





1 Department of Anesthesiology, Children's Hospital of Chongqing Medical University, 400014 Chongqing, China
2 National Clinical Research Center for Child Health and Disorders, 400014 Chongqing, China
3 Ministry of Education Key Laboratory of Child Development and Disorders, 400014 Chongqing, China
4 Chongqing Key Laboratory of Child Neurodevelopment and Cognitive Disorders, 400014 Chongqing, China
5 Department of Anesthesiology, Second Affiliated Hospital of Army Medical University, 400037 Chongqing, China
Abstract
Isoflurane is a commonly used general anesthetic widely employed in clinical surgeries. Recent studies have indicated that isoflurane might induce negative impacts on the nervous system, notably by triggering neuronal apoptosis. This process is pivotal to the development and emergence of neurological disorders; its misregulation could result in functional deficits and the initiation of diseases within nervous system. However, the potential molecular mechanism of isoflurane on the neuronal apoptosis remains fully unexplored. This study aims to investigate the regulatory role of the sirtuin 1-mechanistic target of rapamycin (SIRT1-mTOR) signaling pathway in autophagy during isoflurane-induced apoptosis of fetal rat brain neuronal cells.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay, real-time quantitative polymerase chain reaction (qPCR), and Western blot were utilized to evaluate the apoptotic status of hippocampal tissue cells in fetal mice after sevoflurane exposure. Our further investigation was commenced with flow cytometry, immunofluorescence, qPCR, and Western blot to determine the impact of autophagy on sevoflurane-induced apoptosis in these neurons. On the other hand, we conducted an additional set of analyses, including flow cytometric analysis, qPCR, and Western blot, to further elucidate the neuroprotective potential of autophagy in neural cells of fetal mice subjected to sevoflurane-induced apoptosis.
Our findings indicated that a 3% sevoflurane treatment led to a significant rise in apoptosis among fetal rat hippocampal tissue cells and neurons. Levels of apoptosis-associated proteins, cleaved-caspase-3 and Bcl-2 associated X protein (Bax), were found to be markedly higher, coinciding with an enhancement in autophagy as evidenced by increased microtubule-associated proteins 1A/1B-light chain 3 (LC3) and decreased p62 expression. Concurrently, there was a notable up-regulation of sirtuin 1 (SIRT1) and a down-regulation of mechanistic target of rapamycin (mTOR) expression. In conclusion, our research elucidated the pivotal function of cellular autophagy in an apoptosis induced by sevoflurane in fetal rat nerve cells. Through experimental manipulation, we observed that interference with SIRT1 resulted in a reduction of both cleaved-caspase-3 and Bax levels. This intervention also beget a diminished expression of the autophagy-associated factor LC3 and an up-regulation of p62. Furthermore, inhibition against mTOR reversed the effects induced by SIRT1 interference, suggesting a complex interplay amid these regulatory pathways.
SIRT1 possesses a capacity to modulate apoptosis in the hippocampal neurons of fetal rats triggered by sevoflurane, with mTOR functioning as an inhibitory factor within this signaling pathway.
Keywords
- SIRT1-mTOR signaling pathway
- sevoflurane
- nerve cells autophagy
- cerebral nerve cell apoptosis
Sevoflurane is a widely applied clinical anesthetic known for its rapid onset, quick recovery, and stable therapeutic effects [1]. Nevertheless, studies have indicated that inhalation of anesthetics by neonates would ultimately result in acute neural inflammation and a significant increase in rapid apoptosis of neural cells, subsequently leading to cognitive impairments [2, 3, 4]. Extensive preclinical research has suggested that repeated exposure to sevoflurane in neonatal mice and monkeys brings about cognitive deficits and neurotoxicity [5, 6, 7, 8, 9]. Therefore, delving into the impact and mechanisms of sevoflurane anesthesia on brain neural cells has crucial clinical significance.
Autophagy and apoptosis are two common and crucial cellular biological processes. Under normal circumstances, autophagy serves to eliminate aberrant protein aggregates or damaged organelles within cells, providing protection to neuronal cells [10]. Literature report indicated that autophagy is of significance in neurodegenerative diseases namely Alzheimer’s, Parkinson’s, Huntington’s, neuronal excitotoxicity, and brain ischemia [11]. Apoptosis is instigated via a spectrum of intrinsic and extrinsic mechanisms, encompassing mitochondrial-associated pathways and regulatory cell death pathways. In brain neurons, apoptosis significantly influences neural development, selective neuronal survival, and the occurrence of neurodegenerative diseases. Aberrant regulation of apoptosis may contribute to the development of neurodegenerative diseases, such as Parkinson’s disease, stroke, and brain injury. Autophagy and apoptosis exhibit interrelated and reciprocal biological functions in brain neurons. By clearing harmful components and damaged organelles within cells, autophagy promotes cell survival and functional stability, thereby inhibiting apoptosis. However, dysregulation of autophagy is implicated in exacerbating cellular death and apoptotic events. Concurrently, apoptosis is reciprocally influenced by autophagy, with discrete signaling molecules assuming pivotal roles in the orchestration of these interrelated cellular processes.
