






 , Xiaoyi Fang 2,†, Huayan Liu 1, Qianqian Fan 1
, Xiaoyi Fang 2,†, Huayan Liu 1, Qianqian Fan 11 Department of Neonatology, Longhua District Central Hospital, 518110 Shenzhen, Guangdong, China
2 Department of Neonatology, The Seventh Affiliated Hospital, Sun Yat-sen University, 518107 Shenzhen, Guangdong, China
†These authors contributed equally.
Abstract
Hypoxic-ischemic brain damage (HIBD) is a prevalent brain injury with high mortality and morbidity. It results from hypoxia and ischemia of the brain due to various perinatal factors. A previous study showed that knockdown of programmed cell death factor 4 (PDCD4) could reduce infarction injury resulting from ischemia/reperfusion injury. However, exact mechanism by which PDCD4 acts in HIBD is not yet understood. Our aim in present investigation was to investigate the function and mechanism of PDCD4 in alleviating HIBD.
An HIBD model was developed using neonatal rats. After 48 h of modeling, short-term neurological function was evaluated and the brain tissue removed for assessment of cerebral infarct volume and brain water content (BWC). A cell model of oxygen glucose deprivation/reoxygenation (OGD/R) was also constructed. Overexpression or knockdown of insulin-like growth factor 2 mRNA binding protein 3 (IGF2BP3) or PDCD4 was performed in pretreated cells.
The geotaxis reflex time, cerebral infarct volume, and BWC all increased after HIBD in this neonatal rat model. Additionally, the levels of PDCD4 and of the N6-Methyladenosine (m6A) reader protein IGF2BP3 were increased in HIBD rats and OGD/R-stimulated pheochromocytoma (PC12) cells relative to controls. Moreover, OGD/R-stimulated pheochromocytoma PC12 cells showed decreased cell viability, increased apoptosis, and elevated Interleukin 6 (IL-6), Interleukin 1 β (IL-1β), and tumor necrosis factor-α (TNF-α) contents. These features were reversed after knocking down IGF2BP3. The interaction between IGF2BP3 protein and PDCD4 mRNA was confirmed by RNA immunoprecipitation and RNA pull-down assays. Furthermore, knockdown of IGF2BP3 in OGD/R-stimulated PC12 cells reduced cell damage via down-regulation of PDCD4. Finally, the IGF2BP3/PDCD4 axis alleviated OGD/R-induced cell injury in primary cortical neurons (PCNs).
PDCD4 and m6A reader protein IGF2BP3 were up-regulated in an HIBD neonatal rat model. Knockdown of IGF2BP3 in OGD/R-stimulated PC12 cells or PCNs alleviated cell damage through reducing PDCD4.
Keywords
- IGF2BP3
- PDCD4
- OGD/R
- hypoxic-ischemic brain damage
Hypoxic ischemic brain damage (HIBD) is a prevalent and severe neurological condition affecting neonates, encompassing both full-term and preterm infants [1]. Hypoxic ischemia (HI) is a leading cause of neonatal mortality and long-term disability. Currently, only approved treatment for perinatal HI is therapeutic hypothermia [2]. The pathogenesis of HIBD involves oxidative damage and cell death [3]. Most of the neuroprotective strategies currently under evaluation are for the treatment of HIBD [4, 5]. Nevertheless, curing HIBD remains challenging due to the absence of effective treatments and the incomplete understanding of the intricate molecular mechanisms involved [6]. Therefore, further investigations into HIBD pathogenesis are crucial for the development of effective prevention and treatment strategies.
Programmed cell death factor 4 (PDCD4) is a newly identified tumour suppressor with various roles, such as suppressing cell growth, inhibiting tumor invasion and metastasis, and promoting apoptosis [7]. Increased levels of PDCD4 have been shown to aggravate acute kidney [8], intestinal [9], and heart [10] injuries caused by ischemia-reperfusion. Knockdown of PDCD4 has been found to reduce infarct injury and cortical neuron apoptosis caused by ischemia-reperfusion injury [11], although PDCD4 role in HIBD has yet to be reported. We observed decreased cell viability, increased apoptosis, and upregulated levels of PDCD4 in oxygen glucose deprivation (OGD)-stimulated H19-7 and pheochromocytoma (PC12) cells in an earlier study [12]. These effects could be reversed by intervention with diazoxide. A possible interaction between PDCD4 and insulin-like growth factor 2 mRNA binding protein 3 (IGF2BP3) was predicted by online analysis software catRAPID omics v2.0 (http://service.tartaglialab.com/page/catrapid_omics2_group). As one of the N6-Methyladenosine (m6A) reader proteins, IGF2BP3 has the ability to identify the m6A site on mRNA and regulate protein levels by promoting the stability of mRNA [13, 14]. However, the roles of both IGF2BP3 and PDCD4 in HIBD remain unclear.
Based on the above observations, we hypothesized that IGF2BP3 plays a crucial role in attenuating HIBD in neonatal rats by regulating PDCD4. Our in vivo and in vitro experiments demonstrasted knockdown of IGF2BP3 down-regulates PDCD4 levels to attenuate HIBD. These research findings may improve our understanding of HIBD pathogenesis and offer novel targets and strategies for HIBD treatment.
