



1 College of Basic Medical Sciences, Shaanxi University of Chinese Medicine, 712046 Xianyang, Shaanxi, China
2 Second Clinical Medical College, Shaanxi University of Chinese Medicine, 712046 Xianyang, Shaanxi, China
†These authors contributed equally.
Abstract
Hepatic fibrosis is a major public health problem that endangers human wellbeing. In recent years, a number of studies have revealed the important impact of metabolic reprogramming on the occurrence and development of hepatic fibrosis. Among them, the Warburg effect, as an intracellular glucose metabolism reprogramming, can promote the occurrence and development of hepatic fibrosis by promoting the activation of hepatic stellate cells (HSCs) and inducing the polarization of liver macrophages (KC). Understanding the Warburg effect and its important role in the progression of hepatic fibrosis will assist in developing new strategies for the prevention and treatment of hepatic fibrosis. This review focuses on the Warburg effect and the specific mechanism by which it affects the progression of hepatic fibrosis by regulating HSCs activation and KC polarization. In addition, we also summarize and discuss the related experimental drugs and their mechanisms that inhibit the Warburg effect by targeting key proteins of glycolysis in order to improve hepatic fibrosis in the hope of providing more effective strategies for the clinical treatment of hepatic fibrosis.
Keywords
- Warburg effect
- hepatic fibrosis
- aerobic glycolysis
- hepatic stellate cells
- hepatic macrophages
Hepatic fibrosis (HF) is a chronic process of hepatic tissue repair resulting from multiple forms of persistent damage to the liver. Without timely intervention, hepatic fibrosis may progress to liver failure, leading to patient death [1, 2]. Although some progress has been made in the diagnosis and treatment of hepatic fibrosis, there continues to be a lack of effective treatments, as the morbidity and mortality of hepatic fibrosis continues to increase yearly [3, 4, 5]. Recently, several studies have revealed the influence of metabolic reprogramming on the development of hepatic fibrosis, among which the Warburg effect, as a kind of intracellular reprogramming of glucose metabolism, has attracted increased attention from researchers [6]. The Warburg effect, which is characterized by a shift in the intracellular glucose metabolism pattern from oxidative phosphorylation to aerobic glycolysis, was initially mentioned in the context of cancer research [7, 8]. Subsequent studies have shown that cells present in hepatic fibrosis prefer to utilize aerobic glycolysis for their own energy, and thus the Warburg effect is widespread in fibrotic diseases in tissues and organs such as the kidneys, liver, skin, and lungs [9, 10, 11, 12, 13, 14, 15]. Among them, in hepatic fibrosis, the Warburg effect influences the onset and progression of hepatic fibrosis mainly by regulating the activation of hepatic stellate cells (HSCs) and the polarization of Kupffer cells (KCs) [16]. Secondly, researchers found that targeting the Warburg effect by inhibiting key enzymes and related proteins of glycolysis could effectively reduce hepatic fibrosis [17]. This implies that the Warburg effect may be a new target for the treatment of hepatic fibrosis, which needs to be adequately studied. This review focuses on the specific mechanisms by which the Warburg effect affects the progression of hepatic fibrosis. Meanwhile, relevant drugs targeting the Warburg effect for the treatment of hepatic fibrosis and its corresponding mechanisms are further summarized, hoping to provide a theoretical basis for the clinical development of new strategies for the treatment of hepatic fibrosis.
As the primary energy substance for maintaining homeostasis in the body, glucose has a critical effect on the growth and development of cells [18]. Glucose metabolism refers to the process of energy metabolism and biosynthesis by organisms using substances such as glucose and glycogen. A big part of glucose metabolism is glycolysis. Some of the enzymes and proteins that play a part in this process are hexokinase (HK), phosphofructose kinase (PFK), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), pyruvate kinase (PK), and glucose transporter (GLUT) [19, 20]. Fig. 1 shows how glucose moves from the extracellular space to the cytoplasm with the help of GLUT. The first irreversible step of glycolysis is when HK phosphorylates it to glucose-6-phosphate (G6P), a process that requires ATP. Glucose phosphate isomerase changes G6P into fructose-6-phosphate (F6P). PFK then phosphorylates F6P into fructose-1,6-diphosphate (FDP). This is the second reaction of glycolysis that can not be undone. Moreover, PFK itself is allosterically activated by PFK2-catalyzed fructose-2,6-bisphosphate, suggesting that PFK2 is a key regulator of glycolysis. Following a series of reactions, FDP is changed into phosphoenolpyruvate (PEP). Pyruvate kinase PK [21] then converts PEP into pyruvate (PA), releasing an ATP molecule in the process. This phase represents the ultimate stage in the glycolytic pathway. Normal cells use pyruvate dehydrogenase (PDH) to change PA into acetyl coenzyme A (acetyl CoA). This allows them to enter the tricarboxylic acid (TCA) cycle and use oxidative phosphorylation (OXPHOS) to make a large amount of ATP to meet the energy needs of living beings. However, under hypoxic conditions, cells generate a significant quantity of lactic acid and a small amount of ATP as their main energy source through glycolysis. Lactate is transported from the cytoplasm to the extracellular space with the help of the monocarboxylic acid transporter (MCT). This lowers the pH of the adjacent area [22, 23].
