (20 µg/kg, HY-19414, MedChemExpress, Monmouth Junction, NJ, USA), respectively, via osmotic minipumps (Alzet Osmotic Minipumps, Durect Corporation, Cupertino, CA, USA) 6 h before MV, following protocols described previously [24, 25]. The osmotic minipumps were implanted under anesthesia with ketamine (100 mg/kg) and xylazine (10 mg/kg) and delivered the indicated dose over the 6 h until start of MV. The Ang (1-7) receptor, Mas, was antagonized by subcutaneous injection of A779 (80 µg/kg, HY-P0216, MedChemExpress, Monmouth Junction, NJ, USA) 6 h before MV.
To increase the protein expression of ACE2, 300 µg of pGLV-EF1a-GFP-ACE2 (GenePharma Co., Ltd., Shanghai, China) was instilled into the trachea 24 h before MV. An equal concentration of pGLV-EF1a-GFP was instilled in the trachea of animals of the other groups. In the two groups receiving tracheal instillations 6 or 24 hours before MV, the mice were anesthetized when they received tracheal instillations, then allowed to recover until the MV experiment.
For H&E staining, lung tissue sections were prepared by embedding them in paraffin. The sections underwent serial washing, starting with xylene, ethanol and distilled water. Subsequently, the sections were immersed in hematoxylin solution for 5 minutes, followed by washing with tap water and a treatment with 1% hydrochloric acid alcohol. This was followed by washing with tap water and treatment with 0.6% ammonia water. The sections were finally washed under running water. The sections were immersed in an eosin staining solution for 2 minutes, followed by serial immersion in 95% ethanol, absolute ethanol, and xylene steps for dehydration. Finally, the sections were dried and sealed with neutral gum. The staining was observed under a microscope (BX50, Olympus, Tokyo, Japan).
For IHC, lung samples were initially washed with PBS and incubated with 5% normal goat serum. Subsequently, the samples were incubated with rabbit GSDMD antibody (1:500, ab209845, Abcam, Cambridge, MA, USA) and ACE2 antibody (1:500, ab272500, Abcam, Cambridge, MA, USA) for 1–2 hours at 37 °C. Subsequently, samples were incubated with a biotin-labeled secondary antibody (1:2000, ab207996, Abcam, Cambridge, MA, USA) at 37 °C for 20 minutes, followed by washing with PBS. The samples were further incubated with horseradish enzyme-labeled streptavidin at 37 °C for 20 minutes and washed thrice with PBS. Color development was achieved using diaminobenzidine, and microscopic observation was conducted using the Olympus BX50 microscope.
For ELISA, 100 µL of the BALF supernatant was added per well in a 96-well
plate. ELISA was performed following the manufacturer’s instructions (ELISA kits
for IL-1
The supernatant was collected after centrifuging BALF or culture medium at 1000
A549 (CRM-CCL-185) alveolar type II epithelial cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco’s modified eagle medium (DMEM) (#21068028, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, # A5669801, Gibco, Grand Island, NY, USA), 10 mg/mL of streptomycin, and 10,000 units/mL of penicillin (#P4333, Sigma-Aldrich, Louis, MO, USA). VILI was simulated in vitro by subjecting A549 cells to cyclic stretch (CS). A549 cells were seeded on BioFlex plates (Flexcell Int, Hillsborough, NC, USA). After reaching approximately 50% confluency, the BioFlex plate was mounted on a Flexcell cell stress loading system (FX-5000T, Flexcell Int, Hillsborough, NC, USA). The cells attached to the device were subjected to CS, with a stretch-recovery cycle of 15 times per minute for 4 h [26]. The A549 cells used in the study underwent short tandem repeats (STR) profiling to validate their authenticity and were tested to be free of mycoplasma contamination. The cells were cultured in a humidified incubator at a temperature of 37 °C, with 5% CO2.
Ang (1-7) (HY-12403, MedChemExpress, Monmouth Junction, NJ, USA) was added to the medium at a final concentration of 100 nmol/L for pretreatment over 12 h, with continuous intervention during CS. Tunicamycin (TM, HY-A0098, MedChemExpress, Monmouth Junction, NJ, USA) was added to the medium at a final concentration of 2 µg/mL to induce ERS.
Initially, 100 µL of cells (5000 cells) were seeded into each well of a 96-well plate. Cells were cultured and treated with the indicated drugs. Subsequently, 10 µL of CCK-8 solution (#96992, Sigma-Aldrich, Louis, MO, USA) was added to each well. Blank controls comprised the cell culture medium, drug, and CCK-8 solution (without cells). Cells were incubated in an incubator for 1 h. Following incubation, absorbance at 450 nm was measured using a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
To measure the ROS production in lung tissues, following the harvesting, lungs
were instantly frozen and homogenized using polytron (40 mM ice-cold
Tris
To measure ROS levels in cells, DCFH-DA (Invitrogen, Waltham, MA, USA) was diluted at 1:1000 in a serum-free culture solution. Cells were seeded in 6-well plates and treated with the indicated drugs. The culture solution was removed, and 1 mL of diluted DCFH-DA was added per well. The cells were incubated in the incubator for 20 minutes. Subsequently, the cells were washed thrice with serum-free cell culture solution to eliminate any residual DCFH-DA. Fluorescence intensity was measured using a microplate reader (PerkinElmer, Waltham, MA, USA) with excitation and emission wavelengths of 485 nm and 538 nm, respectively.
TRIzol® reagent was used to extract 1 µg of total RNA from 10 mg mouse lung tissue and reverse transcribed into cDNA using the PrimeScript RT Reagent Kit (RR047A; Takara Bio, Inc. San Jose, CA, USA). SYBR green reagent (Takara Bio, Inc., San Jose, CA, USA) was used for qPCR. The 2-ΔΔCq method was used to normalize the expression levels to those of GAPDH. The following primer sequences were used for qPCR: ACE2 forward (F), 5′- ACTCCGATCATCAAGCGTCA-3′ and reverse (R), 5′-GGCTCCATTCAGTGTTCCAGA-3′; GAPDH F, 5′-GTCGTACCACAGGCATTGTGATGG-3′ and R, 5′-GCAATGCCTGGGTACATGGTGG-3′.
The Minute™ Cytoplasmic and Nuclear Fractionation kit (SC-003, Invent Biotechnologies, Plymouth, MA, USA) was used to separate proteins from the cytoplasmic and nuclear fractions. After separation, the proteins were subjected to western blot analysis using specific antibodies, including those against ACE2 (1/5000, ab108252, Abcam, Cambridge, MA, USA), C/EBP homologous protein (CHOP) (5 µg/mL, ab11419, Abcam, Cambridge, MA, USA), glucose-regulated protein 78 (GRP78) (1 µg/mL. ab21685, Abcam, Cambridge, MA, USA), NLRP3 (1/1000, ab263899; Abcam, Cambridge, MA, USA), cleaved caspase-1 (c-caspase-1) (1/1000, ab256469, Abcam, Cambridge, MA, USA), GSDMD-N (1/1000, ab215203, Abcam, Cambridge, MA, USA), and GAPDH (1/10000, ab181602, Abcam, Cambridge, MA, USA). The membranes were incubated with goat anti-mouse IgG+IgM H&L (HRP) secondary antibody (1/5000, ab47827, Abcam, Cambridge, MA, USA). Bands were visualized using an electrogenerated chemiluminescence (ECL) kit (#PE0010, Solarbio, Beijing, China) and analyzed using Image Pro Plus 6.0 (Media Cybernetics, Inc., Bethesda, MD, USA).