Seven-day-old newborn Sprague-Dawley (SD) rats (randomly male and female) were
obtained from Hunan SJA Laboratory Animal Co., Ltd. The newborn rats were allowed
to nurse freely by female rats in a 12-hour alternating light/dark cycle, with a
temperature of 24
Negative geotropism was tested 48 h after HIBD by two blinded investigators in an unbiased setting, with 18 rats used in each group. The test involved placing the neonatal rats head-down on a tilt board set at a 45° angle, and then recording the time taken by the animal to turn and assume a head-up position. The test was limited to a maximum duration of 60 s [17].
TTC staining was performed 48 h after HIBD in order to evaluate cerebral infarct
volume, with 6 rats being allocated to each group. A coronal section (2 mm
thickness) of the brain was immersed in 1% TTC solution (AWI0490, Abiowell,
Changsha, Hunan, China), stained for 15–30 min at 37 °C, and then photographed
for evaluation. After staining with TTC, the infarct volume of brain tissue was
measured using a computerized pathological image analysis system. Infarct volume
= (volume of the normal hemisphere – non-infarct volume of the infarct
hemisphere)/volume of the hemisphere
The BWC was examined 48 h after HIBD as previously described [18, 19]. Six rats
were used in each group, with the BWC (%) calculated using formula: (wet weight
– dry weight)/wet weight
Neonatal SD rats (within 24 h after birth) were soaked in 75% alcohol, disinfected for 3 min, and then transferred to a sterile operating table. The fetal rat cortical tissue was separated aseptically on a super-clean workbench, cut up with scissors, and placed into a 15 mL centrifuge tube. Pancreatic enzyme digestion solution (AWC0232, Abiowell, Changsha, Hunan, China) was then added (5 mL, 0.25%), mixed well, and allowed to digest the tissue for 15 min at 37 °C. To stop the digestion, Dulbecco’s modified Eagle medium (DMEM, D5796, Sigma, Saint Louis, MO, USA) with 10% fetal bovine serum (FBS, 10099141, Gibco, Grand Island, NY, USA) was added. Mixture was then centrifuged at 300 g for 5 min. Supernatant was discarded. DMEM with 10% FBS was added, and a cell suspension made by gently blowing 20 times with a Pasteur pipette. The filtrate was collected using a 70 µm filter, centrifuged at 300 g for 5 min. Pellet was re-suspended in DMEM with 10% FBS and used to seed 6-well plates that had been pre-coated with poly-lysine. On the second day, the medium was replaced with complete neuron culture medium (PriMed-iCell-005, icellbioscience, Shanghai, China) with 2% B27, with replacement every 2–3 days.
For identification of PCNs, cell crawling tablets were cleaned 2–3 times with phosphate buffered saline (PBS), fixed with 4% paraformaldehyde (AWI0062, Abiowell, Changsha, Hunan, China), and permeated at 37 °C for 30 min with 0.3% Triton X-100 (AWH0299, Abiowell, Changsha, Hunan, China). Cell crawling tablets were blocked with a 5% solution of bovine serum albumin (BSA, V900933, Merck, Beijing, China) for 1 h at 37 °C, and incubated with an appropriately diluted primary antibody for neuron-specific enolase (NSE, AWA10959, Abiowell) overnight at 4 °C. This was followed by incubation with 50–100 µL of anti-rabbit-IgG-labeled fluorescent antibody (AWS0005a, Abiowell) for 90 min. Slides were incubated with 4′,6-diamidino-2-phenylindole (DAPI) working solution (AWC0292a, Abiowell, Changsha, Hunan, China) for 10 min at 37 °C. They were then sealed with buffered glycerin, preserved away from light, and observed with an epi-fluorescence microscope (BA210T, Motic, Fujian, China).
Pheochromocytoma (PC12) cells (undifferentiated, AW-CCR443, Abiowell) were cultured in a humidified atmosphere at 37 °C with 5% CO2 in Roswell Park Memorial Institute (RPMI) 1640 medium (M3817, sigma, Saint Louis, MO, USA) supplemented with 10% horse serum, 5% FBS, and 100 U/mL penicillin-streptomycin. The PC12 cells were authenticated Quantitative polymerase chain reaction (QPCR), and all experiments were conducted using mycoplasma-free cells. For differentiation induction, PC12 cells were treated with 50 ng/mL of nerve growth factor-beta and 1% FBS for 4 days. A cell OGD/reoxygenation (OGD/R) model was first established. PC12 cells or PCNs were cultured at 37 °C for 24 h in serum- and glucose-free medium in the atmosphere of 93% N2, 5% CO2 and 2% O2. After OGD/R, PC12 cells or PCNs were transferred to medium containing glucose, brought back to normal oxygen levels, and incubated at 37 °C with 5% CO2 for 24 h to reoxygenate [20]. In addition, cells were treated with OGD/R following knockdown or overexpression of IGF2BP3 or PDCD4. Short hairpin negative control (sh-NC), overexpression negative control (oe-NC), sh-IGF2BP3, oe-IGF2BP3, sh-PDCD4, and oe-PDCD4 were synthesized by HonorGene (Changsha, Hunan, China). Cells were transfected with Lipofectamine 2000 (11668019, invitrogen, Carlsbad, CA, USA) for 48 h.