Sirtuin type 1 (SIRT1) is the mammalian homolog of yeast silent information regulator 2 (Sir2) and belongs to the third class of histone deacetylases. It has been shown to extend the lifespan of lower organisms, inhibit age-related myocardial diseases, and protect the mouse heart from oxidative stress [12]. Additionally, accumulating evidence demonstrates that SIRT1 participates in neuronal cell autophagy within the brain [13]. SIRT1 regulates autophagy through the interaction with the autophagy-related genes autophagy related 5 (Atg5), autophagy related 7 (Atg7), and autophagy related 8 (Atg8), thereby inducing their deacetylation [14]. Mechanistic target of rapamycin (mTOR) is a suppressor of cellular autophagy, and an inhibition against this molecule is known to enhance autophagic activity [15]. Interplay between SIRT1 and mTOR has been shown in prior research to orchestrate cellular autophagy, thereby contributing to neuroprotective mechanisms within the brain. Chen et al. [16] have discovered that Urolithin A promotes autophagy through activating the SIRT1 signaling pathway and inhibiting the mTOR signaling pathway, which makes for the prevention of D-galactose-induced brain aging by activating the miR-34a-mediated SIRT1/mTOR signaling pathway for neuroprotection. Wang et al. [17] discovered that nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) induces autophagy by regulating the SIRT1/mTOR pathway, thereby protecting aged rats from damage caused by acute ischemic stroke. However, precise impact of the SIRT1/mTOR pathway on cell autophagy and its influence on neuronal apoptosis following sevoflurane inhalation in fetal rats remains to be elucidated. This study demonstrated the mechanism of action of the SIRT1-mTOR signaling pathway in regulating cell autophagy in sevoflurane-induced apoptosis of hippocampal neurons in fetal rats.
Eight male and sixteen female specific
pathogen free (SPF)-grade sprague dawley (SD) rats (aged 7–8 weeks) were
provided by Chongqing Ensville Biotechnology Co., Ltd. (Chongqing, China). All the
rats were maintained under standardized conditions with a temperature of 23
Carefully remove the skull using straight and curved forceps, exposing the brain. Under a stereomicroscope, gently dissect the cerebral cortex using straight forceps to expose the hippocampal tissue. Carefully separate the hippocampal tissue from the surrounding brain tissue. Carefully remove the hippocampal tissue using curved forceps. Place the extracted hippocampal tissue in a container with phosphate buffered saline (PBS) buffer for washing and subsequent experiments.
All aninmal experiments were carried out in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and were approved by the Medical Research Ethics Committee of Children’s Hospital of Chongqing Medical University (No. (142) in 2023).
Extraction of hippocampal neurons from fetal rats was followed by their cultivation, with all procedures executed as detailed below: Hippocampus obtained from the fetal rat were cut up fully with ophthalmic scissors and were added to the 0.125% of trypsin (S310KJ, Beyotime, Shanhai, China) for incubation for 30 min at 37 °C. The digestion was terminated by the addition of dulbecco’s modified eagle medium (DMEM) (L110KJ, Basalmedia, Shanghai, China) containing 10% fetal bovine serum (FBS; AC03L055, Life-iLab, Shanghai, China). The cells were centrifuged, resuspended, and filtered until a uniform cell suspension was formed. Extracted cells were cultured in Neuronbasa medium (21103049, Gibco, Grand Island, NY, USA) supplemented with 2% B27 and 10% FBS (AC03L055, Life-iLab) at 37 °C with 5% CO2. On day 3 of inoculation, 5 µmol/L cytarabine was added to the culture plates to inhibit growth of non-neural cells. The medium was changed every 3 days and neuron cell growth was observed under an inverted microscope (BLD-200, BJCOSSIM, Beijing, China). Flow cytometry analysis was applied to detect and identify fetal rat hippocampal neuron cells (Supplementary Fig. 1). Testing of the neuron cells for mycoplasma was negative.
Healthy fetal rat hippocampal neurons were harvested and digested using
0.25% trypsin solution to collect cells and prepare a cell suspension. Cell
density was adjusted to 2
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining of fetal rat hippocampal tissue was performed upon application of 3,3’-Diaminobenzidine (Streptavidin-Horseradish Peroxidase) Tunel Cell Apoptosis Detection Kit (G1507, Servicebio, Wuhan, China) according to the instructions of the manufacturer. Briefly, those tissue slices were dewaxed and penetrated through Proteinase K working solution at 37 °C for 20 min. Each sample was added into Equilibration Buffer and incubated in Terminal deoxynucleotidyl transferase (TdT) incubation buffer at 37 °C for 1 h. Subsequently, pre-diluted streptavidin-Horseradish Peroxidase (HRP) reaction solution was added into each tissue. Following sample washing, the slides were dyed by Diaminobenzidine (DAB) chromogenic solution and were added into hematoxylin. The slides were sealed with a neutral resin. Finally, images were obtained through the inverted microscope (MF53, Mshot, Guangzhou, China).