Data are presented as the average
To elucidate the association of ACE2 expression with VILI, we established a
mouse model of VILI through prolonged MV. Our assessment revealed notable
alveolar structural damage (Fig. 1A). Thickened alveolar walls (infiltration of
inflammatory cells into the alveolar walls), hyaline membranes (the accumulation
of proteinaceous material in the alveolar spaces), cellular infiltration (massive
infiltration of inflammatory cells), and edema were visualized in VILI-inflicted
tissues. Pulmonary edema (Fig. 1B) and an increase in IL-1
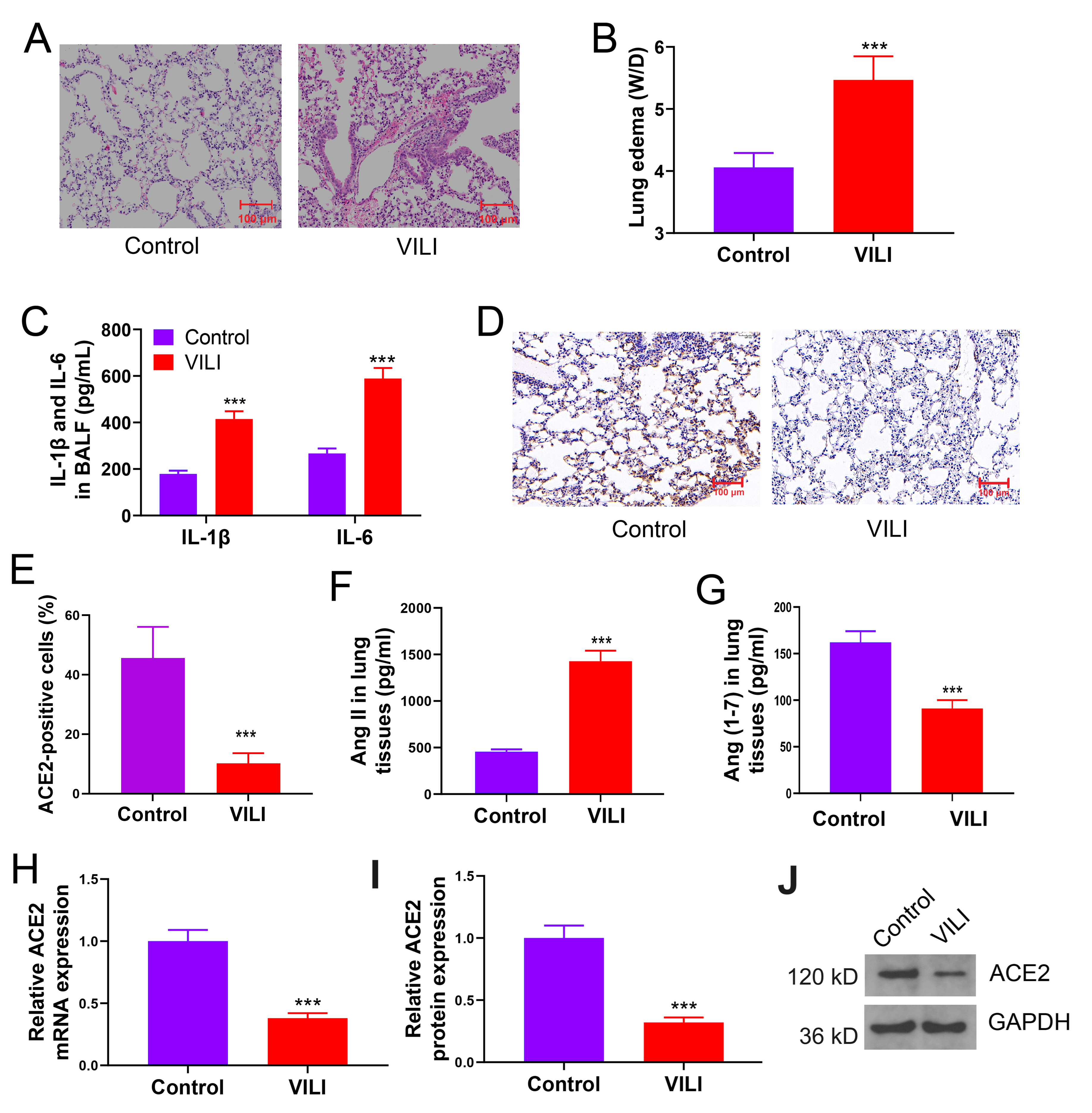 Fig. 1.
Fig. 1.
ACE2/Ang (1-7) levels are downregulated while Ang II is
upregulated in mice with VILI. (A) Lung tissue damage due to MV-induced VILI.
(B) Lung edema in VILI. (C) Results of inflammatory cytokine levels across BALF
samples. (D,E) Expression of ACE2 in lung tissues by immunohistochemistry. (F,G)
Comparison of Ang II and Ang (1-7) levels in tissues. (H–J) Comparison of protein
or mRNA expressions of ACE2 in VILI. VILI, ventilator-induced lung injury;
W/D, wet/dry weight ratio; IL-6, interleukin-6; IL-1
In VILI-inflicted lung tissue, a significant elevation in the levels of CHOP and GRP78 proteins was found, along with the increase in ROS levels, suggesting ERS induction (Fig. 2A–C). Alveolar cells in the VILI group displayed an augmentation in GSDMD, as shown by immunohistochemistry analysis (Fig. 2D,E). An elevation in LDH secretion into BALF was found (Fig. 2F). The levels of NLRP3, c-caspase-1, and GADMD-N proteins in lung tissues were increased significantly (Fig. 2G,H), suggesting ERS induction in VILI and increased pyroptosis in alveolar cells.