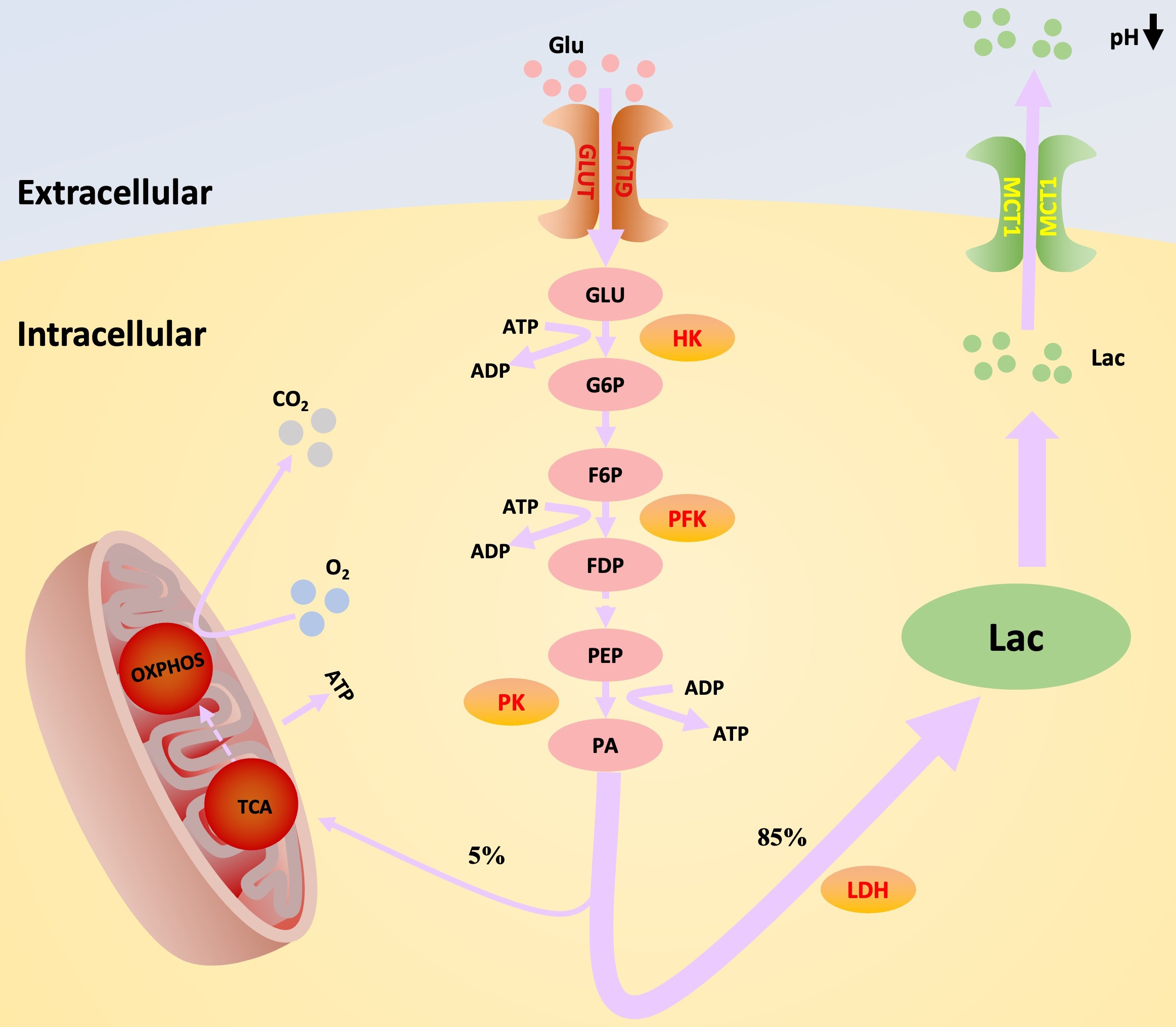 Fig. 1.