Protein extraction was performed from cells and damaged brain tissue (6 rats in
each group) with Radio Immunoprecipitation Assay Lysis buffer (P0013B, Beyotime,
Shanghai, China) and the protein content measured. Proteins underwent separation
on Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with the
use of MB2479 from meilunbio (Dalian, Liaoning, China), followed by transfer onto polyvinylidene fluoride
membranes. Incubation with primary antibody was then conducted overnight at 4 °C
using following antibodies: PDCD4 (ab80590, 1:1000, Abcam, Cambridge, UK),
IGF2BP3 (14642-1-AP, 1:5000, Proteintech, Chicago, IL, USA), Bcl-2 (60178-1-Ig,
1:5000, Proteintech), Bax (ab32503, 1:5000, Abcam), caspase3 (19677-1-AP, 1:1000,
Proteintech), and
RNA was isolated through Trizol (15596018, Invitrogen, Carlsbad, CA, USA) and
converted into cDNA through reverse transcription with the kit (CW2569, CWBIO,
Bejing, China). The cDNA was then amplified with Ultra SYBR Mixture (CW2601,
CWBIO) on ABI 7900 system (ABI, Carlsbad, CA, USA). Gene expression levels were measured through
2-ΔΔCt method with
Cells were trypsinized, counted, and subsequently plated into 96-well plates (1
Cells were detached using ethylenediaminetetraacetic acid (EDTA)-free trypsin, followed by centrifugation for 5 min at 1000 rpm to collect cell pellets. The pellets were resuspended, washed with PBS, and centrifuged again for 5 min at 1000 rpm. The cells were subsequently suspended in 500 µL of binding buffer and incubated with 5 µL of Annexin V-APC (KGA108, KeyGen, Nanjing, Jiangsu, China). Subsequently, 5 µL of propidium iodide (537060, sigma, Saint Louis, MO, USA) was added to mixture, mixed, and then incubated in dark for 10 min. Flow cytometry analysis was conducted with a Beckman instrument (A00-1-1102, Beckman Coulter, Brea, CA, USA) within 1 h.