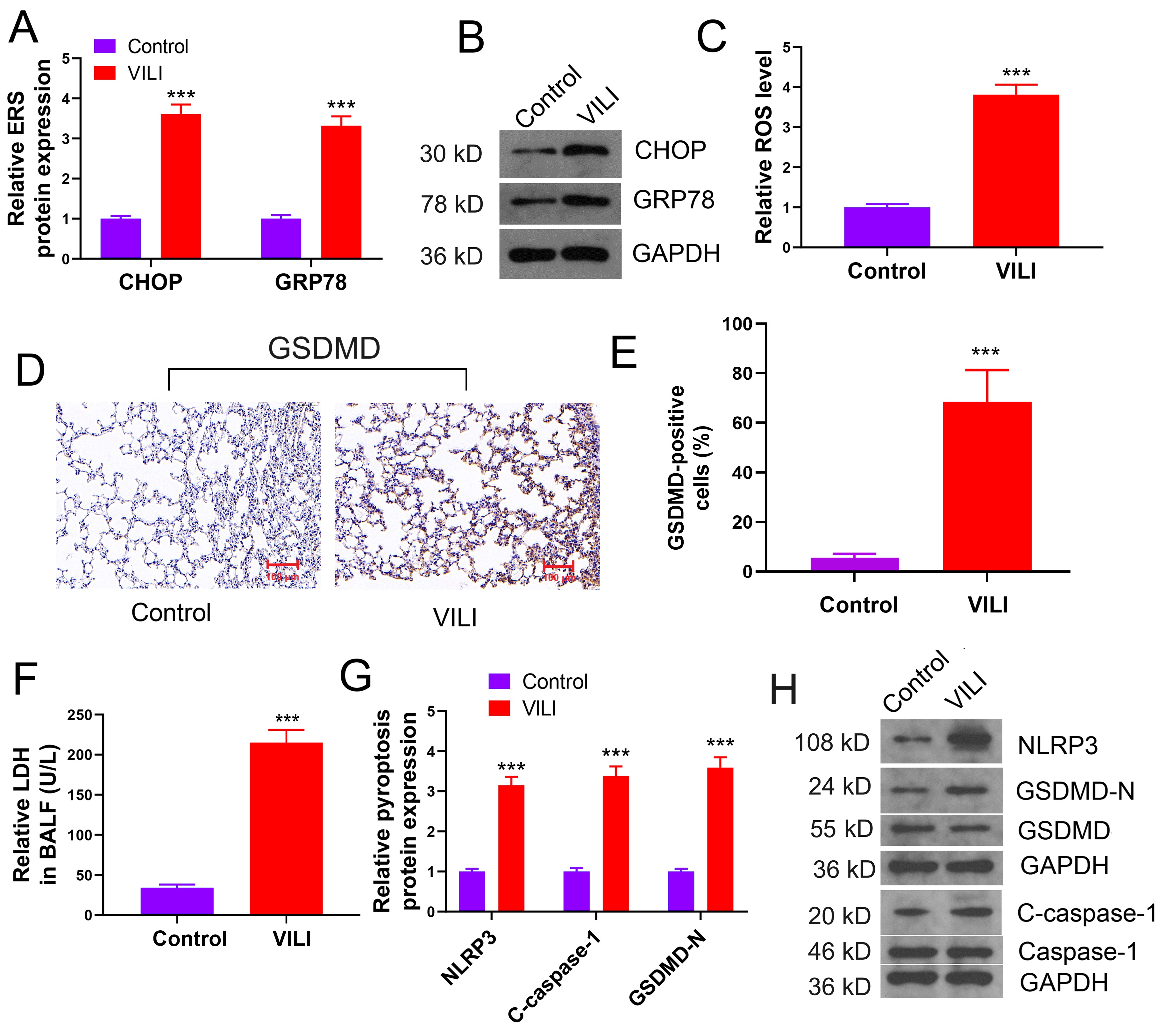 Fig. 2.
Fig. 2.
ERS and pyroptosis are induced in VILI. (A,B) Changes of
ERS-related proteins in VILI. (C) Results of ROS levels in tissues. (D,E) Changes
of expression of the GSDMD protein. (F) Results of LDH concentrations in BALF
samples. (G,H) The levels of pyroptosis-related proteins, NLRP3 and
c-caspase-1. VILI, ventilator-induced lung injury; ERS, endoplasmic reticulum
stress; CHOP, C/EBP homologous protein; GRP78, glucose-regulated protein 78; ROS,
reactive oxygen species; BALF, bronchoalveolar lavage fluid; LDH, lactate
dehydrogenase; NLRP3, NOD-like
receptor protein 3 inflammasome; GSDMD, gasdermin D. Scale bar = 100 µm. ***p
Heightened ROS levels due to ERS activation led to the stimulation of NLRP3, a primary mechanism underlying pyroptosis. To further elucidate the regulatory mechanism of ERS and pyroptosis in VILI, we employed 4-PBA to inhibit ERS and performed MV. Mice were randomly divided into three groups: control, VILI, and VILI+4-PBA. Intervention with 4-PBA significantly alleviated alterations in alveolar structure, lung edema, and inflammation, all induced by VILI (Fig. 3A–C). Moreover, lung tissue of mice of the VILI+4-PBA group displayed substantially lower levels of CHOP and GRP78 proteins compared to those in the VILI group, along with marked reductions in marker proteins of pyroptosis and LDH levels (Fig. 3D–H). Thus, ERS inhibition by 4-PBA significantly alleviated VILI-induced pyroptosis, suggesting that ERS plays a role in inducing pyroptosis in VILI.
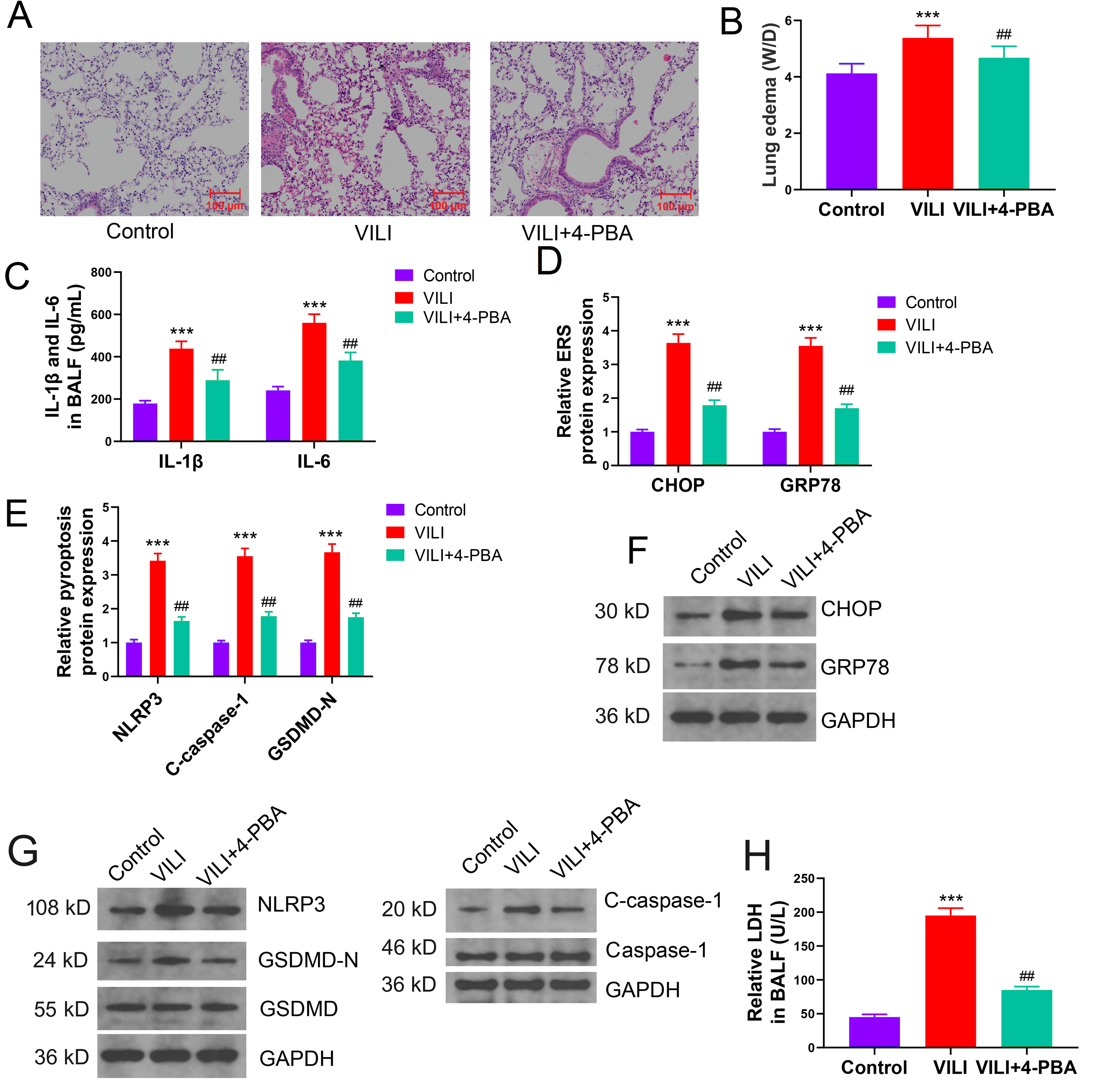 Fig. 3.