Fig. 1.
Diagram of Warburg effect and its key enzymes. When the Warburg effect occurs, the main mode of intracellular sugar metabolism changes from aerobic oxidation to aerobic glycolysis. This process is accompanied by changes in the production and function of a series of key proteins and enzymes in the glycolytic process, as well as a concomitant increase in the production of lactate, the end product of glycolysis. GLU, glucose; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; FDP, fructose-1,6-diphosphate; PEP, phosphoenolpyruvate; PA, pyruvate; Lac, lactic acid; GLUT, glucose transporter; HK, hexokinase; PFK, phosphofructose kinase; PK, Pyruvate kinase; LDH, lactate dehydrogenase; TCA, tricarboxylic acid; OXPHOS, oxidative phosphorylation; MCT1, monocarboxylic acid transporter1; ATP, adenosine triphosphate; ADP, Adenosine Diphosphate.
Different from normal cells, in order to obtain a large amount of metabolites and energy, tumor cells rapidly obtain them through glycolysis to satisfy their fast proliferation and differentiation, both under aerobic and anaerobic conditions [24]. As early as the 1820s, the German biochemist Otto Warburg hypothesized that cancer cells have a preference for the glycolysis pathway even under conditions of sufficient oxygen and finally produce lactic acid to provide energy for themselves. The anomalous metabolic process involving glucose is referred to as the Warburg effect, alternatively known as aerobic glycolysis [25, 26]. A wide range of malignant tumors, including malignant lipoma, hepatocellular carcinoma, colon cancer, breast cancer, and lung cancer exhibit the Warburg effect [27, 28, 29, 30, 31]. Recently, a growing body of research has demonstrated that, in addition to oncological diseases, the key genes of the Warburg effect are abnormally highly expressed in inflammatory and immune diseases [32, 33], heart failure [34], pulmonary hypertension [35], and a variety of non-oncological diseases, such as fibrotic syndromes of renal, pulmonary, and peritoneal tissues [8]. On a per-unit glucose basis, the Warburg effect has a lower efficiency than oxidative phosphorylation, but the glucose metabolism rates are much higher than oxidative phosphorylation, so the energy production per unit of time is almost the same for both metabolic modes [36]. Obviously, the Warburg effect exhibits a more favorable advantage in preempting nutrients when the amount of glucose is limited [37]. It has been demonstrated that the Warburg effect exerts a key role under normoxic conditions because it helps the cell membrane meet its needs for quick energy [38]. In addition, the intermediate metabolites produced during aerobic glycolysis can be used for the synthesis of nucleic acids, proteins, and lipids, which provide a certain material basis for cell proliferation [39]. It can be seen that the Warburg effect confers many advantages on the cells.
The occurrence of hepatic fibrosis involves numerous cellular and molecular biological events, among which the activation of HSCs and polarization of hepatic macrophages are the key events that promote the development of hepatic fibrogenesis [40]. Upon activation, HSCs exhibit heightened functionality and undergo active proliferation. They stimulate the creation of extracellular matrix (ECM) while also impeding its breakdown. This leads to an excessive accumulation of ECM, hence facilitating the advancement of hepatic fibrosis [41, 42]. Hepatic macrophages have the ability to secrete growth factors that stimulate the activation of HSCs. At the same time, they also trigger an inflammatory response, worsening hepatocyte damage and promoting the progression of hepatic fibrosis [43]. Research has demonstrated the presence of the Warburg effect in activated HSCs and polarized macrophages. It has also been observed that alterations in this glucose metabolism mode subsequently stimulate the activation of HSCs and the polarization of hepatic macrophages. These processes are crucial for the advancement of hepatic fibrosis [7] (Fig. 2).
