







1 Cancer Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
2 Center for Genome Analysis, Wuhan Ruixing Biotechnology Co., Ltd., 430075 Wuhan, Hubei, China
3 Center for BioBigData Analysis, ABLife BioBigData Institute, 430075 Wuhan, Hubei, China
§Present address: Wuhan Bio-salt technology Co., Ltd., 430075 Wuhan, Hubei, China.
Abstract
The fate and functions of RNAs are coordinately regulated by RNA-binding proteins (RBPs), which are often dysregulated in various cancers. Known as a splicing regulator, RNA-binding motif protein 6 (RBM6) harbors tumor-suppressor activity in many cancers; however, there is a lack of research on the molecular targets and regulatory mechanisms of RBM6.
In this study, we constructed an RBM6 knock-down (shRBM6) model in the HeLa cell line to investigate its functions and molecular targets. Then we applied improved RNA immunoprecipitation coupled with sequencing (iRIP-seq) and whole transcriptome sequencing approaches to investigate the potential role and RNA targets of RBM6.
Using The Cancer Genome Atlas dataset, we found that higher expression of RBM6 is associated with a better prognosis in many cancer types. In addition, we found that RBM6 knockdown promoted cell proliferation and inhibited apoptosis, demonstrating that RBM6 may act as an anti-oncogenic protein in cancer cells. RBM6 can regulate the alternative splicing (AS) of genes involved in DNA damage response, proliferation, and apoptosis-associated pathways. Meanwhile, RBM6 knockdown activated type I interferon signaling pathways and inhibited the expression of genes involved in the cell cycle, cellular responses to DNA damage, and DNA repair pathways. The differentially expressed genes (DEGs) by shRBM6 and their involved pathways were likely regulated by the transcription factors undergoing aberrant AS by RBM6 knockdown. For iRIP-seq analysis, we found that RBM6 could interact with a large number of mRNAs, with a tendency for binding motifs GGCGAUG and CUCU. RBM6 bound to the mRNA of cell proliferation- and apoptosis-associated genes with dysregulated AS after RBM6 knockdown.
In summary, our study highlights the important role of RBM6, as well as the downstream targets and regulated pathways, suggesting the potential regulatory mechanisms of RBM6 in the development of cancer.
Keywords
- RBM6
- RNA interaction
- DNA damage response
- type I interferon
- alternative splicing
The oncogenesis and development of cancers are very complex processes that are regulated at different molecular layers and by multiple kinds of molecules, including RNAs [1] and RNA binding proteins (RBPs) [2, 3]. RBPs can interact with RNAs and influence all of the biological processes of RNAs from generation to degradation, and are often misexpressed in various cancers leading to functional consequences [4, 5, 6, 7, 8]. A previous study showed that 15 RBPs significantly influenced the prognostic results of patients with head and neck squamous cell carcinoma (HNSCC) [9]. RBM3, one RNA-binding motif protein, can reduce cell apoptosis and promote the radio resistance of nasopharyngeal carcinoma by affecting the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/B-cell lymphoma-2 (Bcl-2) signaling pathway [10]. Moreover, RBPs are dysregulated in various cancers, leading to dysfunctional gene splicing and tumor-specific dependencies [11]. For example, it has been shown that RNA Binding Fox-1 Homolog 2 (RBFOX2)-regulated alternative splicing genes are enriched Ras Homolog Family (RHO) GTPase pathways, through which RBFOX2 reduces the metastatic potential of pancreatic ductal adenocarcinoma cells [12]. However, there has been a lack of research on the role of RBPs in cancer, especially the regulatory mechanism of splicing factors in post-transcriptional processes.
RBM6, a canonical alternative splicing (AS) factor, is frequently mutated in human cancers [13, 14, 15]. RBM6 contains several domains that are associated with AS regulation, including two RNA recognition motifs [16, 17, 18, 19]. RBM6 is often localized in the nucleus and forms foci corresponding to splicing speckles [20]. RBM6 can regulate gene expression and AS, which are involved in tumorigenesis [21]. As a tumor-suppressor, RBM6 inhibits the growth and progression of laryngocarcinoma [22]. In hepatocellular carcinoma (HCC), RBM6 can promote cell apoptosis, and inhibit proliferation, migration, and invasion, thereby inhibiting the progression of HCC [23]. A previous study reported that RBM6 negatively regulates the proliferative capacity of cancer cells by displaying positional effects on AS regulation with the cooperation of RBM5 and RBM10 [24]. These findings demonstrate that RBM6 has important functions in human tumorigenesis. Thus, it is worthwhile to further study the regulatory functions of RBM6 in tumors and its downstream targets to identify novel potential therapeutic targets.
To this end, in the current study, we assessed the expression patterns and survival effects of RBM6 on 24 cancer types available in The Cancer Genome Atlas (TCGA) database. The results revealed that patients with higher RBM6 expression levels often had better overall survival in most cancer types. To further investigate how RBM6 regulates gene transcription and AS, and how RBM6 is involved in cancer progression, we constructed an RBM6 knockdown model in the HeLa cell line. Furthermore, we used RNA-seq to study global transcriptome profile in order to investigate the downstream RNA targets of RBM6 in HeLa cells and analyze the alterations of signaling pathways caused by RBM6 knockdown. We also identified the RBM6-regulated AS events (RASEs). Moreover, we identified the genes bound by RBM6 using the improved RNA immunoprecipitation and sequencing approach (iRIP-seq). The results of this study highlight the important regulatory roles and downstream targets, as well as the potential regulatory mechanisms of RBM6 in the pathogenesis or progression of tumors.
We downloaded and obtained the gene expression (RNA-seq) and survival information datasets of 24 cancer types from TCGA project, including tumor and normal samples, from the UCSC XENA database (https://xenabrowser.net/datapages/). Then we analyzed the expression levels of RBM6. The survival package in R (version 4.2.3) was used to perform survival modeling and Kaplan-Meier (KM) analyses.
The short hairpin RNA (shRNA) duplexes of RBM6 were purchased from GenePharma (Suzhou, China). The negative control shRNA (shNC) sequence was: 5′-TTCTCCGAACGTGTCACGT-3′ (sense), whereas the shRNA targeting RBM6 (shRBM6) sequence was: 5′-TTCAGGTTCCTTATCAGCTGG-3′ (sense).