Fig. 3.
Inhibition of ERS alleviates pyroptosis in VILI in
vivo. (A) Effect of 4-PBA (ERS inhibitor) intervention on VILI. (B) Effect of
ERS inhibition on VILI-inflicted lung edema. (C) Effect of ERS inhibition on
inflammatory cytokine levels in BALF of mice with VILI. (D–G) Comparison of ERS
and pyroptosis-related protein levels in each group. (H) Effect of ERS inhibition
on LDH concentrations in BALF samples from mice with VILI. VILI,
ventilator-induced lung injury; ERS, endoplasmic reticulum stress; IL-6,
interleukin-1; IL-1
ACE2’s primary role is to facilitate the conversion of Ang II to Ang (1-7). To elucidate whether the inhibitory effects of Ang (1-7) on pyroptosis in VILI are associated with ERS, VILI was emulated in vitro by CS, and TM was employed to induce ERS. Consequently, A549 cells were divided into the following four groups: control, CS, CS+Ang (1-7), and CS+Ang (1-7) +TM. Prolonged in vitro CS significantly reduced cell viability, and Ang (1-7) markedly restored the compromised cell viability induced by CS. Notably, cell viability in the CS+Ang (1-7) +TM group was significantly lower compared to the CS+Ang (1-7) group (Fig. 4A). CS in vitro upregulated the protein expressions of CHOP and GRP78 in A549 cells. Ang (1-7) substantially mitigated ERS during CS, and TM effectively induced ERS in vitro (Fig. 4B,C). In line with the trend of ERS, in vitro CS enhanced LDH efflux, NLRP3 activation, and GSDMD-N levels. Ang (1-7) significantly ameliorated CS-induced pyroptosis, with its levels being markedly higher in the CS+Ang (1-7) +TM group compared to the CS+Ang (1-7) group (Fig. 4D–F). Ang (1-7) could alleviate ERS and pyroptosis during CS. Moreover, upon continuous ERS, the inhibitory effect of Ang (1-7) on pyroptosis was impeded, suggesting that Ang (1-7) mitigates alveolar pyroptosis in VILI through its ERS-inhibiting role.
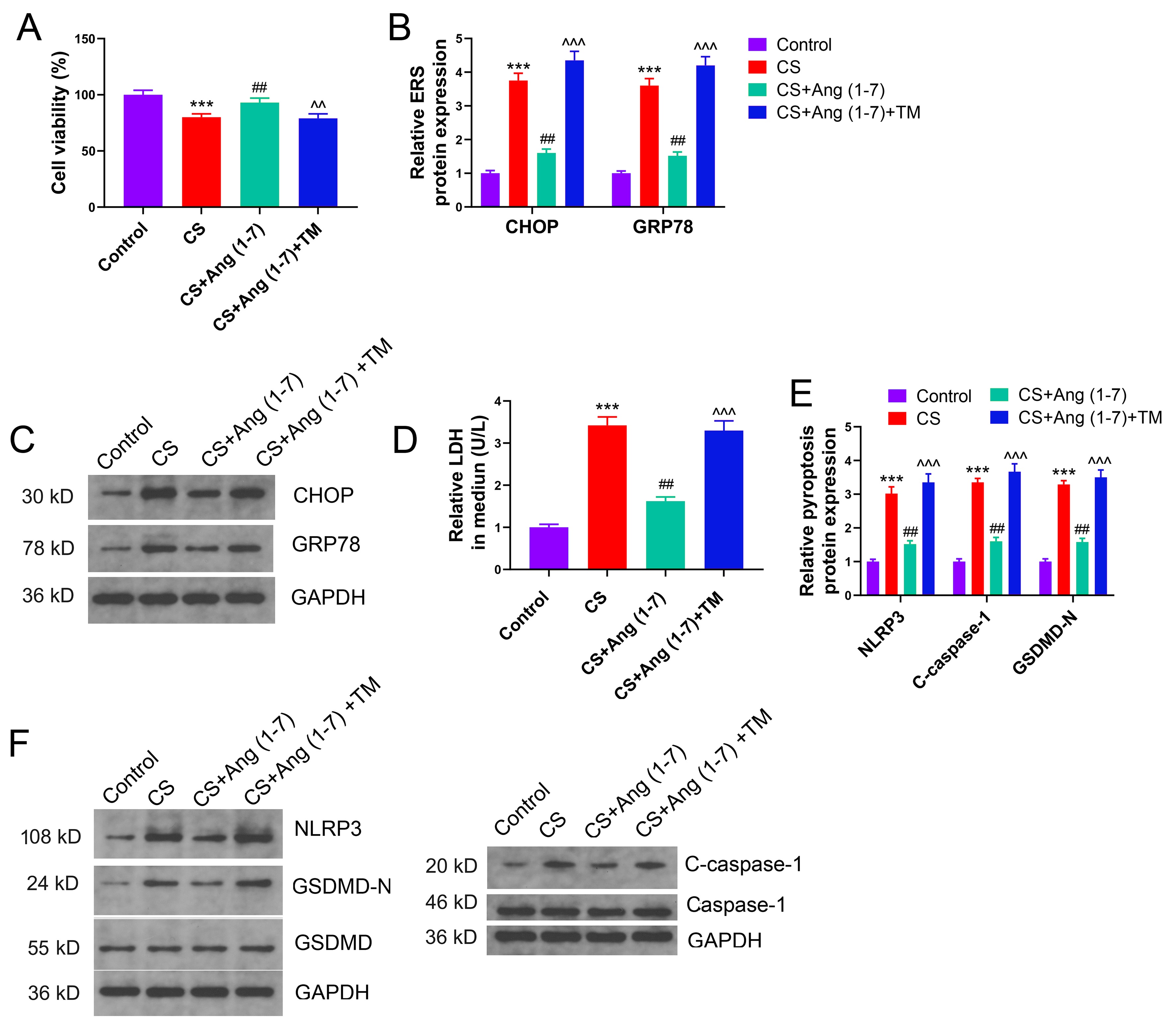 Fig. 4.
Fig. 4.
Ang (1-7) inhibits pyroptosis by alleviating endoplasmic
reticulum stress (ERS) under CS in vitro. (A) Effect of Ang (1-7) and
ERS activator on the viability of alveolar cells under CS in vitro.
(B,C) Comparison of protein expressions of ERS markers in cells of each group.