Concentration and purity of RNA were measured in each sample via
spectrophotometry after total RNA extraction. Subsequently, a reverse
transcription kit (TSK302M, Tsingke, Beijing, China) was used for cDNA synthesis,
and fluorescence quantitative PCR detection was performed through 2
| Primers (organism: rat) | Sequences |
| cleaved-caspase-3-F | GAGCTTGGAACGCGAAGAAA |
| cleaved-caspase-3-R | TTGCGAGCTGACATTCCAGT |
| Bax-F | CGTCTGCGGGGAGTCACG |
| Bax-R | AGCCATCCTCTCTGCTCGAT |
| Sirt1-F | CTACCGAGACAACCTCCTGTT |
| Sirt1-R | GTCACTAGAGCTTGCGTGTG |
| Mtor-F | GCAATGGGCACGAGTTTGTT |
| Mtor-R | AGTGTGTTCACCAGGCCAAA |
| p62-F | GAGGCACCCCGTAAAGTCAT |
| p62-R | GATTCAACCGCCATGTGCTT |
| Lc3-F | CCAGGATTCAGCACCAGATGT |
| Lc3-R | CCTTGGCTACCTGGACTGTT |
| Gapdh-F | TGGTGCTGAGTATGTCGTGG |
| Gapdh-R | GGCGGAGATGATGACCCTTT |
Sirt1, Sirtuin type 1; Bax, Bcl-2-associated X protein; Mtor, Mammalian target of rapamycin; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; Lc3, Microtubule-associated protein 1A/1B-light chain 3.
After extracting 500 µg of total proteins from the
samples and mixing them with 5
After fixation, cell coverslips were washed with PBS three times, 5 min per round. A suitable amount of 0.3% Triton X-100 (ST795, Beyotime, Beijing, China) permeabilization solution was added to coverslips and incubated. These coverslips were then rinsed three times. Goat serum (C0265, Beyotime, Beijing, China) was added for blocking and then removed without rinsing. Primary antibody against SIRT1 (1:200, A0230, ABclonal, Wuhan, China) was used for incubation overnight at 4 °C. After washing three times with PBS, the specimens were cultured with secondary antibodies Cy3 Goat Anti-Rabbit IgG (H+L) (1:50, AS008, ABclonal, Wuhan, China) and FITC Goat Anti-Rabbit IgG (H+L) (1:50, AS011, ABclonal, Wuhan, China) in the dark, and followed by another three rounds of phosphate buffered saline (PBS) washing. 