The HeLa cell line (CL-0101; Procell, Wuhan, China) was validated by STR profiling and tested negative for mycoplasma, and then cultured at 37 °C with 5% CO2 in RPMI-1640 medium (CM-0063; Procell) supplemented with 10% fetal bovine serum (10091148; Gibco, Beijing, China), 100 µg/mL streptomycin, and 100 U/mL penicillin (SV30010; Hyclone, Logan, UT, USA). Then shRBM6 and shNC were transfected into HeLa cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. After 48 h, the HeLa cells were collected for subsequent experiments and analysis.
The cell proliferation assay was performed as previously described [25] using the Cell Counting Kit-8 assay (40203ES76; Yeasen, Shanghai, China). Briefly, shRBM6- and shNC-transfected HeLa cells were seeded in culture plates at a density of 10,000 cells/well. After 24, 48, and 72 h of incubation, 10 µL CCK-8 solution was added to the cells followed by incubation for an additional 3 h at 37 °C. Then the optical density (OD) was measured at an absorbance of 490 nm using a microplate reader (ELX800; Biotek, Winooski, VT, USA). The OD values were used to calculate the proliferation rate.
Apoptotic cells were stained using the Annexin V-PE/7-AAD Apoptosis Detection
Kit (KGA-1017; KeyGEN, Nangjing, China) after 72 h of transfection. Then the
shRBM6- and shNC-transfected cells (2
DNA was removed by RQ1 DNase (Promega Corp., Madison, WI, USA) to extract total RNA. After quality and quantity assessment of RNA by the SmartSpec Plus spectrophotometer (BioRad Laboratories, Inc., Hercules, CA, USA), we used 1 µg integral RNA for RNA-seq library construction with the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (#E7420; New England Biolabs, Ipswich, MA, USA) by purifying polyadenylated RNAs. Then the RNAs were fragmented and converted into cDNAs. After end repair and A tailing, the DNAs were ligated with the Diluted NEBNext® Adaptor (New England Biolabs, Ipswich, MA, USA). The ligation products corresponding to 300–500 base pairs and digested with heat-labile uracil-DNA glycosylase, were processed and stored at –80 °C for sequencing. For next-generation sequencing, the libraries were applied to the Illumina NovaSeq 6000 sequencing system (Novogene, Beijing, China) for 150 nucleotide (nt) paired-end sequencing.
We initially discarded raw reads with N bases, removed adaptors and low-quality bases using FASTX-Toolkit (version 0.0.13, https://github.com/Debian/fastx-toolkit), and finally kept reads longer than 16 nt. The quality filtered reads were aligned to the GRCh38 genome by HISAT2 [26]. Mapped reads with only one genomic location were used to calculate the read count of genes and the normalized fragments per kilobase of transcript per million fragments mapped (FPKM).
We used the splicing site usage variation analysis
(SUVA, version 1.0.0,
https://github.com/ablifedev/SUVA) pipeline [27] to calculate and quantify the AS
events (ASEs) and RASEs (p-value
The differentially expressed genes (DEGs) were predicted using the
DEseq2 software package (version 1.42.0,
https://github.com/thelovelab/DESeq2) [28], which analyzes the differential
expression of genes. A two-fold change (FC) and false discovery rate (FDR)
The iRIP-seq technique was conducted as previously described [29]. Briefly,
after irradiation at 400 mJ/cm2, the HeLa cells were lysed in cold wash
buffer supplemented with RNase inhibitor (2313U; Takara Bio Inc., Shiga, Japan)
and protease inhibitor cocktail (B14001; Bimake, Shanghai, China) on ice for 30
min. Then the RNA/protein mixture was vibrated vigorously and centrifuged to
remove cell debris. Then RNAs were digested by MNase and RNaseT1 (Thermo Fisher
Scientific, Waltham, MA, USA). Ethylenediamine tetra-acetic acid
(EDTA, Sangon Biotech., Shanghai, China) was
added to stop the digestion. The supernatant was incubated with 13 µg RBM6
antibody (14360-1-AP; Proteintech, Hongkong, China) and control IgG-antibody
(AC005; ABclonal, Wuhan, China) overnight at 4 °C. After applying the
mixture to the magnet and removing the supernatants, the beads were sequentially
washed twice with lysis buffer. Next, the suspension was incubated in a heat
block to release the immunoprecipitated RBPs with crosslinked RNAs and vortexed
for 30 min. Proteinase K (Sangon Biotech., Shanghai, China) was added to the 10%
input (without being immunoprecipitated) and immunoprecipitated RBM6 with
crosslinked RNAs. The RNAs were purified with phenol: chloroform: isopentyl
alcohol (25:24:1 pH
The cDNA libraries were prepared with the KAPA RNA Hyper Prep Kit (KK8541, Roche, Basel, Switzerland) according to the manufacturer’s instructions. Then the libraries were prepared following the manufacturer’s instructions and applied to the Illumina NovaSeq sequencing system for 150 nt paired-end sequencing.
We aligned the quality filtered reads onto the genome by HISAT2 (version 2.2.1, https://github.com/DaehwanKimLab/hisat2) [26], kept the uniquely aligned reads, and removed the PCR duplicates with the same aligning position. Then, Piranha was used to perform peak calling between RBM6-IP and input samples [30]. The target genes of RBM6 were finally determined by the peaks. The binding motifs of RBM6-bound peaks were called by HOMER software (version 4.10, https://github.com/javrodriguez/HOMER) [31].