(D) Comparison of LDH secretion in each group. (E,F) Effect of Ang (1-7) and ERS
activator on the expression of pyroptosis-related proteins in alveolar cells
under CS. CS, cyclic stretch; Ang, angiotensin; CHOP, C/EBP homologous protein;
GRP78, glucose-regulated protein 78; NLRP3, NOD-like receptor protein 3
inflammasome; GSDMD, gasdermin D; LDH, lactate dehydrogenase. ***p
To elucidate the effects of ACE2 in VILI, we employed RES to activate ACE2’s
function, while MLN-4760 was used to inhibit it. VILI was induced in the model.
RES alleviated lung injury significantly in the mouse model of VILI, evidenced by
reduced lung edema (W/D) and lowered IL-6 and IL-1
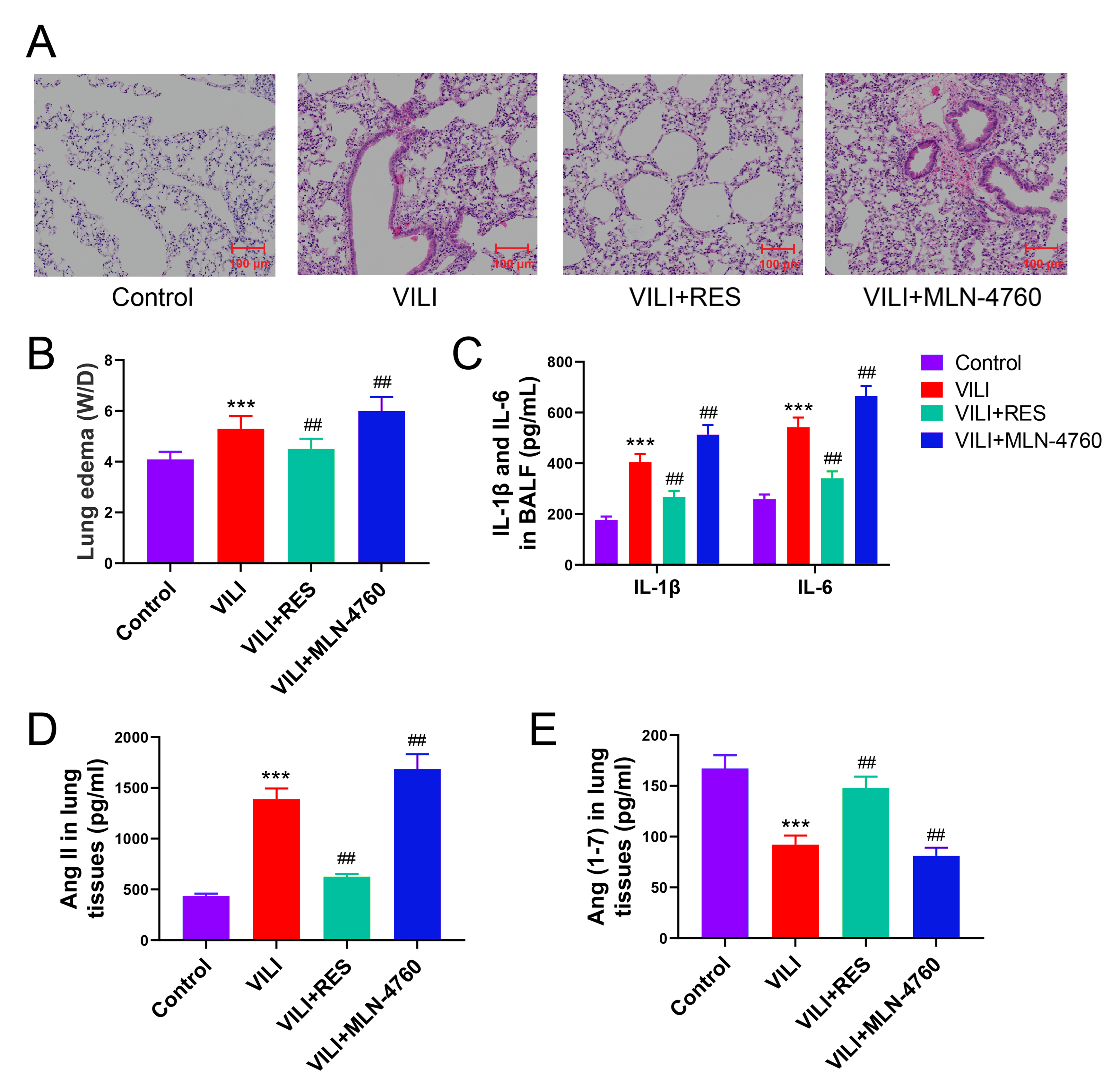 Fig. 5.
Fig. 5.
ACE2 is important in promoting Ang (1-7) production and lung
injury in mice with VILI. (A) Effect of activator (RES) and inhibitor (MLN-4760)
of ACE2 on lung injury in mice with VILI demonstrated by H&E staining. (B,C)
Effect of activation or inhibition of ACE2 on lung edema and inflammation in
VILI. (D,E) Comparison of Ang II and Ang (1-7) levels in the lung tissue. VILI,
ventilator-induced lung injury; RES, resorcinolnaphthalein; W/D, wet/dry weight
ratio; IL-6, interleukin-6; IL-1
We examined the effects of RES and MLN-4760 on ERS and pyroptosis in VILI. RES significantly suppressed the expression of CHOP and GRP78 proteins in VILI, and ACE2 inhibition further aggravated ERS (Fig. 6A,B). ACE2 activation attenuated ROS accumulation and LDH efflux in lung tissue and BALF with VILI induction, whereas ACE2 inhibition exacerbated pyroptosis (Fig. 6C,D). GSDMD expression in lung tissues was significantly inhibited by RES but enhanced remarkably by MLN-4760, as shown by the results of immunohistochemistry (Fig. 6E,F). Taken together, ACE2 reduction inhibits the conversion of Ang II to Ang (1-7), contributing further to ERS and pyroptosis induction in VILI.