4’,6-diamidino-2-phenylindole (DAPI) (C1005, Beyotime, Beijing, China) was provided and the cells were cultured again to stain nuclei. Excess DAPI was removed by washing with PBS three times. The coverslips were sealed with an anti-fluorescence quencher. Image acquisition was performed via an Mshot MF53 inverted microscope (MF53, Mshot, Guangzhou, China).
Annexin V Fluorescein Isothiocyanate/Propidium Iodide (FITC/PI)-apoptosis kit (40302ES50, YEASEN, Shanghai, China) was applied to detect the cell apoptosis by flow cytometry. Cells were seeded in the 6-well plate and incubated in the CO2 incubator. When the cell density reached 70%, cells were added into FITC-dextran (1 mg/mL) and incubated for 2 h. Subsequently, cells were treated with the following procedures: PBS washing, trypsin digestion, and centrifugation after neutralizing with serum-containing medium. The supernatant was abandoned and cell pellets were resuspended in PBS. Finally, flow cytometry was performed using a CytoFLEX flow cytometer (CytoFLEX, Beckman, Brea, CA, USA).
Data analyses and plotting were performed using the GraphPad Prism 8.01 software
(GraphPad Software Inc., San Diego, CA, USA). The data are presented as mean
Outcomes of TUNEL assay (Fig. 1A) showcased a notable increase in the rate of
cell apoptosis within the hippocampal tissue of fetal rats exposed to 3%
sevoflurane, when compared to the control. Results from qPCR and western blot
(Fig. 1B,C) revealed that in 3% sevoflurane treatment group, the mRNA and
protein expression of cleaved-caspase-3, Bax, and SIRT1 were substantially
elevated (p