Quantitative PCR (qPCR) was conducted as previously described [32]. Briefly, cDNAs were synthesized on a thermocycler (T100; Bio-Rad, Hercules, CA, USA) using a Reverse Transcription kit (R323-01; Vazyme Biotech, Nanjing, China), and qPCR was performed on the ABI QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific) following the standard operational process. The concentration of each transcript or splicing event was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and their levels were calculated using the 2-ΔΔCT method [33]. The unpaired and two-tailed Student’s t-test were used to compare the significant differences of each transcript or splicing event, the primer sequences are presented in Supplementary Table 1. Each sample had three technical replicates.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genome (KEGG) databases
were used to calculate functions and enriched score and significance for each
functional pathway using the KOBAS 2.0 server [34]. The
statistical significance of each pathway was defined using hypergeometric tests
and the Benjamini-Hochberg FDR controlling procedure (corrected p
The two-tailed unpaired Student’s t-test was used for other comparisons
that were not defined above between the shRBM6 and control groups. The threshold
for a statistically significant difference was set as p
Previous studies have demonstrated that RBM6 can serve as a tumor suppressor and
repress tumor growth and progression [22, 23, 35]. We compared RBM6
expression in tumor vs. adjacent normal samples in 24 cancer types from
TCGA. Among the 24 cancer types,
RBM6 expression was upregulated (p
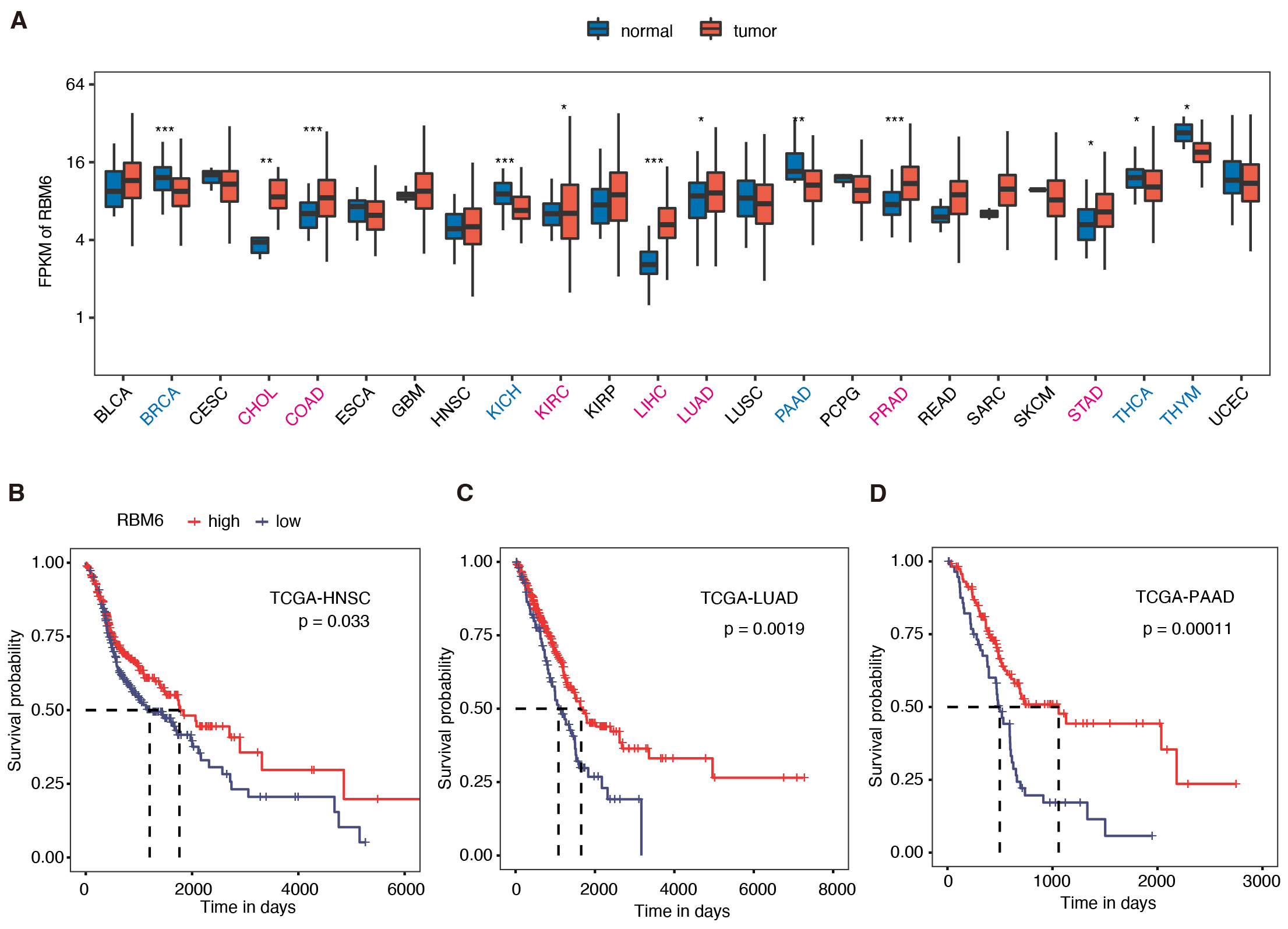 Fig. 1.
Fig. 1.
Higher RBM6 expression was associated with better
prognosis in several tumor types by pan-cancer analysis. (A) The boxplot shows
RBM6 expression pattern in tumor vs. normal tissues in the 24 cancer
types from TCGA. RBM6 expression was up-regulated in seven cancer types, and
down-regulated in five cancer types. * p
To further investigate the functions of RBM6 in tumor cells, we constructed an
RBM6 knockdown (shRBM6) cell model by transfecting an RBM6-shRNA
sequence into HeLa cells; HeLa cells transfected with an empty vector were served
as the NC. We assessed the knockdown efficiency of shRBM6 by the real-time
polymerase chain reaction (RT-PCR) and found that RBM6 was significantly down
regulated in the shRBM6 samples (p
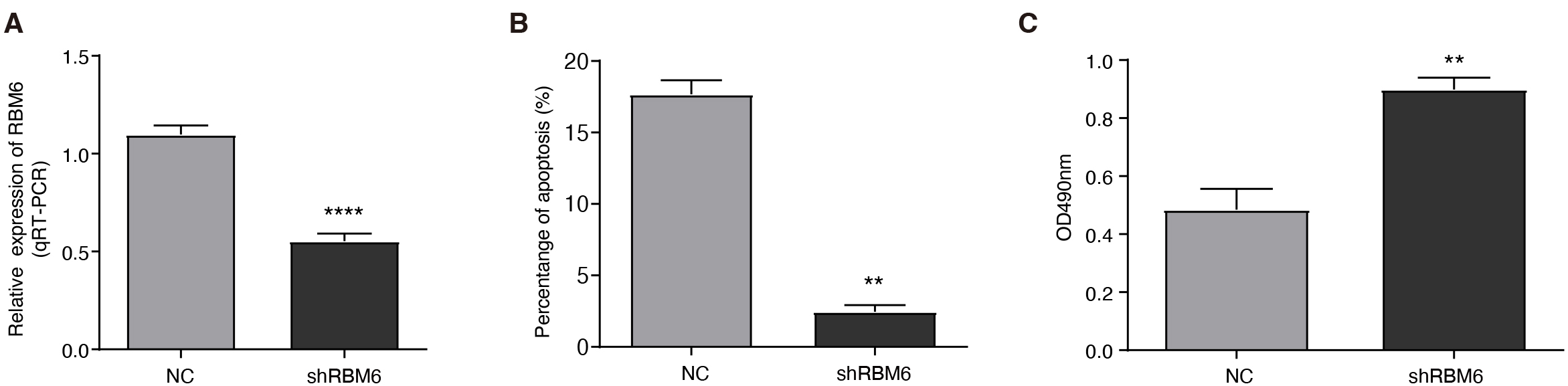 Fig. 2.