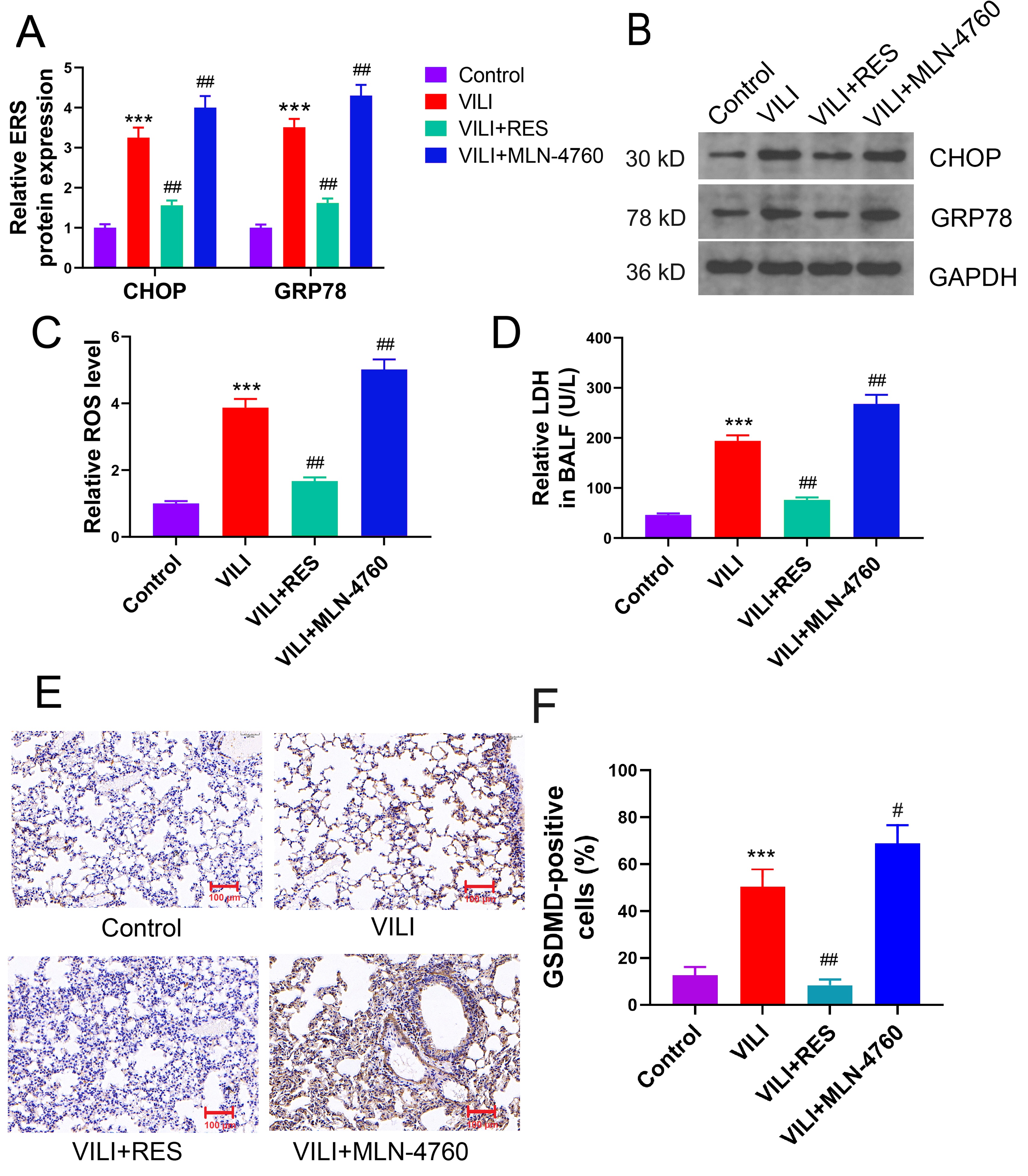 Fig. 6.
Fig. 6.
ACE2 mitigates ERS and pyroptosis in VILI by promoting Ang (1-7)
production. (A,B) Effects of ACE2 activator and inhibitor (MLN-4760) on ERS in
lung tissues of mice with VILI. (C) ROS levels in lung tissues. (D) Levels of LDH
secretion in BALF. (E,F) Effects of activation or inhibition of ACE2 on GSDMD
expression in VILI. ERS, endoplasmic reticulum stress; VILI, ventilator-induced
lung injury; CHOP, C/EBP homologous protein; GRP78, glucose-regulated protein 78;
RES, resorcinolnaphthalein. Scale bar = 100 µm. ***p
To further substantiate the mechanism through which ACE2 mitigates ERS and pyroptosis in VILI, rescue experiments were performed. ACE2 protein was overexpressed by transfection, while A779 was used to antagonize Mas, the receptor for Ang (1-7). The mice were categorized into the four following groups: control, VILI, VILI+ACE2, VILI+ACE2+A779. Augmenting ACE2 protein expression effectively curbed lung tissue damage, ameliorated lung edema (W/D), and attenuated inflammation. Notably, while A779 had no significant effect on the ACE2 protein expression, it markedly reduced the ameliorative effects of ACE2 overexpression on VILI (Fig. 7A–C). A779 did not significantly change the ACE2 overexpression in lung tissues, as shown by the results of immunohistochemistry (Fig. 7D,E). Furthermore, in vivo overexpression of ACE2 reduced the accumulation of Ang II in lung tissues while promoting the production of Ang (1-7). The Mas inhibitor had no statistically discernible effect on Ang II and Ang (1-7) (Fig. 7F,G). The substantial attenuation of VILI by ACE2 protein was markedly hindered upon blocking of the Ang (1-7) receptor. ACE2’s mechanism for inhibiting VILI is intrinsically linked to the catalytic generation of Ang (1-7).
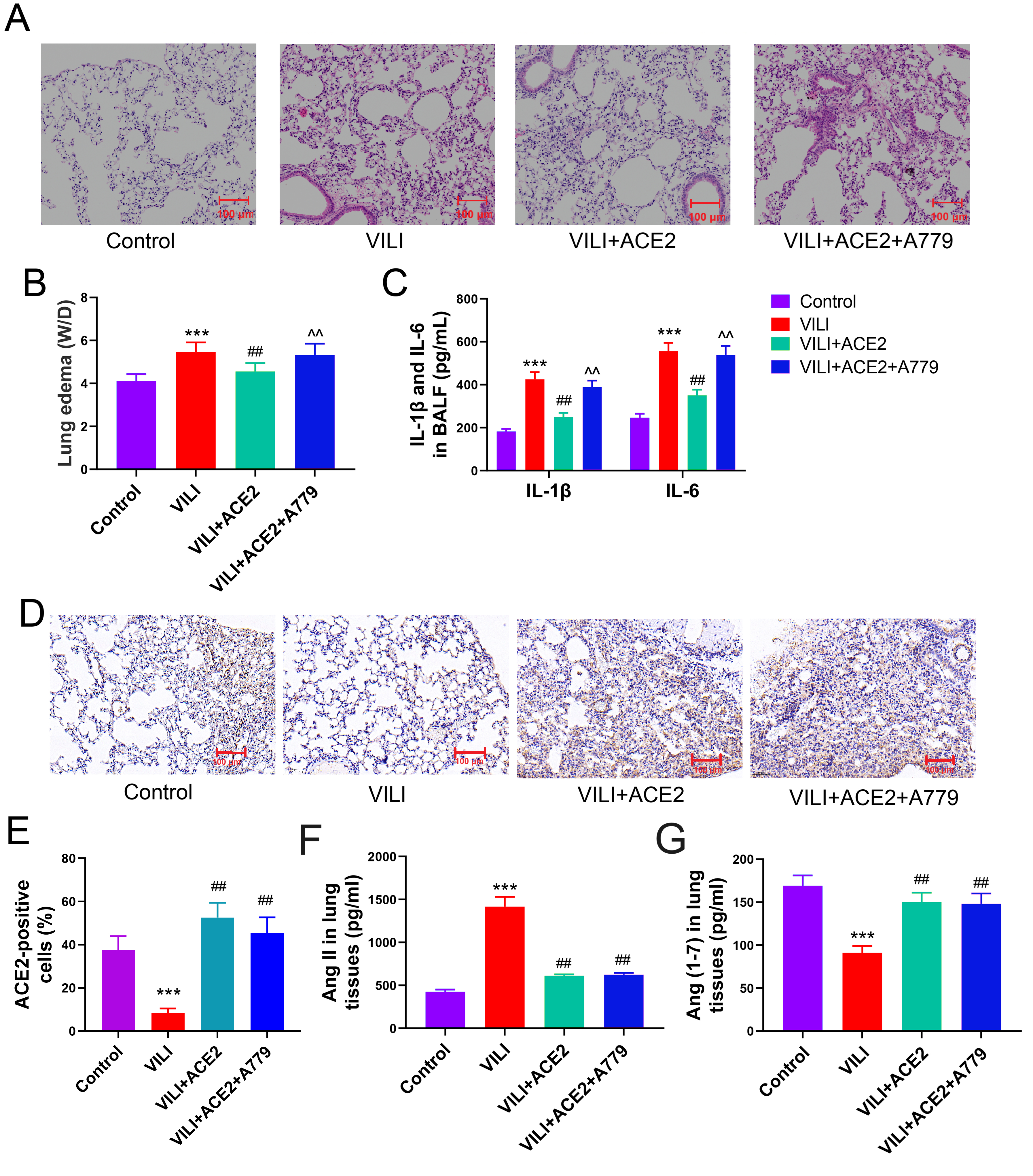 Fig. 7.