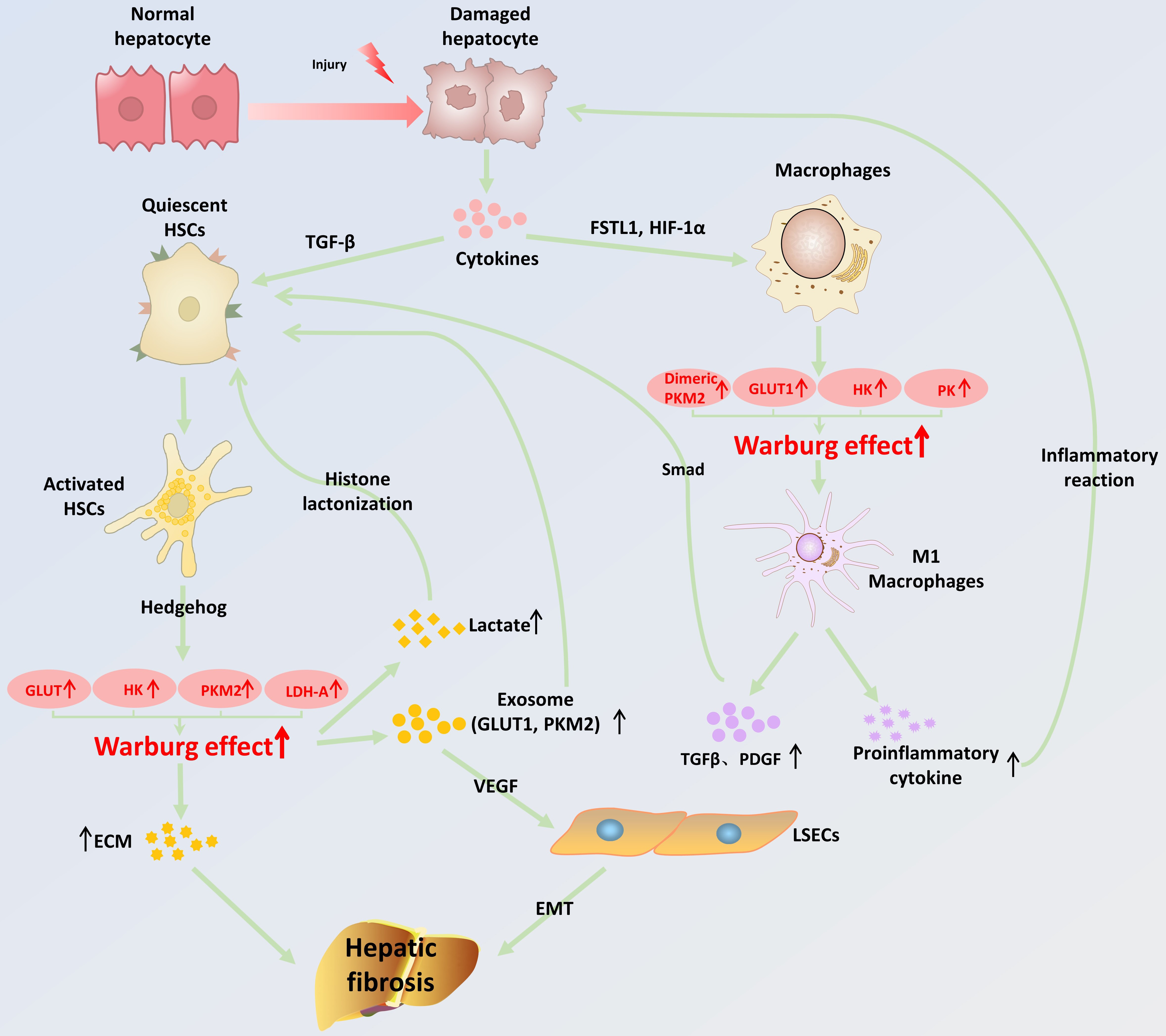 Fig. 2.
Fig. 2.
Schematic representation of the mechanisms by which the Warburg
effect promotes the onset and progression of hepatic fibrosis. When hepatocytes
suffer from various chronic injuries, cytokines are released that promote
activation of HSCs and polarization of hepatic macrophages. The increased energy
demand of activated HSCs promotes the synthesis of glycolysis-related enzymes
through the activation of the Hedgehog pathway, resulting in an enhanced
intracellular Warburg effect. When the Warburg effect is enhanced in HSCs, it
promotes the release of lactate and exosomes, which further promotes the
activation of quiescent HSCs, leading to massive deposition of ECM. In addition,
the exosome-activated HSCs will highly express VEGF, causing the EMT process in
LSECs. All of the these together promote the occurrence and development of
hepatic fibrosis. Meanwhile, induced by FSTL1 and HIF-1
In the perisinusoidal interstitium of the liver, HSCs are a distinct subtype of
mesenchymal cells. They make up about one-third of the liver’s nonparenchymal
cells [44]. HSCs in a healthy liver are in a quiescent condition, meaning they
are not actively dividing or proliferating. When the liver sustains an injury,
various cells, such as hepatocytes and hepatic macrophages, can release a
significant amount of pro-inflammatory factors, pro-fibrotic factors, and growth
factors. This release of factors prompts the transformation of HSCs from a
dormant to an active status. Activated HSCs undergo rapid proliferation. As a
result, the levels of vitamin A, glial fibrillary acidic protein (GFAP), and
peroxisome proliferator-activated receptor-
Activated HSCs also produce a series of glycolytic intermediates under the
influence of the Warburg effect that promote the activation of quiescent HSCs. A
study by Wan and his colleagues [50] confirmed that exosomes containing two key
proteins for glycolysis, GLUT1 and PKM2, could be
released when the Warburg effect was enhanced in activated HSCs.