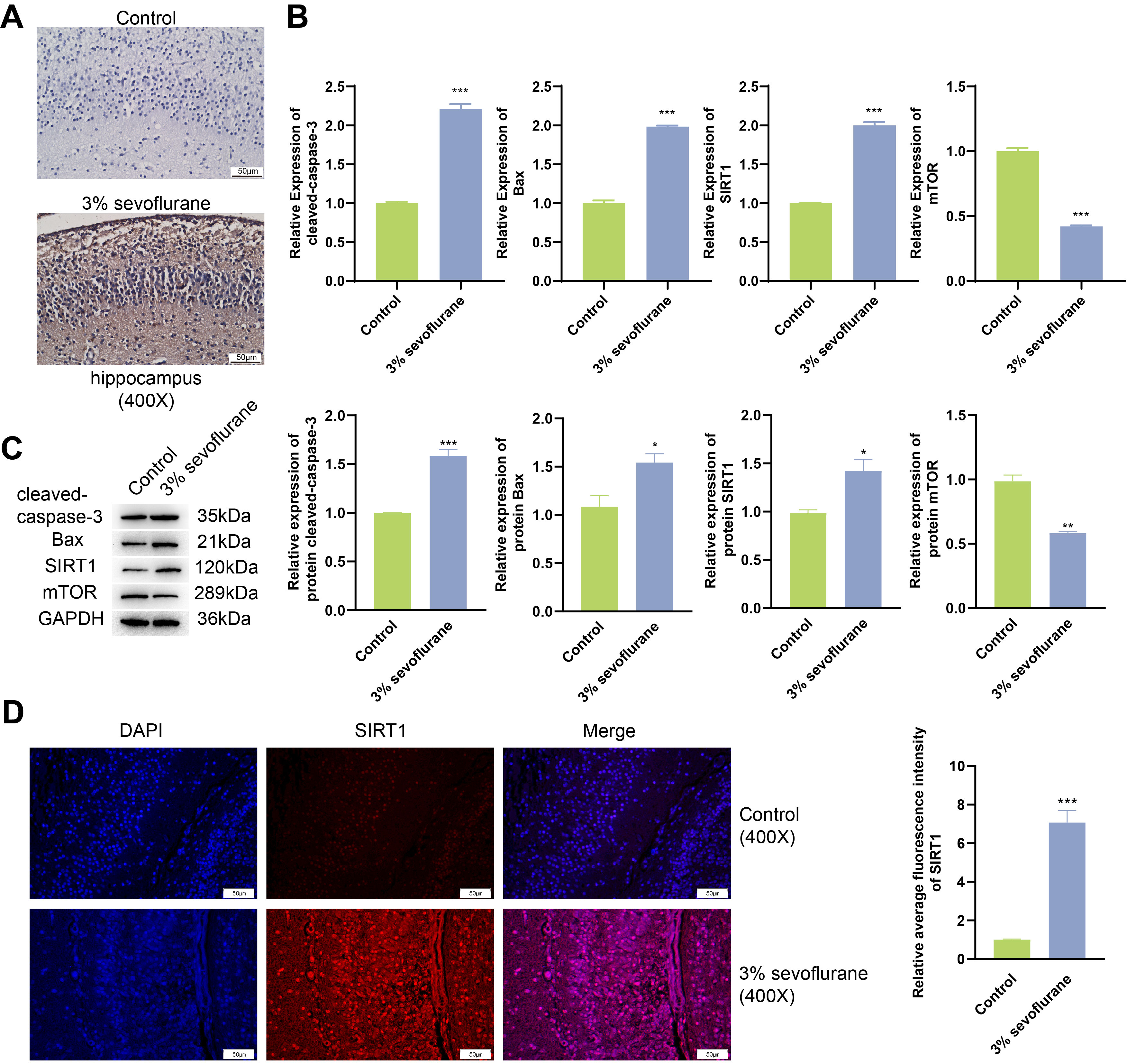 Fig. 1.
Fig. 1.
Inhalation of sevoflurane begets apoptosis of hippocampal tissue
in fetal rats. (A) Fetal rat hippocampal tissuel apoptosis was assessed through
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)
analysis. Scale bars, 50 µm. (B) Real-time quantitative polymerase chain
reaction (qPCR) analysis was conducted on fetal rat hippocampal tissue for
cleaved-caspase-3, Bax, Sirtuin 1 (SIRT1), and mechanistic target of rapamycin
(mTOR). (C) Western blot analyses of fetal rat hippocampal tissue for
cleaved-caspase-3, Bax, SIRT1, and mTOR. (D) Immunofluorescence combined with
4’,6-diamidino-2-phenylindole (DAPI) staining was utilized to examine expression
of SIRT1 in fetal rat hippocampal tissue. Scale bars, 50 µm. *p
In 3% sevoflurane treatment group, apoptotic levels of fetal rat hippocampal
neurons were markedly higher than the control, as indicated by the flow cytometry
results (Fig. 2A). This suggested that sevoflurane treatment could lead to an
increase in neuronal apoptosis, potentially exerting adverse effects on cognitive
functions. Immunofluorescence detection results (Fig. 2B) revealed an apparent
elevation in SIRT1 protein expression in 3% sevoflurane treatment group. SIRT1
functions as an essential regulator of cellular autophagy, and its increased
expression during sevoflurane therapy likely signifies the activation of
autophagic processes. Further qPCR and WB analysis results (Fig. 2C,D)
demonstrated a substantial increase (p
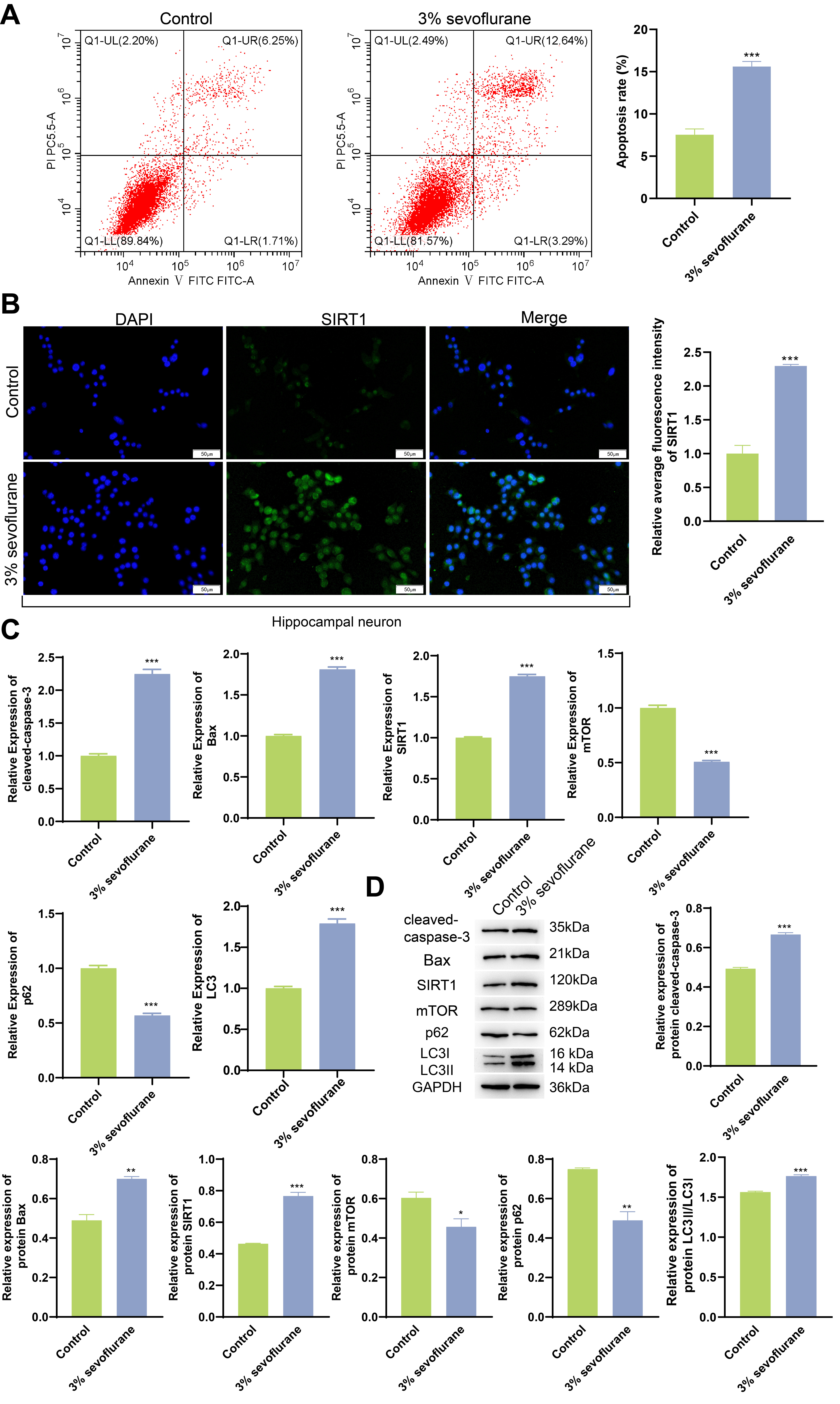 Fig. 2.
Fig. 2.
Promoting effect of sevoflurane on apoptosis and autophagy of
fetal rat hippocampal neurons. (A) Changes in apoptosis of fetal rat hippocampal
neuron cells after 3% sevoflurane treatment. (B) The expression of SIRT1 was