Interleukin 6 (IL-6), Interleukin 1
The interaction between IGF2BP3 protein and PDCD4 mRNA was studied with EZMagna RIP kit (17-701, Merck Millipore, Darmstadt, Germany). PC12 cells were dissolved in RIP solution and incubated with beads coupled to Ago2 or anti-rabbit immunoglobulin G (IgG, negative control). Precipitated RNA was assessed using qRT-PCR [21].
To confirm the interaction between IGF2BP3 protein and PDCD4 mRNA, RNA pull-down detection was performed using previously described methods [22, 23]. Specifically, the cell extract was mixed with biotinylated RNA and then added to rinsed streptavidin agarose beads and incubated. The microbeads were subsequently washed and boiled, and the proteins analyzed by SDS-PAGE and Western blot.
Statistical analysis of data was performed using Graphpad Prism 8.0 software
(GraphPad Software, Inc., San Diego, CA, USA), and results were presented as the
mean
We first constructed a neonatal rat model of HIBD. At 48 h after HIBD, the geotaxis reflex time, infarct volume and BWC of Model group were all higher compared to Sham group (Fig. 1A–C). We next investigated the possible mechanism of action of PDCD4 in HIBD. catRAPID online analysis predicted that PDCD4 interacts with IGF2BP3. PDCD4 and IGF2BP3 levels were evaluated both in vivo and in vitro. In neonatal rats, PDCD4 and IGF2BP3 levels were higher in Model group than Sham group (Fig. 1D). In vitro, PDCD4 and IGF2BP3 levels were also elevated in OGD/R group than Control group (Fig. 1E). These results demonstrated PDCD4 and of the m6A reader protein IGF2BP3 levels were up-regulated in HIBD neonatal rats.