Co-incubation of this exosome with resting HSCs resulted in high expression of
GLUT1 and PKM2 [50]. Increased GLUT1 expression contributes to an increased rate
of glycolysis to promote the activation of HSCs. In addition, the dimeric form of
PKM2 can work as a protein kinase, starting the transcription of certain genes
like MYC and CCND1 in the nucleus by controlling the change of
histone H3. This activates the Wnt/
Hepatic macrophages, often referred to as Kupffer cells (KCs),
are mononuclear macrophages that originate from the bone marrow and are located
in the liver. They make up approximately 90% among all macrophages in the body’s
tissues and display considerable heterogeneity. Hepatic macrophages can be
categorised into two distinct categories according to their activation
mechanisms, cell surface markers, and the cytokines they secrete: conventionally
activated (M1) macrophages and alternatively activated (M2) macrophages [56].
Lipopolysaccharide and interferon (IFN) are primarily responsible for activating
M1 macrophages, which are also referred to as pro-inflammatory macrophages. They
play a role in the T helper cell 1 (Th1) and Th17 immune responses [57].
Interleukin-4 (IL-4) and IL-13 are the main substances that make M2-type
macrophages work. These are also called inflammation-suppressing macrophages, and
they are part of Th2-type immune responses [58]. Due to their distinct
polarisation characteristics, they fulfil diverse functions in the progression of
hepatic fibrosis. In the initial and developmental stages of hepatic fibrosis,
damage-related pattern molecules and apoptotic cells activate Kupffer cells in
the Disse space and cause their phenotype to change, as the phenotype of hepatic
macrophages at this time is dominated by the M1 phenotype. Activated Kupffer
cells can promote hepatic fibrosis in the ways shown in Fig. 2: (1) Kupffer cells
produce pro-fibrotic factors, such as TGF-
Research has shown that modifying the metabolic procedure of glycolysis in KCs
is essential for stimulating its polarization [63]. The primary metabolic pathway
in M1-type macrophages is aerobic glycolysis, whereas M2-type macrophages
predominantly depend on oxidative phosphorylation [64]. Follicle suppressor-like
egg 1 (FSTL1) is a cytokine with several effects. Rao and colleagues [65] found
that the glycolysis-related protein PKM2 can directly interact with FSTL1. This
interaction leads to the activation of M1-type macrophages and an increase in the
Warburg effect within these macrophages. It starts a metabolic reprogramming
process that changes hepatic macrophages from an M1-type phenotype to an M2-type
phenotype when the sugar metabolism in KCs changes from glycolysis to oxidative
phosphorylation. Xu and colleagues [66] found that when membrane-bound protein A5
and PKM2 come into direct contact with certain amino acid residues (Asp101,
Leu104, and Arg106), it stops Tyr105 from being phosphorylated and makes it
easier for PKM2 tetramers to form. PKM2 tetramers suppress the Warburg effect,
leading to a change in cellular characteristics towards the M2 phenotype. This
change ultimately improves conditions such as steatosis, inflammation, and
hepatic fibrosis in the non-alcoholic steatohepatopathy (NASH) model.
Additionally, HIF-1
Given the importance of macrophages with different polarization phenotypes in the occurrence and progression of hepatic fibrosis, as well as the induction effect of the Warburg effect on liver macrophage phenotype, inhibiting the Warburg effect to promote macrophage phenotype conversion from M1 to M2 is a novel method to alleviate inflammation and treat hepatic fibrosis.