Fig. 2.
RBM6 knockdown promoted proliferation and
inhibited apoptosis. (A) The bar plot shows reverse transcription and quantitative polymerase chain reaction (RT-qPCR) validation of RBM6
knockdown efficiency. (B) The bar plot shows the decreased cellular apoptosis
rate in the shRBM6 group. (C) The bar plot indicates the increased cellular
proliferation level in the shRBM6 group. The values are presented as the
mean
To further elucidate how RBM6 modulates the synthesis and processing of RNA in HeLa cells, we performed unbiased RNA-seq analysis for shRBM6 and normal controls (NC) HeLa cells by capturing the total polyadenylated RNAs, with three biological replicates for each group. After quality filter and genome alignment of the raw RNA-seq data, more than 95% of the aligned reads had a uniquely aligned location in the genome and were used for further analyses (Supplementary Table 2). By calculating the read count and FPKM value for each gene, we detected a total of 28,218 expressed genes from the six RNA-seq samples (Supplementary Table 3). We presented the FPKM values of RBM6 and confirmed its successful knockdown from the RNA-seq analysis results (Fig. 3A).
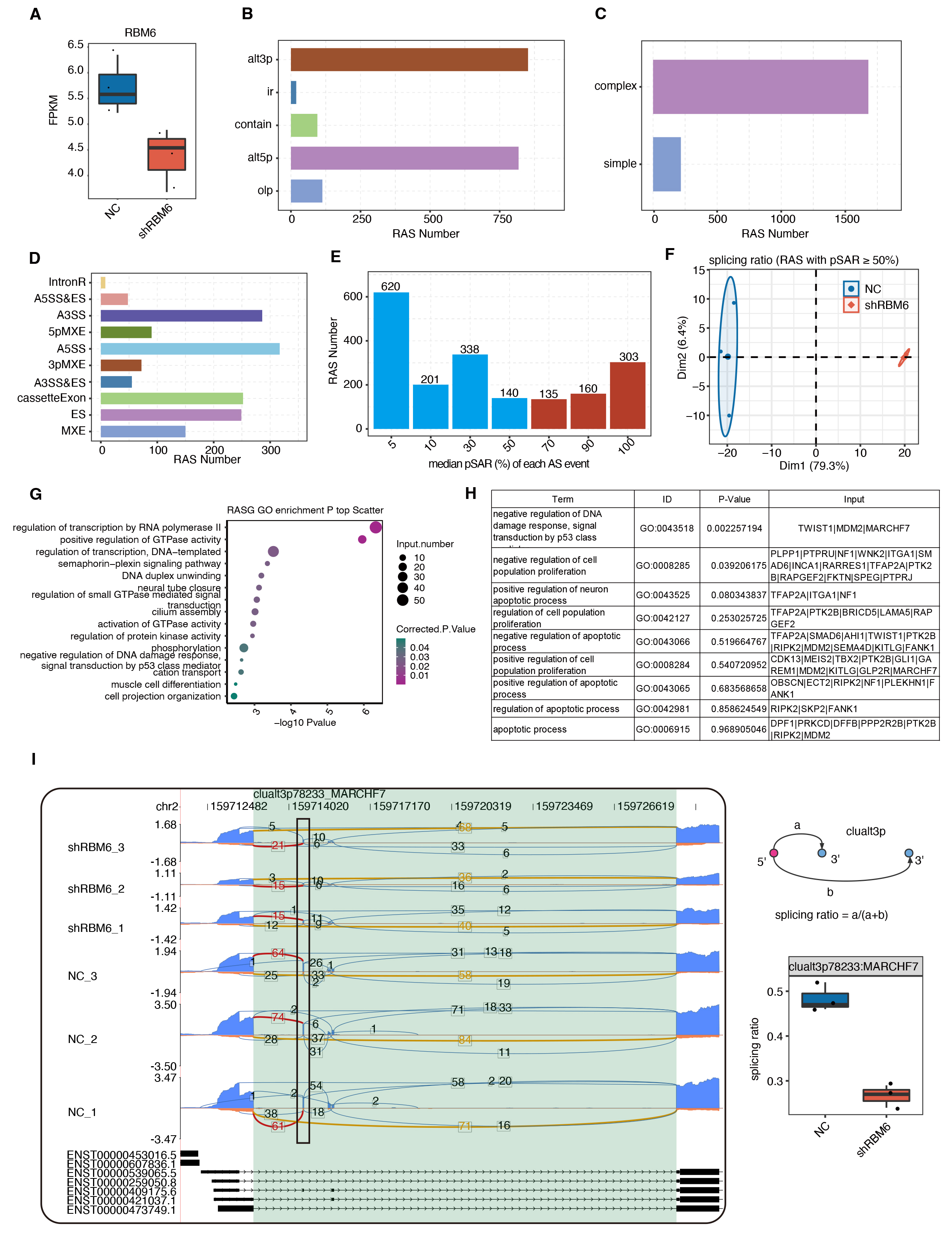 Fig. 3.