detected by immunofluorescence. Scale bars, 50 µm. (C) qPCR analysis of
cleaved-caspase-3, Bax, SIRT1,
mTOR, p62, and LC3II in fetal rat hippocampal neurons.
(D) Western blot analyses of cleaved-caspase-3, Bax, SIRT1, mTOR, p62, and microtubule-associated protein light chain 3Ⅱ (LC3II)
in fetal rat hippocampal neurons. *p
In this section, we employed strategies of SIRT1 disruption and mTOR suppression
to evaluate effects of autophagy on sevoflurane-induced apoptosis in fetal rat
hippocampal neurons. Flow cytometric analysis revealed (Fig. 3A) that hippocampal
neuron cell apoptosis decreased in 3% sevoflurane + SIRT1 inhibitor group
compared to 3% sevoflurane group, suggesting that SIRT1 interference is likely
to produce a protective effect against sevoflurane-induced neuronal apoptosis. In
our mTOR inhibition experiment, rapamycin was employed to suppress mTOR activity;
this approach resulted in a higher incidence of hippocampal neuron apoptosis in
3% sevoflurane + SIRT1 inhibitor + rapamycin group compared to 3% sevoflurane +
SIRT1 inhibitor group (Fig. 3A). This observed increase implies that mTOR
suppression potentially negates the neuroprotective impact of SIRT1 knockdown,
thereby intensifying sevoflurane-induced neuronal apoptosis. Immunofluorescence
detection (Fig. 3B) implied an apparent decrease in the expression of SIRT1 in
both 3% sevoflurane + SIRT1 inhibitor and 3% sevoflurane + SIRT1 inhibitor +
rapamycin groups. Moreover, qPCR and WB results (Fig. 4A,B) further supported
these findings. In 3% sevoflurane + SIRT1 inhibitor group, expression of SIRT1
significantly decreased, along with a significant decrease in cleaved-caspase-3,
Bax, and LC3II expression (p
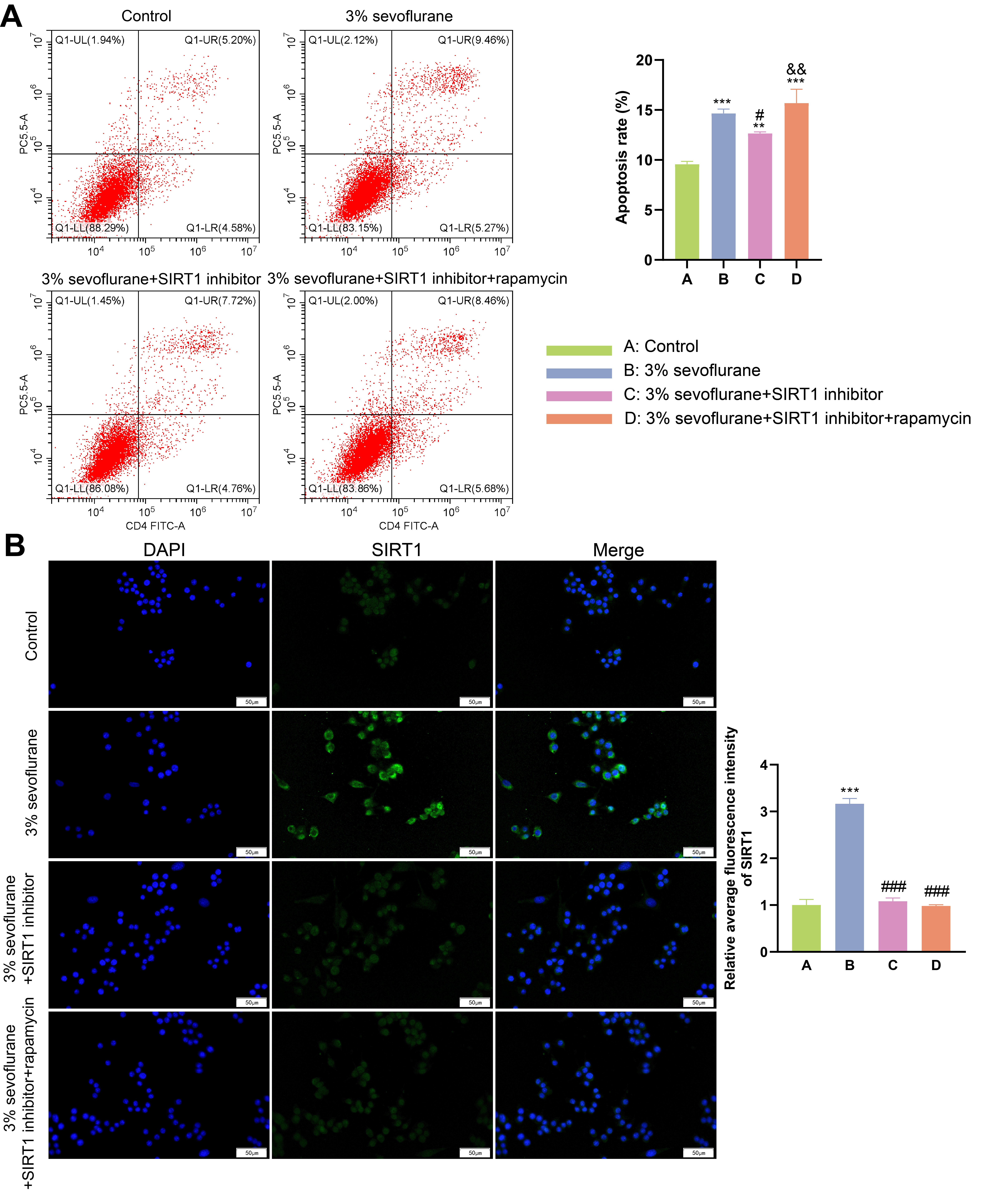 Fig. 3.
Fig. 3.