The Warburg effect is significantly involved in the development of hepatic fibrosis, mainly by upregulating the expression of some glycolysis-related proteins, regulating the activation of HSCs, and polarizing M1-type macrophages. Recent research has found that targeting glycolysis-related proteins such as GLUT, PKM2, PFK1, HK, and lactate Dehydrogenase A (LDH-A) can inhibit the Warburg effect and produce antifibrotic effects [69, 70]. In addition, many drugs have been demonstrated in animal and cellular experiments to alleviate hepatic fibrosis by targeting key proteins in the Warburg effect (Table 1, Ref. [49, 51, 53, 65, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83]).
| Name of therapeutic drug | Strategy | Test subject | Therapeutic dose | Mechanism | Target | Reference |
|---|---|---|---|---|---|---|
| Phloretin | Modulation of hepatic stellate cells (HSCs) activation | CCl4-injected C57 black 6 (C57BL/6) and primary mouse HSCs | Animal model: 10 mg/kg; Cell model: 50 µM | Smad, p38 mitogen-activated protein kinase (MAPK), and Phosphatidylinositol 3-kinase-Protein Kinase B (PI3K/AKT) pathway | Glucose transporter 1 (GLUT1) | [49] |
| Galloflavin | Modulation of HSCs activation | Lieming Xu-2 (LX-2) cells and HSC-T6 cells | 20 µM | Hedgehog pathway | Lactate dehydrogenase (LDH) activity, Hexokinase 2 (HK2), and GLUT1 | [71] |
| Curcumin | Modulation of HSCs activation | Primary rat HSCs and immortalized human hepatocytes (IHHs) | 0–30 µM | P38 MAPK pathway | GLUT2 | [72] |
| Modulation of HSCs activation | Primary rat HSCs and IHHs | 0–30 µM | p38 MAPK pathway | GLUT4 | [73] | |
| Modulation of HSCs activation | Primary rat HSCs | 20 µM | Peroxisome proliferator-activated receptor- |
HK and PFK2 | [74] | |
| Deoxyelephantopin | Modulation of HSCs activation | Primary rat HSCs | 2.5, 5, 10 and 20 µM | Hedgehog pathway | HK, Phosphofructose kinase 2 (PFK2), and GLUT4 | [75] |
| Costunolide | Modulation of HSCs activation | Primary rat HSCs | 10, 20 and 30 µM | Hindering de novo synthesis | HK2 | [53] |
| Fucoxanthin | Modulation of HSCs activation | Primary mouse HSCs | 2 or 3 µM | - | GLUT1, HK1 and HK2 | [76] |
| EGCG, taurine and genistein | Modulation of HSCs activation | HSC-T6 | - | - | HK2 | [77] |
| Alamandine | Modulation of HSCs activation | Bile duct ligation (BDL)–induced hepatic fibrosis rodent model | Animal model: 50 µg/kg; Cell model: 1 nM, 0.1 and 10 µM | Sequestosome1 (SQSTM1)-mediated lysosomal pathway | PFK liver-type (PFKL) | [78] |
| 3PO | Modulation of HSCs activation | Primary rat HSCs, LX-2 cells and BDL-induced C57BL/6 | 5, 7.5 and 10 µM | - | PFKFB3 | [79] |
| Shikonin | Modulation of HSCs activation | CCl4-injected C57BL/6, primary mouse HSCs and LX-2 cells | Animal model: 10 mg/kg; Cell model: 0.2 and 0.5 µM | Wnt/ |
PKM2 | [51] |
| Celastrol | Modulation of macrophage polarisation | RAW264.7 | 0.05, 0.1 and 0.2 µM | Forming a covalent bond with PKM2 | PKM2 | [80] |
| DASA-58 | Modulation of macrophage polarisation | Bone marrow-derived macrophages (BMDMs) | 50 µM | Promoting the formation of PKM2 tetramers | PKM2 | [65] |
| XAV-939 | Modulation of HSCs activation | CCl4-injected C57BL/6 and LX-2 cells | Animal model: 10 mg/kg; Cell model: 10 µM | Wnt/ |
LDH-A | [81] |
| Oroxylin A | Modulation of HSCs activation | CCl4-injected C57BL/6 and LX-2 cells | Animal model: 40 mg/kg; Cell model: 20, 30 and 40 µM | - | LDH-A | [82] |
| Curcumol | Modulation of LSECs angiogenesis | CCl4-injected C57BL/6 and Rat LSECs | Animal model: 30 mg/kg; Cell model: 10, 20 and 40 µM | Affects the transcriptional function of Kruppel-like factor 5 (KLF5) | LDH-A | [83] |
GLUT is mainly responsible for glucose transport in the human body and participates in the glycolytic process [84]. Numerous studies have shown that increased levels of expression of GLUT1, GLUT2, and GLUT4 among GLUTs in hepatic fibrosis can induce reprogramming of glucose metabolism to occur within HSCs and enhance the Warburg effect, which promotes HSCs activation and ultimately leads to the onset and progression of hepatic fibrosis [85, 86]. Targeted inhibition of the various GLUTs can reduce the Warburg effect in HSCs and achieve antifibrotic therapy in the liver.