Fig. 3.
RBM6 modulated the AS pattern of genes enriched in DNA
damage response- and cell survival-related pathways. (A) Boxplot shows the Fragments per kilobase per million mapped (FPKM)
of RBM6. (B) Barplot shows the number of RASEs detected by SUVA between shRBM6
and NC. (C) Barplot presents the number of RASEs by dividing them into
complex and simple ASEs using SUVA. (D) Barplot shows the corresponding classical
ASE types for the RASEs detected by SUVA. (E) Barplot shows the number of RASEs
with increased pSAR values. RASEs with pSAR
As an AS factor, RBM6 is supposed to modulate the tumorigenesis or development
probably by playing an essential role in regulating the AS profile. Since cancers
undergo complex splicing events [36], we detected thousands of ASEs in RNA-seq
using SUVA [27] (Supplementary Fig. 3A). Among these ASEs, 1897
high-confidence RASEs were identified by applying stringent cutoffs of p
These 598 RBM6-RASEs were distributed in 463 genes, which were enriched in GTPase and transcription pathways (Fig. 3G). We found that RBM6 regulates the AS of many genes involved in proliferative and apoptotic processes (Fig. 3H). The reads distribution diagram showed an RASE in the membrane associated ring-CH-type finger 7 (MARCHF7) gene, and the splicing model showed the significantly dysregulated splicing pattern of this event (Fig. 3I). In such a complex AS region, SUVA identified the RASE that was significantly altered between the shRBM6 and control samples. As shown in Fig. 3I, the exons shown as black boxes were skipped after knocking down RBM6. Another RASE, located in PRKCD, showed a tendency for intermediate exons (Supplementary Fig. 3D). These results indicate that RBM6 may affect the survival and development of cancer cells by regulating variable splicing of genes involved in cell proliferation or apoptosis.
To investigate RBM6-mediated transcriptional regulation, the DESeq2 package was
used to identify the DEGs between shRBM6 and NC samples using FC
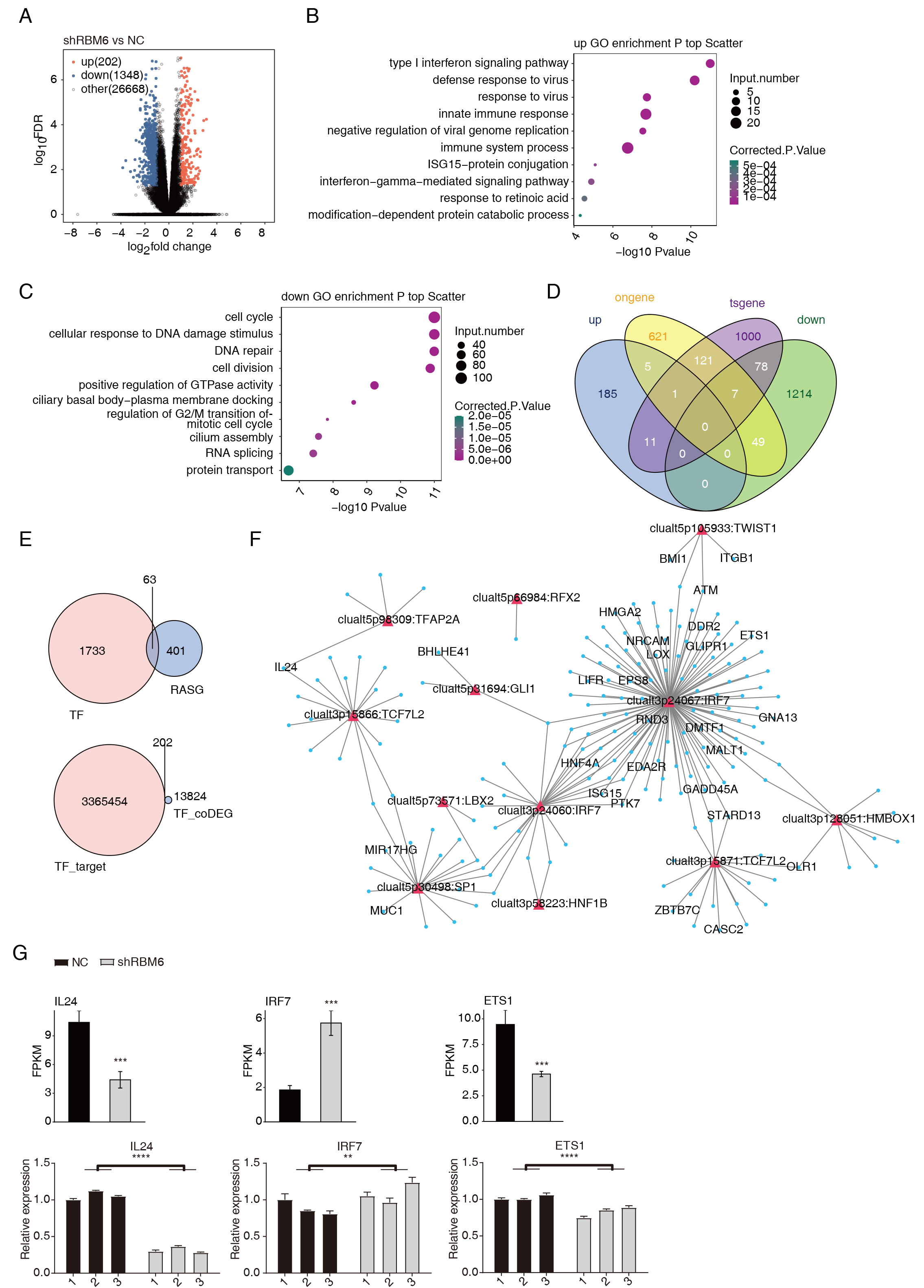 Fig. 4.
Fig. 4.