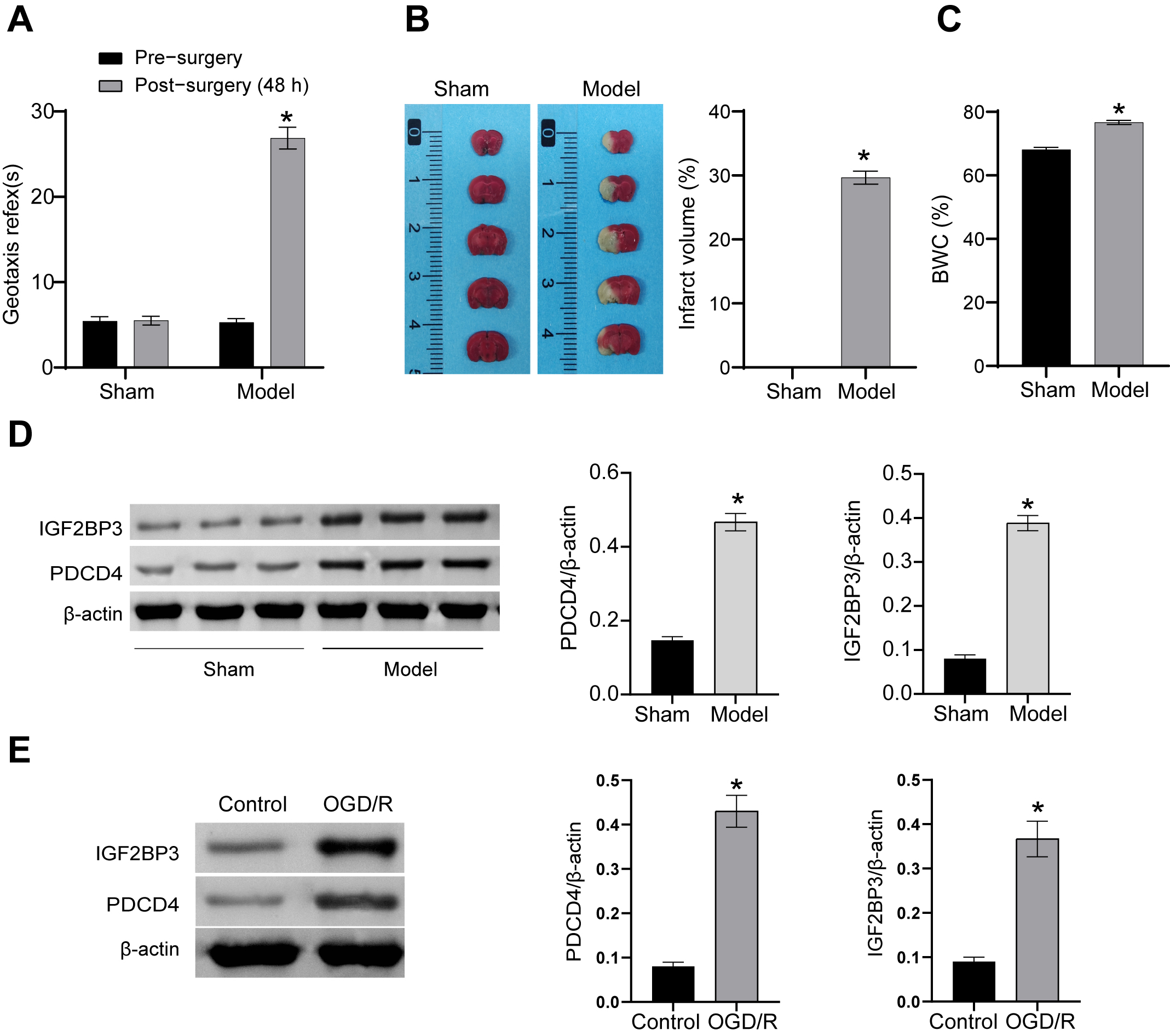 Fig. 1.
Fig. 1.
Upregulation of PDCD4 and m6A reader protein IGF2BP3 in HIBD
neonatal rats. (A) Assessment of short-term neurological function (geotaxis
reflex time). (B) TTC staining for cerebral infarct volume. (C) Assessment of
BWC. (D) Western blot analysis of PDCD4 and IGF2BP3 levels in Sham Sand
model groups. (E) Western blot detection
of PDCD4 and IGF2BP3 levels in Control and OGD/R groups. *p
Knockdown of IGF2BP3 was performed to study its function. First, the efficiency
of knockdown was verified (Fig. 2A). IGF2BP3 mRNA and protein levels in
OGD/R+sh-IGF2BP3 group were lower than OGD/R+sh-NC group, indicating successful
knockdown of IGF2BP3. Moreover, cell viability and Bcl-2 levels were repressed in
OGD/R group than control group, whereas apoptosis was increased. Bax,
cleaved-caspase3, IL-6, IL-1
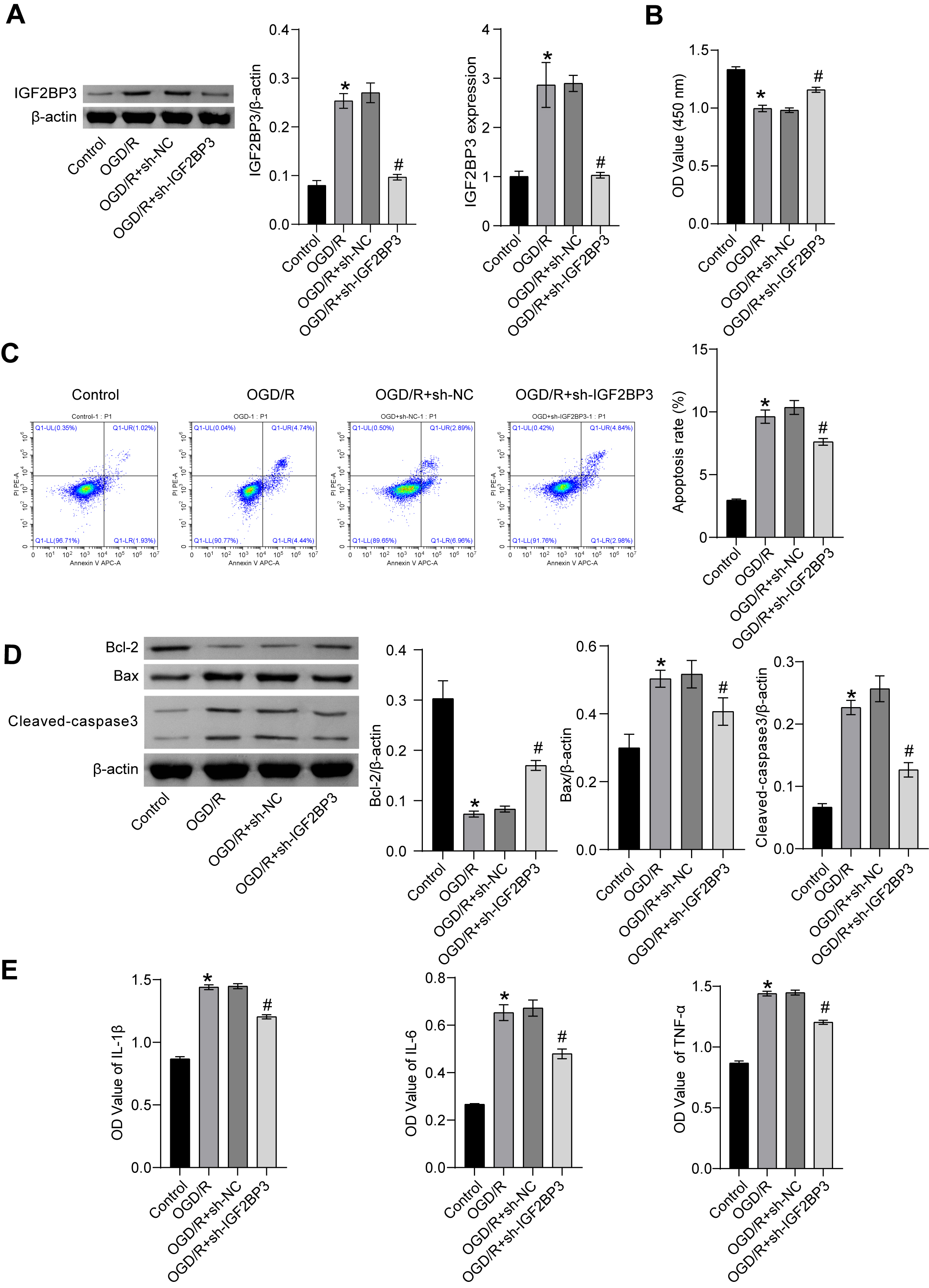 Fig. 2.
Fig. 2.
Knockdown of IGF2BP3 reduced OGD/R-induced injury in
pheochromocytoma PC12 cells. (A) IGF2BP3 mRNA and protein levels. (B) Cell
viability was determined by means of the CCK-8 assay. (C) Flow cytometry
detection of apoptosis. (D) Cellular Bax, Bcl-2, and cleaved-caspase3 protein
levels. (E) Evaluation of IL-6, IL-1
PDCD4 protein levels were higher OGD/R group than control group, but decreased following knockdown of IGF2BP3 (Fig. 3A). Furthermore, RIP and RNA pull-down revealed an interaction between IGF2BP3 protein and PDCD4 mRNA (Fig. 3B,C). These results indicated that IGF2BP3 interacted with PDCD4 mRNA to regulate PDCD4 levels.
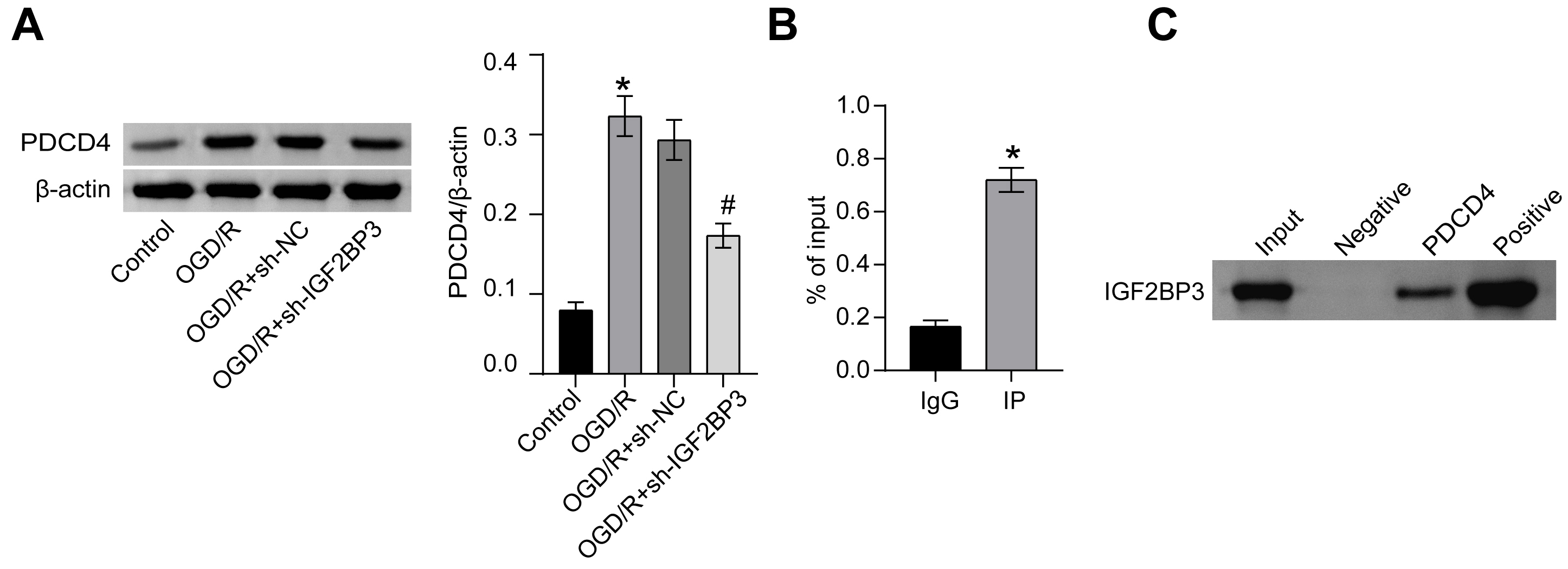 Fig. 3.