The most widespread glucose transporter protein in the human body, GLUT1, is
primarily engaged in the regulation of glucose metabolism, which impacts cell
proliferation and growth [87, 88]. The activation of HSCs and aerobic glycolysis
can be enhanced by TGF-
In conclusion, LGLUT acts as in essential role during the evolution of hepatic fibrosis, and the Warburg effect of HSCs can be inhibited by targeting LGLUT to achieve anti-fibrosis. GLUT may therefore be a future prospective target for hepatic fibrosis treatment.
The initial step that restricts the rate of glycolysis is facilitated by the enzyme HK, which effectively hinders the exit of glucose from the cell for the purpose of energy metabolism [92]. There exist 4 isoforms of HK, namely HK1, HK2, HK3, and HK4. However, the majority of normal tissues exclusively express HK1. However, HK2 is more potent in promoting aerobic glycolysis and is highly expressed in hepatic fibrosis. Recent studies have shown that HK2-induced lactate promotes histone lactylation, which controls HSCs activation and leads to hepatic fibrosis. Inhibition of histone lactylation by HSCs-specific or systemic HK2 deletion attenuates HSCs activation and hepatic fibrosis, providing a new potential target for effective treatment of hepatic fibrosis [53, 93].
Costunolide, a naturally occurring compound belonging to the sesquiterpene
lactone class, has demonstrated hepatoprotective properties [94]. Ban et al. [95] discovered that costunolide hindered the production and functioning of
HK2 through de novo synthesis, impeded the aerobic glycolysis process in HSCs,
and decreased the expression of type 1 collagen in the extracellular matrix. This
reduction in type 1 collagen expression is particularly beneficial for treating
hepatic fibrosis. Fucoxanthin (FCX) is a carotenoid similar to lutein that is
found in high concentrations in brown algae and diatoms [96]. Prior research has
shown that FCX may effectively block the activation of Smad3 and decrease the
buildup of reactive oxygen species (ROS) in HSCs, resulting in antifibrotic
actions [97]. In an experiment conducted by Bae and colleagues [76], they
discovered that treatment with FCX led to a reduction in the expression of HK1
and HK2 in activated HSCs. This decrease significantly reduced the occurrence of
aerobic glycolysis in HSCs. Additionally, FCX also reduced the respiration of
mitochondria by modulating the functioning of genes associated with this process.
As a result, FCX exerted a dual inhibitory effect on the activation of HSCs.
Moreover, curcumin, a natural substance, increases the expression of
PPAR-
In summary, it has been established that the activity of HK2 is pivotal in the remodeling of glucose metabolism in the context of hepatic fibrosis. Inhibition of aerobic glycolysis via HK2 targeting exhibits considerable promise as a therapeutic approach for hepatic fibrosis.
PFK1 is a crucial enzyme that plays a significant role in the second step of glycolysis, acting as a rate-limiting factor. It is found in human tissues primarily in three different forms: muscle-type (PFKM), liver-type (PFKL), and platelet-type (PFKP). PFK1 is vital for ensuring the stability of the glycolytic process [98]. Prior studies have demonstrated that the stimulation of PFK1 can enhance the Warburg effect, leading to increased proliferation of tumor cells. Conversely, inhibiting PFK1 activity hinders tumor cell growth and metastasis. Consequently, PFK1 has become a vital focus for treating tumors by targeting the Warburg effect [99, 100, 101, 102, 103]. Recent investigations have revealed the promising potential of PFK1 in treating hepatic fibrosis. Alamandine is an endogenous heptapeptide that can be formed from angiotensin A via angiotensin-converting enzyme-2 or directly by decarboxylation of angiotensin-(1–7) [104]. Previous studie has shown that alamandine inhibits NOX4-dependent ROS-induced autophagy in HSCs, resulting in a reduction in HSCs migration and collagen production. This has a beneficial impact on the treatment of hepatic fibrosis [105]. A study by Zhang et al. [78] found that alamandine could also enhance the sequestosome1 (SQSTM1)-mediated lysosomal pathway to promote PFK1 degradation by down-regulating the expression level of Caveolin-1, which reduced the Warburg effect in HSCs and then inhibited the further activation of HSCs and alleviated the symptoms of hepatic fibrosis. The activity of PFK1 is regulated by PFKFB3, the enzyme responsible for the synthesis of fructose 2,6-bisphosphate. This molecule acts as a variable activator of PFK1, leading to a substantial increase in its catalytic activity [106]. Thus, PFKFB3 is considered a crucial target for modulating the Warburg effect [107]. CPEB4 is a protein that binds to cytoplasmic polyadenylation elements (CPEs) [108, 109]. Mejias and colleagues [79]demonstrated that CPEB4 expression increased during the initial phase of HSCs activation. They also found that CPEB4 directly interacts with the 3′-non-translated region of the PFKFB3 transcript and the CPE origin of the 3′-untranslated region. This promotes cytoplasmic polyadenylation of PFKFB3 mRNA, leading to an increase in the expression of the PFKFB3 protein and the upregulation of the Warburg effect. This further activates HSCs, worsening the severity of hepatic fibrosis. Subsequent to administering a small amount of the PFKFB3 inhibitor 3PO in cellular experiments, it was observed that the glycolysis level of HSCs decreased by 51% and the activation of HSCs was markedly suppressed.