Autophagy regulates apoptosis of hippocampal neurons in fetal
rats induced by sevoflurane inhalation anesthesia. (A) Changes in apoptosis of
fetal rat hippocampal neuron cells after different treatment. (B) The expression
of SIRT1 was detected by immunofluorescence after different treatment. Scale
bars, 50 µm. Statistical significance compared to the control group:
**p
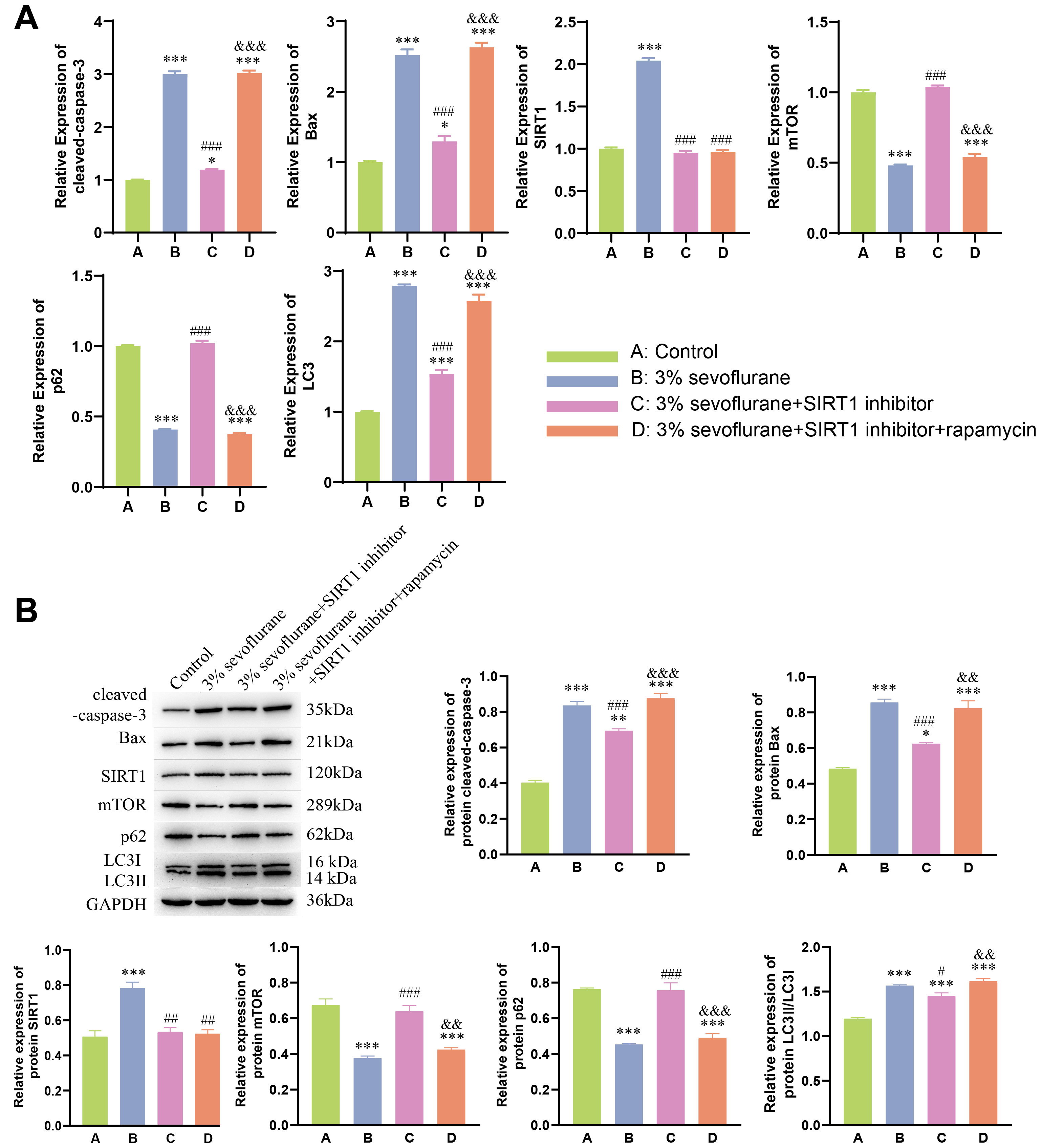 Fig. 4.
Fig. 4.
Neuroprotective effect of autophagy on fetal rat nerve cell
apoptosis induced by sevoflurane inhalation anesthesia. (A) qPCR analysis of
cleaved-caspase-3, Bax, SIRT1, mTOR,
p62, and LC3II in fetal rat hippocampal neurons. (B) Western
blot analysis of cleaved-caspase-3, Bax, SIRT1, mTOR, p62, and LC3II in fetal rat
hippocampal neurons. Statistical significance compared to the control group:
*p
Sevoflurane, a commonly used anesthetic in clinical settings, has detrimental effects on animal nervous system when inhaled at high concentrations for extended periods. Reports have suggested that prenatal exposure to sevoflurane inhibits the proliferation of fetal neural stem cells [18], neuronal migration, and axonal growth [19]. This inhibition leads to neuronal apoptosis [20] and a sustained imbalance between excitatory and inhibitory neurons in the medial prefrontal cortex [21], resulting in learning and memory deficits as well as anxiety-like behavior. Our study findings indicated an increase in apoptosis of fetal rat hippocampal neurons following sevoflurane treatment, potentially serving as a significant factor contributing to cognitive dysfunction. Apoptosis, a normal physiological process in the body, serves as a self-regulated programmed death mechanism, allowing to eliminate damaged or unnecessary cells to maintain normal structure and function of the tissues and organs. However, an abnormal activation of apoptosis leads to various diseases, including neurological disorders. In this study, we initially observed that inhalation of sevoflurane during fetal development resulted in increased apoptosis of hippocampal neurons in fetal rats. TUNEL experiment results showed a significantly higher apoptosis level in sevoflurane-treated group compared with the control group, supporting detrimental impact of sevoflurane on neural cells in fetal rats. Additionally, qPCR and western blot revealed a significant up-regulation in the expression of cleaved-caspase-3 and Bax in the 3% sevoflurane-treated group. Xu et al. [22] demonstrated increased levels of cleaved-Caspase-3 and Bax in HCN-2 cells after sevoflurane treatment. Similarly, Areias et al. [23] found a notable increase in cleaved-caspase-3 after sevoflurane treatment at 1.63 [1.56–1.77]% of brain volume. The results suggested that the developing brain is highly susceptible to anesthesia exposure, leading to an increase in cell apoptosis, as evidenced by a significant increase in SIRT1 expression and a marked decrease in mTOR expression.