Fig. 3.
IGF2BP3 interacted with PDCD4 mRNA to regulate the PDCD4 level.
(A) PDCD4 protein levels. (B) RIP identification of interaction between
IGF2BP3 protein and PDCD4 mRNA. (C) RNA
pull-down was utilized to confirm the interaction between IGF2BP3 protein and
PDCD4 mRNA. *p
We conducted further mechanistic investigations by knocking down IGF2BP3 and
overexpressing PDCD4. Compared with the sh-NC group, PDCD4 levels decreased
following knockdown of IGF2BP3. In contrast, PDCD4 levels increased following the
overexpression of PDCD4 (Fig. 4A). In comparison to sh-NC group, sh-IGF2BP3 group
showed increased cell viability, reduced apoptosis, lower levels of Bax and
cleaved-caspase3, higher levels of Bcl-2, as well as decreased IL-6,
IL-1
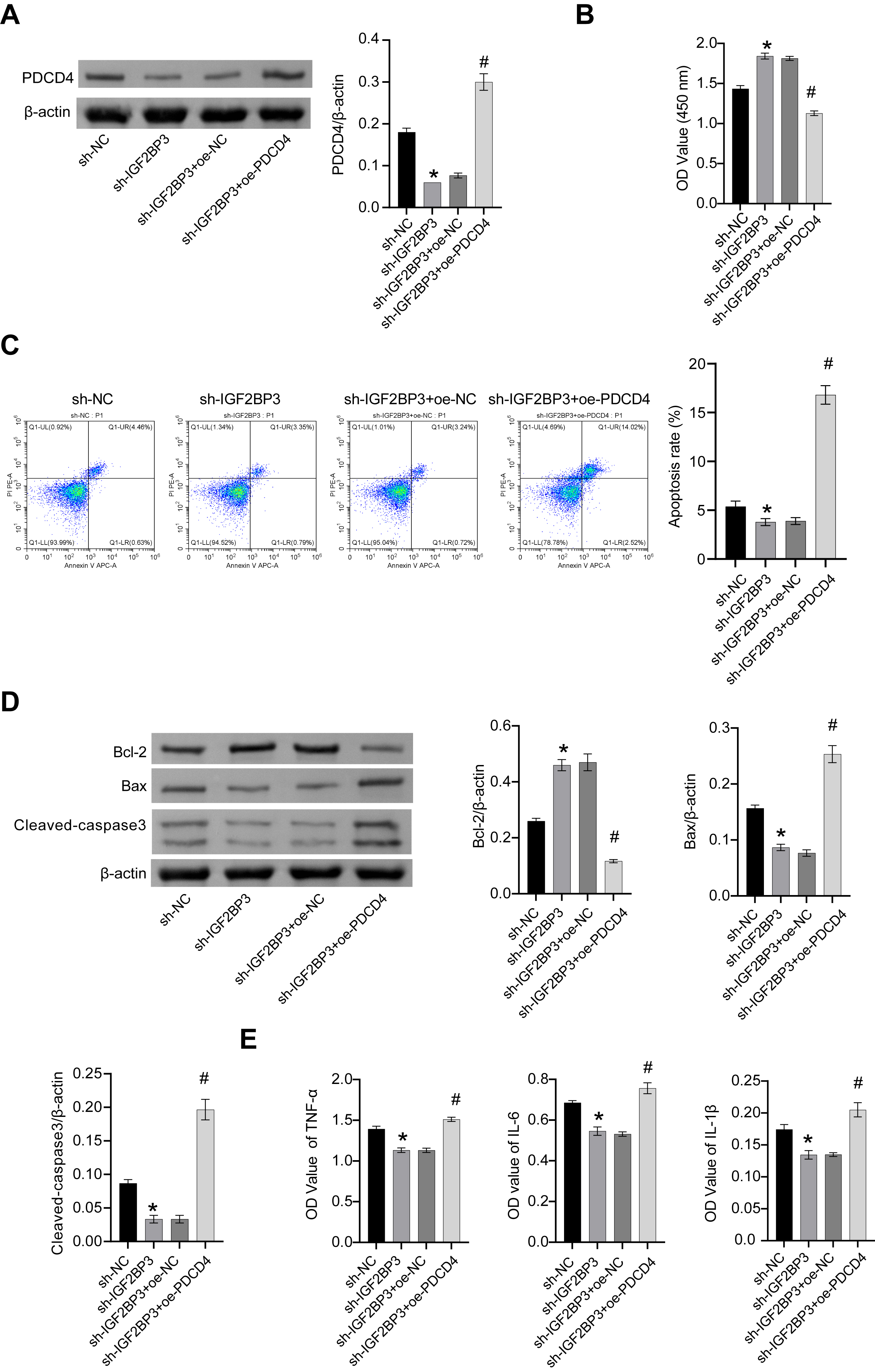 Fig. 4.
Fig. 4.