RBM6 significantly altered the expression of genes associated
with tumor development, which may be mediated by TFs from RBM6-RASGs. (A)
Volcano plot presents the upregulated- and downregulated-DEGs in the shRBM6
vs. NC groups using DESeq2. (B) The top 10 enriched GO pathways of
up-regulated DEGs in the shRBM6 group. (C) The top 10 enriched GO pathways of
down-regulated DEGs in the shRBM6 group. (D) Venn diagram shows the upregulated-
and downregulated-genes and the known oncogenes or tumor suppressor genes. (E)
Venn diagram shows the overlap of TFs and RASGs. Venn diagram showing the overlap
of TF-targets and DEGs (above). Venn diagram shows TF-targets and TF-related DEGs
overlay (below). (F) Network showing TF-target pairs were screen out and the
co-disturbed. (G) Bar plot showing the RT-qPCR results. TF, ranscription factors;
DEGs, differentially expressed genes (** p
To further identify the potential functions of these DEGs, we performed GO (Supplementary Table 6) and KEGG (Supplementary Table 7) enrichment analyses. The top 10 upregulated- and downregulated GO terms and the top 10 upregulated- and downregulated KEGG pathways were shown in Fig. 4B,C and Supplementary Fig. 4B,C, respectively. The up-regulated genes in the shRBM6 cells are primarily enriched in type I interferon (IFN) signaling pathways, defense response to virus, and the innate immune response (Fig. 4B). Type I IFN is tightly associated with viral infection and has double-edged effects on tumor development and resistance to treatment [37, 38]. Interestingly, the down-regulated DEGs were mainly associated with cell cycle and DNA damage response (DDR) pathways, including cellular responses to DNA damage stimulus, cell cycle, and DNA repair. Positive regulation of GTPase activity, RNA splicing, and cilium assembly were also enriched in the down-regulated DEGs (Fig. 4C). Cellular responses to DNA damage stimulus are closely related to apoptosis in multiple cancers [39, 40]. These results suggest that shRBM6 can activate the type I IFN signaling pathway and suppress the DDR, ultimately inhibiting the apoptosis of cancer cells. Meanwhile, we also found that the expression of many oncogenes or tumor suppressor genes was detected in the DEGs caused by shRBM6 (Fig. 4D and Supplementary Fig. 4D).
RBM6 is an important AS regulator. The genes involve RBM6-RASE (RASGs) were
enriched in transcriptional regulatory pathways (Fig. 3G). We speculate that the
AS of many transcription factors (TFs) is regulated by RBM6, which in turn has an
impact on the transcriptome. In total, 63 of 464 RASGs were identified as TFs
(Fig. 4E). We conducted a correlation analysis of the expressions of DEGs and the
splicing ratios of RASEs from these 63 TFs (Pearson’s correlation coefficient
To study the RBM6-RNA interactions and the mechanism underlying the function of RBM6, we obtained the global RBM6-RNA interaction profile in HeLa cells by iRIP-seq [29, 41]. We used ultraviolet cross-linking and RNA digestion to capture the exact RNA-protein interaction locations. The iRIP cDNA libraries were sequenced on the Illumina platform, generating abundant iRIP-seq reads for RBM6 IP and input control samples with two biological replicates for each (Supplementary Table 8). We processed and aligned the cDNA sequence reads to the human GRCh38 genome using HISAT2. The fractions of the aligned reads were 78.14–86.99%, from which we selected the uniquely aligned reads for following analyses (Supplementary Table 8). By classifying the uniquely mapped reads to the genomic location, we found that the RBM6-associated reads showed a higher percentage in the 5′UTR and intergenic regions but lower percentage in the intron and coding sequence (CDS) regions (Fig. 5A). The normalized reads density along the uniformed gene region showed that RBM6 had a stronger tendency for the 5′UTR regions compared with the CDS and 3′UTR regions (Fig. 5B).
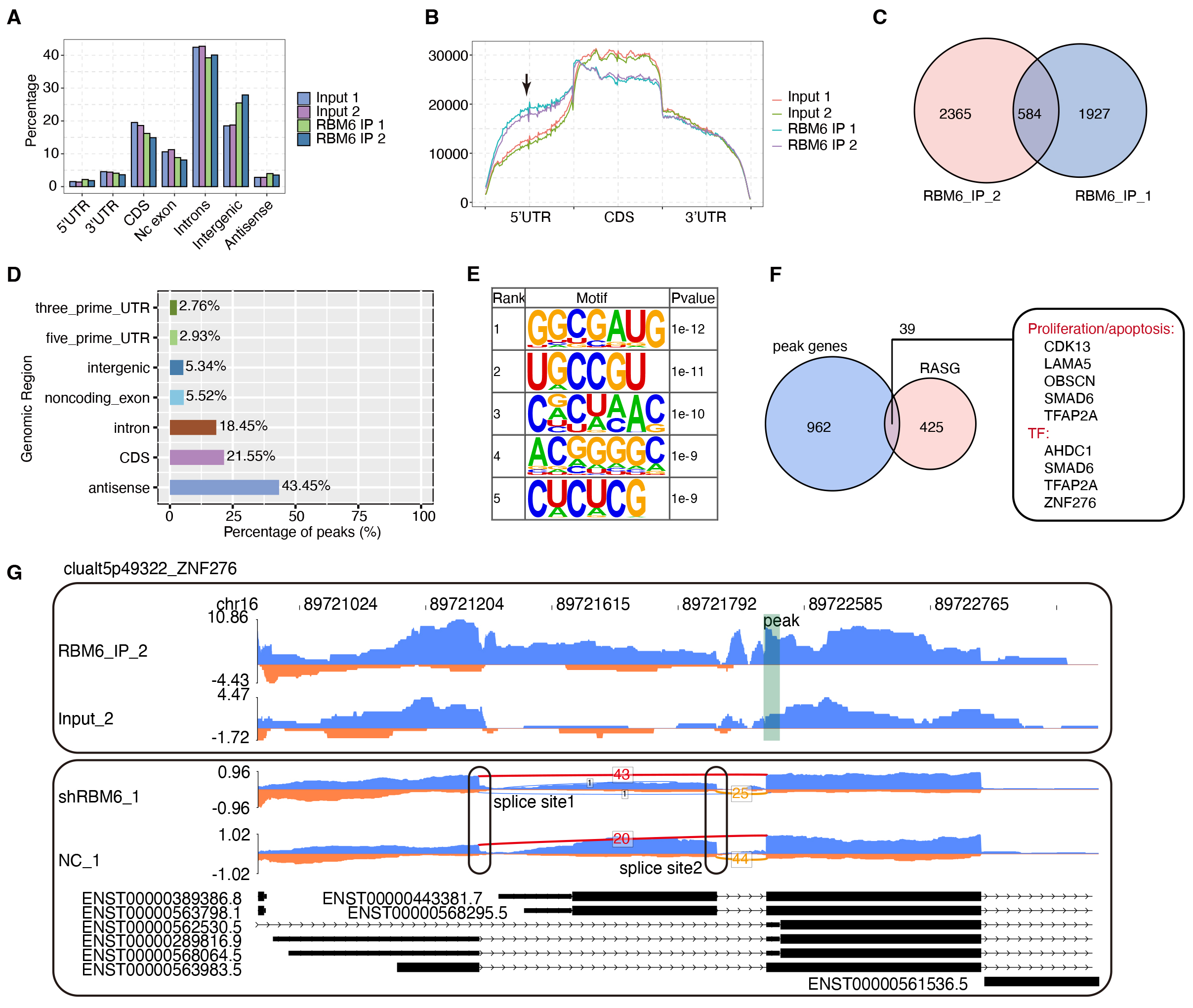 Fig. 5.