Fig. 7.
Inhibition of the Ang (1-7) receptor blocks ACE2’s effects on
lung injury in mice with VILI. (A) Effect of ACE2 overexpression and inhibitor
of the Ang (1-7) receptor, MAS1 (A779) on lung injury in mice with VILI. (B,C)
Comparison of lung edema and inflammation in each group. (D,E) Comparison of ACE2
protein levels in the lung tissue. (F,G) Comparison of Ang II and Ang (1-7)
levels in the lung tissue. VILI, ventilator-induced lung injury; ACE2,
angiotensin-converting enzyme 2; RES, resorcinolnaphthalein; W/D, Wet/Dry weight
ratio; IL-6, interleukin-1; IL-1
The effects of overexpression of ACE2 protein and inhibition of the Ang (1-7) receptor on ERS and pyroptosis were assessed. With enhanced ACE2 protein levels, the expression of CHOP and GRP78 proteins and ROS production were substantially suppressed. ERS in the VILI+ACE2+A779 group was notably higher than that in the VILI+ACE2 group (Fig. 8A–C). Furthermore, ACE2 overexpression effectively reduced LDH efflux into BALF and suppressed NLRP3 activation, along with the generation of GSDMD-N. The level of pyroptosis in the VILI+ACE2+A779 group was significantly higher than that in the VILI+ACE2 group (Fig. 8D–F). Ang (1-7) receptor blockade significantly inhibited the alleviation of ERS and pyroptosis by ACE2 protein in VILI. ACE2’s mechanism for inhibiting ERS and pyroptosis in VILI is closely associated with its catalytic generation of Ang (1-7).
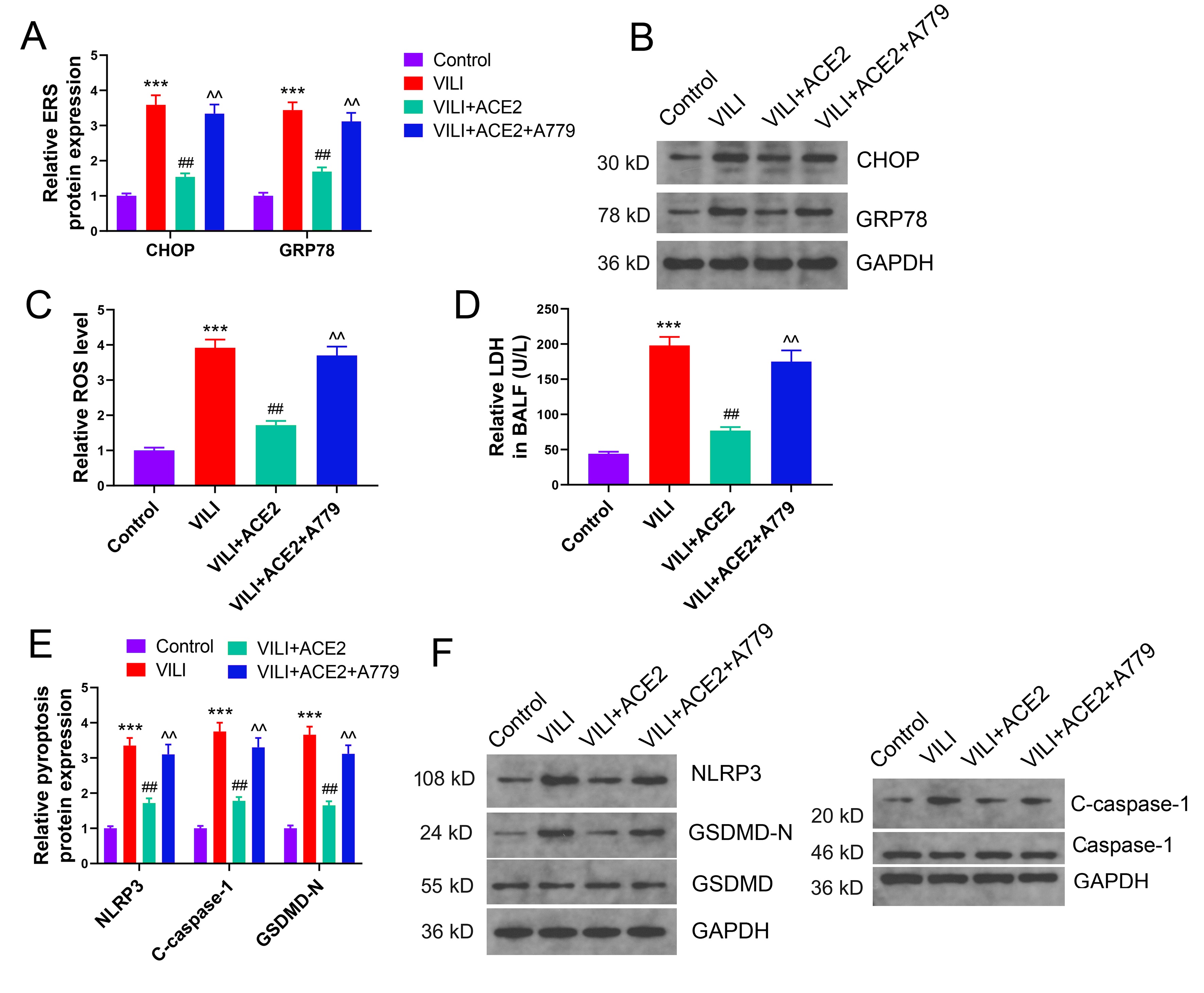 Fig. 8.
Fig. 8.
Ang (1-7) receptor antagonist blocks the inhibitory effects of
ACE2 on ERS and pyroptosis in VILI mice. (A–C) Effect of overexpression of ACE2
and inhibition of the Ang (1-7) receptor, MAS1 (A779) on ERS and ROS levels in
mice with VILI. (D) Levels of LDH efflux in BALF. (E,F) Effect of ACE2
overexpression and MAS1 inhibitor (A779) on pyroptosis. ERS, endoplasmic
reticulum stress; CHOP, C/EBP homologous protein; GRP78, glucose-regulated
protein 78; VILI, ventilator-induced lung injury; ACE2, angiotensin-converting
enzyme 2; ROS, reactive oxygen species; LDH, lactate dehydrogenase; BALF,
bronchoalveolar lavage fluid; NLRP3, NOD-like receptor protein 3 inflammasome;
GSDMD, gasdermin D. ***p
Ventilators are indispensable in modern medical practice, playing a pivotal role in providing MV for patients [27]. This vital medical device is instrumental in preventing and treating respiratory failure, thereby reducing potential complications and contributing to saving and extending patients’ lives. The significance of ventilators lies in their capacity to substitute natural breathing, ensuring the delivery of oxygen to the patient’s body which enables them to overcome respiratory challenges, and thus, crucially supporting patients in critical conditions, thereby enhancing their chances of recovery and ensuring their survival [28]. However, the prolonged usage of ventilators can induce VILI. An in-depth analysis of the underlying mechanisms of VILI is necessary to improve patient outcomes [29, 30].