Knockdown of IGF2BP3 reduced OGD/R-induced cell damage via
PDCD4. (A) PDCD4 protein levels. (B) Cell viability assessment through CCK-8
assay. (C) Flow cytometry detection of apoptosis. (D) Cellular Bax, Bcl-2, and
cleaved-caspase3 protein levels. (E) Assessment of IL-6, IL-1
Next, we investigated the roles of IGF2BP3 and PDCD4 in PCNs. PCNs were first
isolated and identified, as shown in Fig. 5A. Following the knockdown of IGF2BP3
in PCNs, PDCD4 levels were lower compared to sh-NC group. However, PDCD4 levels
increased in PCNs following the overexpression of PDCD4. PDCD4 levels in PCNs
increased following overexpression of IGF2BP3 compared to oe-NC group. PDCD4
levels decreased in PCNs following knockdown of PDCD4 (Fig. 5B). Furthermore,
compared to sh-NC group, cell apoptosis was decreased, Bax and cleaved-caspase3
levels were supressed, Bcl-2 levels were elevated, and IL-6, IL-1
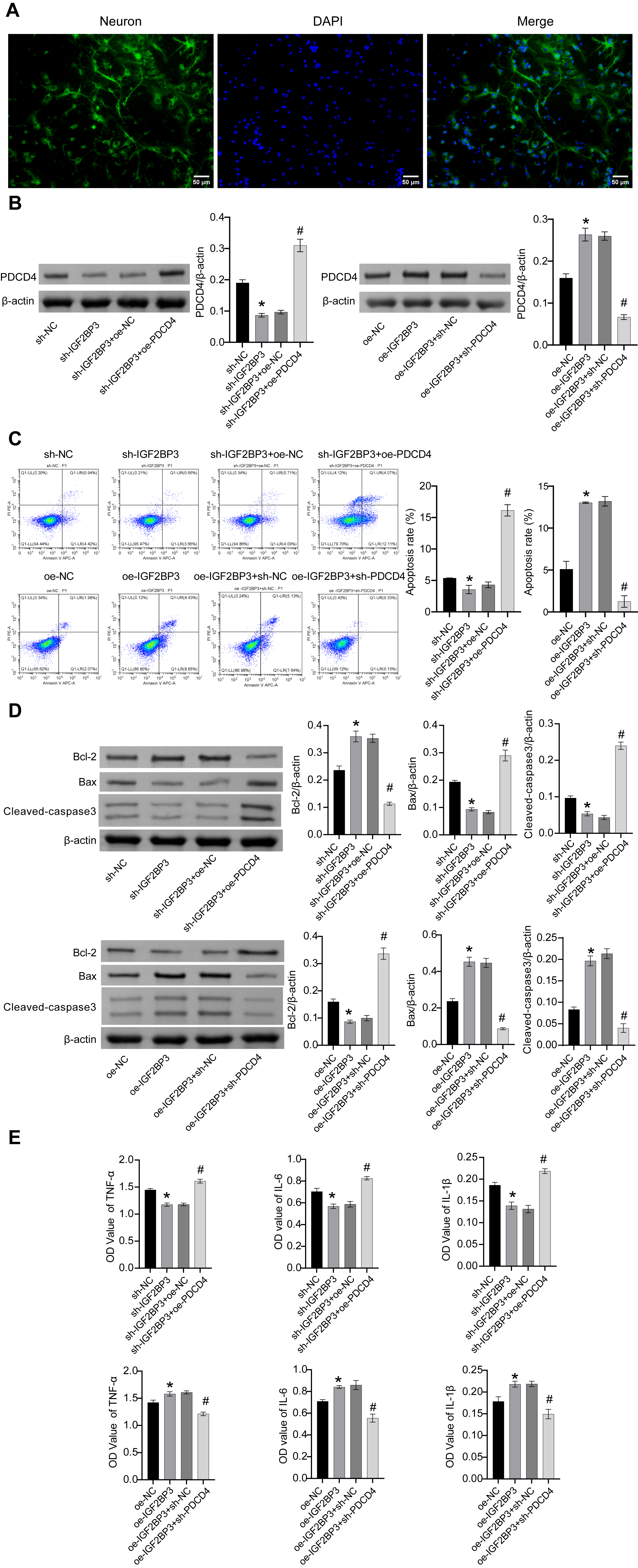 Fig. 5.
Fig. 5.
The IGF2BP3/PDCD4 axis reduced OGD/R-induced cell damage in
PCNs. (A) Identification of PCNs. Scale bar: 50 µm. (B) PDCD4 protein levels. (C) Flow cytometry
for the analysis of apoptosis. (D) Cellular Bax, Bcl-2, and cleaved-caspase3
protein levels. (E) IL-6, IL-1
This study investigated the underlying mechanism of PDCD4 in HIBD through a combination of in vitro and in vivo assays involving IGF2BP3. PDCD4 and m6A reader protein IGF2BP3 were up-regulated in HIBD neonatal rats in vivo. Knockdown of IGF2BP3 in vitro reduced OGD/R-induced cell damage by downregulating PDCD4 levels. This is the first report to our knowledge of the IGF2BP3/PDCD4 axis in HIBD.
With regard to cerebral ischemia-reperfusion injury, Zheng et al. [24] demonstrated that neuroprotective effect of miR-340-5p in OGD/R-induced injury could be partially reversed via overexpression of PDCD4. In our prior work, we had observed PDCD4 was highly expressed in OGD-stimulated H19-7 cells and PC12 cells, and this could be reversed by intervention with diazoxide [12]. Hence, in this research, our aim was to further explore the mechanism of PDCD4 in HIBD. Using catRAPID online analysis, we predicted that PDCD4 could interact with IGF2BP3. Through subsequent in vivo and in vitro experiments, we found PDCD4 and the m6A reader protein IGF2BP3 were both up-regulated in HIBD neonatal rats and in OGD/R-stimulated PC12 cells.