Fig. 5.
RBM6-mRNA interaction map in HeLa cells. (A) Barplot shows the distribution of sequencing reads on different genome regions. (B) Coverage intensity analysis of iRIP-seq reads with the length of the transcription unit was carried out. The 5′UTR, CDS, and 3′UTR of the gene were divided into 100 evenly, each of which was called a bin. The sum of the average reads in each bin was calculated to obtain the overall reads coverage. (C) Venn diagram shows the common and unique peaks in two IP replicate treatment groups by using the Piranha method. The peaks with overlapping positions are clustered in the two replicates, and the overlapping peaks are classified under the same cluster. (D) The proportion of peak reads in different regions of the genome. (E) The top five enriched motifs in the overlapped RBM6 peaks. (F) RASGs co-expressing with RBM6 were screened. Venn diagram shows the peaks bound by RBM6-RASGs. (G) Reads plot shows one RASE in the ZNF276 and RBM6 binding profiles. iRIP-seq, RNA immunoprecipitation coupled with sequencing; CDS, coding sequence; IP, immunoprecipitation; ZNF276, zinc finger protein 276.
To further elucidate the characteristics of the RBM6-RNA interaction, we called the RBM6-binding peaks from iRIP-seq reads using Piranha. In total, 584 shared peaks between the two replicates were identified (Fig. 5C). The genomic locations of these peaks showed that most of these peaks came from the antisense, CDS and intron regions (Fig. 5D). Further analysis showed that RBM6 binding reads were slightly and specifically enriched in the transcription start site, whereas peak reads were significantly enriched in the intron region (Supplementary Fig. 5A,B). Next, we applied the HOMER algorithm to identify the enriched motif sequences over-represented in the RBM6 peaks. Consistent with a previous report [21], the CUCU motif was enriched. However, the top motif was GGCGAUG, indicating that the motif selection could be cellular heterogeneity (Fig. 5E).
Functional clustering by GO was performed to annotate the functions of the RBM6-bound genes containing at least one RBM6 peak; the enrichment fell into the cell cycle, cell differentiation, and cell migration (Supplementary Fig. 5C). As shown in Fig. 5F, 39 RASGs overlapped with RBM6-bound genes, indicating that RBM6 might directly bind to the RNAs of these targets and regulate their AS pattern. In addition, several genes were found to be involved in proliferation or apoptosis, including cyclin-dependent kinase 13 (CDK13), laminin subunit alpha 5 (LAMA5), obscurin (OBSCN), SMAD Family Member 6 (SMAD6), and transcription factor AP2 (TFAP2A). Four TFs, AT-Hook DNA Binding Motif Containing 1 (AHDC1), SMAD6, TFAP2A, and zinc finger protein 276 (ZNF276), were also directly bound by RBM6. Visualization of the reads profile showed an RASE in ZNF276 and RBM6-binding profiles (Fig. 5G). RBM6 specifically binds to the first 5′ exon at the proximal end, and this exon is more selected in the control group, whereas the first 5′ exon at the distal end is more selected after RBM6 knockdown.
In this study, we investigated whether RBM6 can exercise functions through transcriptional or post-transcriptional regulation in HeLa cells. According to pan-cancer data from TCGA, RBM6 was upregulated in 7 of 24 cancer types and downregulated in 5 cancer types and higher RBM6 expression was positively correlated with a better prognosis. These results indicate that RBM6 may have an important role in tumor development. Based on this hypothesis, the HeLa cell model was used to analyze the cellular phenotypes and molecular alterations by knocking down RBM6 with shRNA. ShRBM6 promoted cell proliferation and inhibited cell apoptosis. Furthermore, using RNA-seq, we found that shRBM6 regulated AS pattern of genes involved in cell proliferation and apoptosis pathways, as well as genes involved in transcriptional regulation. The up-regulated DEGs were enriched in the Type I IFN pathway and the down-regulated DEGs were enriched in the DDR pathway. These results suggest that as a splicing factor, RBM6 may regulate the splicing of TF-associated genes. To further investigate how RBM6 plays a role using its RNA binding activity, we analyzed the binding peaks of RBM6 by iRIP-seq reads using the Piranha pipeline [30]. The results showed that RBM6 could bind to genes related to cell apoptosis, cell proliferation, and TFs.
RBM6 is a tumor suppressor gene in multiple cancers. For example, RBM6 can repress the progression of laryngeal carcinoma [22] and HCC [23]. Moreover, overexpression of RBM6 can inhibit cell proliferation and promote the apoptosis of HepG2 cells and HCC tissues [42]. Through pan-cancer analysis, we found that RBM6 expression differs in cancer and adjacent tissues. Meanwhile, higher expression of RBM6 was associated with a better prognosis in most cancers, consistent with previous reports. However, it should be noticed that RBM6 can also promote the progression of tumors in specific cancer types. In this study, we found that higher expression of RBM6 was correlated with a better prognosis, suggesting that RBM6 primarily plays a tumor-suppressor role in multiple cancer types. Meanwhile, we found that knocking down RBM6 promoted cell proliferation and inhibited cell apoptosis, which illustrated the tumor-suppressor function of RBM6 in HeLa cells. The result was consistent with the prognostic analysis. At the same time, additional experiments such as cellular invasion, migration, and scratch assays are also necessary to perform in future studies to confirm the tumor-suppressor function of RBM6.