ACE2, an essential converting enzyme, catalyzes the cleavage of Ang II into the vasodilatory Ang (1-7), thus playing a multifaceted role in regulating blood pressure, inflammation, proliferation, and fibrosis [31, 32, 33]. ACE2’s ability to ameliorate lung injuries is well established; for instance, exogenous ACE2 intervention is promising in mitigating lung injury induced by lipopolysaccharide [34]. ACE2’s function has been implicated in alleviating disease and regulating immunity in virus-induced lung injuries [35, 36]. ACE2’s potential involvement in VILI has been indicated previously [37]. Activation of ACE2 in VILI mice can promote Ang (1-7) production, thereby relieving symptoms [19]. Initial analysis revealed a decrease in ACE2 protein levels after VILI induction, alongside a decline in Ang (1-7) levels and accumulation of Ang II. Interestingly, Ang II receptor antagonists can reduce alveolar edema and inhibit inflammatory cells in VILI, while intravenous administration of Ang (1-7) can alleviate lung injuries in rodents [38, 39, 40]. These findings tentatively suggest the involvement of ACE2 in VILI.
During MV, passive CS and abnormal oxygen levels in alveolar cells directly induce ERS, subsequently resulting in increased ROS levels, which trigger inflammation [9, 41]. NLRP3, an inflammasome responsive to ROS levels, exacerbates inflammation and promotes c-caspase-1 activation, eventually inducing pyroptosis [42, 43]. A recent study has unveiled the occurrence of pyroptosis in VILI, with elevated levels of GSDMD in the BALF of mice with VILI, and oxytocin suppresses inflammation and alleviates pyroptosis [44]. Similarly, caspase-1 levels are elevated in VILI exosomes, leading to pyroptosis in other tissues [45]. The present study showed significant ERS induction in mice with VILI, as evidenced by elevated LDH levels in BALF and increased GSDMD-N expression in tissues which underscores ERS and pyroptosis induction in VILI. To further substantiate whether ERS induces pyroptosis in VILI, rescue experiments were performed using 4-PBA to inhibit ERS. ERS inhibition significantly mitigated pyroptosis under VILI, suggesting that VILI-induced pyroptosis is due to ERS.
Ang II/Ang (1-7) can also modulate ERS and pyroptosis. For instance, Ang II has been implicated in obesity whereby it induces ERS in adipocytes [46]. Ang (1-7) can protect HK2 cells from hydrogen peroxide-induced damage by inhibiting ERS [47]. However, whether ACE2, which plays a key role in facilitating the conversion of Ang II into Ang (1-7), can alleviate pyroptosis by regulating ERS in VILI, remains unclear. Consequently, cellular experiments have been conducted to confirm the effect of Ang (1-7) on pyroptosis in VILI through ERS. In vitro, VILI was simulated by CS of alveolar cells. Sustained ERS was induced by TM, and Ang (1-7) was added to the medium for intervention. Ang (1-7) could alleviate ERS and pyroptosis under conditions simulating VILI. Notably, when ERS was continuously activated, the ability of Ang (1-7) to inhibit pyroptosis was compromised, suggesting that Ang (1-7) can alleviate pyroptosis by inhibiting ERS in VILI.
Ang (1-7) is enzymatically catalyzed by ACE2, making it imperative to comprehensively analyze the effect of ACE2 on ERS and pyroptosis in VILI. Furthermore, investigating whether ACE2’s ability to alleviate ERS and pyroptosis in VILI is intricately linked to Ang (1-7) generation in vivo is necessary. To address this query, RES and MLN-4760 were employed to activate and inhibit ACE2, respectively. The results unequivocally demonstrated that RES effectively promoted the conversion of Ang II to Ang (1-7), restored the suppressed Ang (1-7) levels in VILI, mitigated lung injury, and concurrently inhibited ERS and pyroptosis. Conversely, inhibiting ACE2’s function in the context of VILI hampered the generation of Ang (1-7) and accentuated Ang II accumulation, thus exacerbating ERS and pyroptosis, and ultimately worsening lung injury. Interestingly, the deletion of ACE2 increases ERS in mice, while ACE2 activation alleviates ERS and improves lipid metabolism [48]. ACE2 can ameliorate ERS in endothelial cells exposed to a high-glucose environment [49]. ACE2 mitigates post-traumatic brain neuronal pyroptosis [50]. Furthermore, mesenchymal stem cells that overexpress ACE2 reduced lung injury from COVID-19 by suppressing pyroptosis [51]. Collectively, these findings strongly suggest that reduction in ACE2 levels is pivotal in the induction of ERS and pyroptosis.
To gain further insight into the mechanism by which ACE2 mitigates VILI and its role in catalyzing Ang (1-7) production, the Mas receptor of Ang (1-7) was inhibited along with ACE2 protein overexpression. The Mas antagonist had no significant effect on the expression of ACE2 protein or the production of Ang (1-7), yet remarkably, it significantly impeded the inhibitory effects of ACE2 on ERS and pyroptosis in VILI. Furthermore, blocking Mas affected the protective effect of ACE2 on lung tissue. This compellingly reaffirms that the mechanism underlying ACE2’s ameliorative effect on ERS and pyroptosis in VILI is inseparably linked to Ang (1-7).
This study has some limitations that warrant further consideration. Primarily, it is an experimental investigation based on animal and cell models, and the unavailability of clinical samples from patients with VILI precluded the generalizability of these findings to humans. Consequently, research with clinical samples should be conducted when such resources become accessible. Second, while this study provides valuable insights, the intricate details of the mechanisms through which ACE2 alleviates ERS and pyroptosis warrant further detailed studies. Third, only male mice were used for experiments, which might have caused some bias. Finally, no other component of the renin-angiotensin system, such as ACE1, was subjected to investigation. Further studies on these aspects are required.
In conclusion, the present study demonstrated that reduced ACE2 expression in VILI is involved in ERS and pyroptosis-related injury. ACE2 can alleviate ERS in alveolar cells by catalyzing the production of Ang (1-7), thus inhibiting pyroptosis in VILI. These findings may provide us a novel potential target for VILI treatment.
The data used to support the findings of this study are available from the corresponding author upon request.
XL provided the funding, conducted the experiments, and prepared the manuscript; YZ analyzed the data and revised the manuscript; FG designed, supervised the study and revised the manuscript. All authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Approval for all experimental protocols was granted by the Animal Care Committee of Fuzhou University Affiliated Provincial Hospital (the original name: Shengli Clinical Medical College of Fujian Medical University) (No. 2019-0313).
Thanks to all the peer reviewers for their opinions and suggestions.
This work was supported by Fujian Provincial Health Technology Project (No. 2019-CX-12).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2909334.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.