To investigate the mechanism of neuronal cell death following hypoxic-ischemic injury and identify potential protective agents, an in vitro cell culture model using PC12 cells was established to simulate hypoxic-ischemic-induced cell death through OGD [25]. In our earlier study, we constructed a hypoxic-ischemic encephalopathy cell model using PC12 cells stimulated by OGD [12]. Other reports on the use of PC12 cells to construct in vitro HIBD models have also been published [26, 27]. PC12 cells are a valuable in vitro model for studying the molecular mechanisms underlying hypoxia resistance and oxygen-sensing characteristics [28, 29]. Therefore, in the research, we utilized OGD/R model in PC12 cells to mimic HIBD in vitro.
m6A RNA methylation is most common internal chemical modification of mRNA and is crucial in the development of different tumors through dynamically regulating m6A RNA methylation regulators [30]. In terms of mechanism, the m6A reader IGF2BP3 binds directly to m6A-modified region of mRNA, thereby enhancing mRNA stability and expression [31]. Li et al. [32] reported that exosomal miR-34a-5p secreted from mesenchymal stem cells could attenuate intestinal ischemia/reperfusion injury via METTL3/IGF2BP3-mediated m6A modification of pre-miR-34A. The present study further investigated this mechanism by knocking down IGF2BP3. This was found to reduce OGD/R-induced damage to PC12 cells via IGF2BP3 protein interaction with PDCD4 mRNA and subsequent regulation of PDCD4 levels. Our results confirmed that IGF2BP3 plays a vital role in HIBD via its interaction with PDCD4 mRNA. We further investigated the IGF2BP3/PDCD4 mechanism in HIBD by overexpressing PDCD4. Overall, our study demonstrated that knockdown of IGF2BP3 reduced OGD/R-induced cell injury via PDCD4. Additionally, IGF2BP3/PDCD4 axis was confirmed to reduce OGD/R-induced cell injury in PCNs.
For constructing HIBD model in neonatal rats, Zhang et al. [33] chose male neonatal rats to investigate the role and mechanism of ferroptosis in HIBD. They found ferrostatin-1 attenuated HIBD in neonatal rats by inhibiting ferroptosis. Bao et al. [34] revealed that MR imaging and outcome in neonatal HIBD models were correlated with sex, and hypoxic ischemia affected anxiety, cognition, and locomotion in a sex-dependent manner, with males being more vulnerable. These studies suggest that males are the high-risk factors for HIBD in newborn rats. However, other researchers have conducted studies using neonatal SD rats without sex preference. Mo et al. [35] used neonatal SD rats with undifferentiated gender to investigate the effects of neonatal HIBD on early-stage neuro-motor function, cerebral blood flow, and the neurovascular unit. Xu et al. [36] also used neonatal rats without sex preferences to study protective mechanisms of quercetin in neonatal rat brain injury induced by HIBD. They found that quercetin exerted neuroprotective effects in a rat model of HIBD by regulating NLR family member X1 (NLRX1) and autophagy. In this study, we also used neonatal rats without sex preferences. Whereas important sexual dimorphism exists in several aspects of animals. Males are the high-risk factors for HIBD in newborn rats [33, 34]. Therefore, we should evaluate the sex of animals in the study. This is our limitation. In the future, with sufficient time and funding, we will further explore the impact of gender on HIBD.
In conclusion, the current findings demonstrate that PDCD4 and the m6A reader protein IGF2BP3 are up-regulated in HIBD neonatal rats. Knockdown of IGF2BP3 reduces OGD/R-induced cell damage in vitro by reducing PDCD4 levels. These results revealed IGF2BP3 might be a novel target and approach for HIBD treatment. In future research, our group will explore potential drugs for antagonizing IGF2BP3 and its mechanism in HIBD.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
HIBD, Hypoxic-ischemic brain damage; PDCD4, programmed cell death factor 4; BWC, brain water content; OGD/R, oxygen glucose deprivation/reoxygenation; HI, Hypoxic ischemia; IGF2BP3, insulin-like growth factor 2 mRNA binding protein 3; m6A, N6-Methyladenosine; SD, Sprague-Dawley; TTC, 2,3,5-Triphenyl tetrazolium chloride; DMEM, Dulbecco’s modified eagle medium; FBS, fetal bovine serum; PBS, phosphate buffered saline; NSE, neuron specific enolase; sh-NC, Short hairpin negative control; oe-NC, overexpression negative control; PCNs, primary cortical neurons; qRT-PCR, Quantitative real-time PCR; CCK-8, Cell Counting Kit-8; RIP, RNA immunoprecipitation; IgG, immunoglobulin G; ANOVA, analysis of variance.
YC and XF contributed to conceptualization, data curation, investigation, methodology, validation, writing of the original draft and review. HL and QF contributed to conceptualization, funding acquisition, project administration, supervision, and review. All authors read and approved the final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
All experimental procedures and animal handling were performed with the approval of the Animal Care and Use Committee of Guangdong Medical University (No. GDY1902250), in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and studies involving laboratory animals follows the ARRIVE guidelines.
Not applicable.
This work was supported by the Basic Research Project of Shenzhen Science and Technology Plan Project in 2020 (No. JCYJ20190808154015736, Y.C.; No. JCYJ20190809145409829, X.F.).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2909329.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.