As a splicing factor, RBM6 participates in the regulation of AS [16, 17, 18, 19]. RBM6
regulates the AS-coupled nonstop decay of a positive homologous
recombinational repair (HRR) regulator, Fe65/amyloid beta A4 precursor
protein-binding family B member 1, and RBM6 knockdown significantly reduces Fe65
protein levels, consequently impairing the HRR of DNA double strand breaks [24].
It is also known that RBM6 modulates the proliferation, migration, and apoptosis
of tumor cells by regulating RNA metabolism [43]. This is the first study to
analyze the transcriptome regulated by RBM6 at the genome-wide level in HeLa
cells through high-throughput sequencing. We found that RBM6 regulates the AS of
many proliferation- and apoptosis-related genes, such as MARCH7, which
promotes cell proliferation and invasion through VAV2-RAC1-CDC42 pathway in
cervical cancer cells [44]. Moreover, TFAP2A regulates the growth and survival of
nasopharyngeal carcinoma by regulating the hypoxia inducible factor-1
Interestingly, knocking down the expression of RBM6 also up-regulates the expression of genes involved in the type I IFN pathway. Type I IFNs have been extensively studied for their association with tumor progression [48]. They reportedly contribute to immune cell exhaustion and other harmful effects by prolonged stimulation, thus directly or indirectly allowing cancer cells to escape immune clearance [49]. In the present study, up-regulated genes caused by shRBM6 were mostly enriched in the type I IFN signaling pathway, which suggests that RBM6 knockdown may enhance the ability of cancer cells to escape immune clearance. Furthermore, we found that genes associated with up-regulated type I IFNs include OAS1, OAS2, IFITM1, IFI6, and IRF7. These 5 up-regulated genes are associated with tumor initiation and progression [50, 51, 52, 53, 54, 55], indicating the beneficial effect of RBM6 on the apoptosis of cancer cells.
DDR is an essential function in the maintenance of genome stability in cancer cells [56]. Since aberrant pericellular development is a basic mechanism of tumorigenesis, the cell cycle regulation has become a key target for anti-cancer therapy [57]. In this study, we found that after RBM6 knockdown, genes involved in DDR and cell cycle pathways were significantly down-regulated. Inhibition of DDR genes by shRBM6 may inhibit the activation of p53, thus inhibiting the apoptosis of cancer cells [58]. DDR- and cell cycle-related genes include GADD45A, INO80, EID3, and BCCIP. GADD45A is a p53 effector gene that has protective effects on cancer [59] by mediating cell cycle arrest and inducing apoptosis. INO80 is required for cell cycle control [60]. EID3 has effects on cell proliferation, cell cycle, cell apoptosis, and promotes treatment resistance to the maintenance of cancer stem cell characteristics [61]. The high expression of BCCIP indicates a poor prognosis and promotes the proliferation and migration of LUAD cells [62]. It has been reported that the TFs involved in myogenic transcription are regulated by the AS of their transcripts. The AS of many transcripts is aberrant in neuromuscular pathology [63]. As a splicing factor, RBM6 may regulate transcription by regulating the AS of TFs. In this study, we constructed a co-disturbed network between the DEGs and AS of TFs regulated by RBM6.
RBM6 reportedly controls the proliferation of HeLa cancer cells, and the crosslinking-immunoprecipitation and high-throughput sequencing (CLIP-seq) has shown that RBM6 has a binding preference for the CUCUGAA sequence [21]. In this study, we used the iRIP-seq dataset to identify both directly and indirectly interacting RNA targets with RBM6. The results demonstrated that RBM6 interacted with many mRNAs and pre-mRNAs, with an mRNA binding preference and genomic location towards the 5′UTR. Motifs analysis revealed that RBM6-bound RNAs are enriched in GGCGAUG and CUCU motifs, which may be caused by the different binding effects of RBM6 on different cells. RBM6-bound mRNAs/pre-mRNAs, which involve regulated AS, are enriched in proliferation and apoptosis pathways (CDK13, LAMA5, OBSCN, SMAD6, TFAP2A) and the TFs (AHDC1, ZNF276, SMAD6 and TFAP2A). CDK13promotes DNA damage and cell death in triple-negative breast cancer, thus serving as a therapeutic target [64]. LAMA5 could be a potential prognostic factor in ovarian cancer [65]. Patients with gastric adenocarcinoma with OBSCN mutations have good treatment effects and prognoses [66]. These TFs binding to RBM6 could promote the growth of tumor cells [67, 68, 69, 70]. Together, these results support the hypothesis that RBM6 may play a role in tumor growth by encoding its RNA binding activity either directly or indirectly.
In summary, our study analyzed the RNA binding properties and molecular functions of RBM6 in HeLa cells. RBM6 regulation of the transcriptome was tightly associated with its cellular functions in HeLa cells. In future research, we plan to further investigate how RBM6 binds to the TFs and genes associated with proliferation and apoptosis and the mechanisms underlying the role of RBM6 in cancer.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) are: Gene Expression Omnibus (GEO), accession number: GSE223199. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
DL conceived and designed the experiments and revised the manuscript; PP, WS, and JH performed the experiments; QY, CC, and HG analyzed the data; PP wrote the paper. All authors have read and approved the final manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We are very grateful to technicians from Wuhan Ruixing Biotechnology Co. for their help in generating data presented in this study.
This work was supported by grants from the Youth Project of the National Natural Science Foundation of China (grant number 81904019).
The authors declare no conflict of interest. Qingqing Yin and Hao Guo are employed by Wuhan Ruix- ing Biotechnology Co., Ltd, and Chao Cheng is employed by ABLife BioBigData Institute, the judgments in data interpretation and writing were not influenced by this relationship.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2909330.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.