Additionally, this study has elucidated the involvement of autophagy in neuronal apoptosis triggered by sevoflurane treatment. Autophagy functions as an essential intracellular process for the degradation and recycling of cellular components, crucial for maintaining metabolic equilibrium within the cell. Our experimental results indicated an up-regulation of LC3 expression and a simultaneous down-regulation of p62 expression in sevoflurane-treated group. Wang et al. [24] found increased levels of LC3II/LC3I and decreased p62 in hippocampal neurons treated with sevoflurane. Zhou et al. [25] suggested an increase in the expression of autophagy marker protein LC3 and a decrease in the protein p62 in H4 human glioma cells after 6 h of exposure to 4.1% sevoflurane. These findings confirmed that sevoflurane exposure induces endoplasmic reticulum stress and activates autophagy in H4 human glioma cells, providing negative feedback to inhibit sevoflurane-induced cell apoptosis. Additionally, SIRT1 functions as a pivotal modulator of autophagy [26], with its augmentation being possibly linked to heightened autophagic activity. Conversely, mTOR operates as an inhibitory factor in autophagy regulation, and its suppression is likely to further stimulate an upsurge in autophagic processes. Autophagy in sevoflurane-induced neuronal apoptosis was verified through SIRT1 knockdown and mTOR inhibition. Our results indicated a decreasing trend in the apoptosis of fetal rat hippocampal neurons in the SIRT1 interference experiment. Contrary to expectations, the mTOR inhibition experiment revealed an upward trajectory in rate of hippocampal neuronal apoptosis, indicating that mTOR suppression might potentially counteract the neuroprotective influence conferred by SIRT1 modulation and exacerbate sevoflurane-triggered neuronal cell death. This observation intimated that SIRT1 governs autophagic processes, thereby imposing a mitigating impact on neuronal apoptosis incited by sevoflurane, in opposition to mTOR’s antagonistic regulatory stance. Therefore, autophagy and the SIRT1-mTOR pathway are potential targets in treating anesthesia-induced neuronal apoptosis and cognitive impairment.
In conclusion, our research delineated that exposure to sevoflurane via inhalation in fetal rats precipitates an escalation in neuronal apoptosis, with the modulatory factors SIRT1 and mTOR assuming critical factors in this cascade. Over-expression of SIRT1 is implicated in the sevoflurane-induced augmentation of cellular autophagy, thereby conferring a protective mechanism against neuronal apoptosis. In contrast, mTOR imparts an antagonistic regulatory influence within this scenario. Our research elucidated the ramifications of anesthetic agents on neuronal cells, thereby providing a foundation for the formulation of future investigative endeavors focused on neuroprotective strategies. However, more investigations remain necessary to figure out detailed regulatory mechanisms of cellular autophagy in sevoflurane-induced neuronal apoptosis, and to validate the clinical significance and application potential of these findings. Firstly, further exploring the precise molecular interactions within the SIRT1-mTOR pathway and its role in neuronal apoptosis induced by sevoflurane exposure is essential. Subsequently, future research should focus on the role of key autophagy-related genes such as Beclin1 and LC3 to investigate the relationship between autophagy and sevoflurane-induced apotopsis in fetal rat hippocampal neurons. Additionally, large-scale clinical studies are required to assess the relevance of these findings in human patients undergoing sevoflurane anesthesia. These comprehensive investigations will not only expand our understanding of sevoflurane neurotoxicity but also pave the way for the development of novel therapeutic strategies for preventing or treating sevoflurane-induced apoptosis in neural cells.
Sevoflurane treatment incites apoptosis in hippocampal neurons of fetal rats, wherein cellular autophagy wields a pivotal influence on the proceedings. Empirical research corroborates the notion that SIRT1 orchestrates apoptotic response of these neurons to sevoflurane exposure, concurrently with mTOR fulfilling an inhibitory regulatory function within this framework. These results hold critical significance for a thorough understanding of the mechanism of action of sevoflurane on neural cells and the future development of relevant neuroprotective strategies.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
SIRT1, Sirtuin type 1; Bax, Bcl-2-associated X protein; mTOR, Mammalian target of rapamycin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; LC3, Microtubule-associated protein 1A/1B-light chain 3.
YHL conceived, designed, and performed the experiments, and wrote the original draft. CX and YT analyzed and interpreted the data. SJ designed the research, formal analysis, writing review and editing, and supervision. All authors contributed to the editorial changes in the manuscript. All authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experiments proposed in this study have been approved by the Medical Research Ethics Committee of Children’s Hospital of Chongqing Medical University (No. (142) in 2023). The protocol for animal research outlined in this study complies with ethical guidelines and regulations regarding the humane and responsible use of animals in scientific research.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2909324.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.