It can be seen that regulating PFK1 or its variant activator, PFKFB3, can effectively reduce the Warburg effect within HSCs and inhibit HSCs activation, which can be a new target for future antifibrotic therapy.
PKM2, a pyruvate kinase, is widely recognized as the enzyme that controls the speed of glycolysis. It facilitates the transfer of phosphate groups from phosphoenolpyruvate to ADP, resulting in the production of pyruvate in the cytoplasm. There are two structural variations of PKM2. The most active PKM2 tetramer encourages glucose oxidative phosphorylation for full oxidative catabolism to produce ATP, whereas its dimer encourages the Warburg effect of aerobic glucose fermentation [110]. PKM2 primarily exists as a tetramer in physiological conditions, but mostly forms dimers in hepatic fibrosis. The Warburg effect is enhanced in hepatic macrophages and HSCs, leading to the activation of HSCs and polarization of M1-type macrophages [111]. This process contributes to the advancement and progression of hepatic fibrosis. One potential treatment target for hepatic fibrosis could be the inhibition of PKM2 dimer expression.
Shikonin, which is a natural compound isolated from the Chinese medicine
Dendrobium, has the effect of inhibiting the activity of PKM2 [112]. Zheng
et al. [51] found that the application of shikonin in in vitro
experiments resulted in the blocking of the pyruvate kinase activity of
tetrameric PKM2 and certain functions of dimeric PKM2, the Warburg effect was
attenuated, HSCs activation was inhibited, and hepatic fibrosis appeared to
subside. In the Wnt/
Therefore, targeted inhibition of PKM2 activity or induction of PKM2 dimer to PKM2 tetramer conversion may be a potential strategy for anti-fibrosis treatment.
LDH-A, a crucial enzyme, serves as the ultimate catalyst in the glycolysis
pathway, facilitating the conversion of pyruvate into lactate. Lactate, the final
product of glycolysis, has long been recognized as a waste product of metabolism,
but recent studies have revealed that lactate can exert a crucial role in the
aerobic glycolysis process [117]. Heightened expression or augmented activity of
LDH-A leads to an upsurge in intracellular aerobic glycolysis, which subsequently
fulfills the energy needs of rapidly dividing cells [118]. LDH-A and
HIF-1
The studies cited reveal the promise of targeting LDH-A for the management of hepatic fibrosis and offer fresh possibilities for future clinical antifibrotic therapy.
To summarize, the Warburg effect is critical for the development of hepatic
fibrosis. The key glycolytic proteins GLUT, PKM2, PFK1, HK, and LDH-A are very
promising potential targets to inhibit the Warburg effect and treat hepatic
fibrosis. Nevertheless, the current research focused on targeting the Warburg
effect to treat anti-hepatic fibrosis remains constrained and lacks sufficient
depth in its content. Firstly, hepatic fibrosis can arise from a multitude of
factors, which include cholestatic hepatic disease, non-alcoholic
steatohepatitis, and viral hepatitis. In the future, researchers can create
diverse cellular and animal models based on the specific cause of the disease.
These models will be used to collectively assess the efficacy of the targeted
Warburg effect in treating hepatic fibrosis. Subsequently, the optimal dosage,
administration route, and timing of treatment can be determined to identify
successful, precisely targeted therapies for hepatic fibrosis caused by different
factors. Secondly, researchers should focus on possible off-target effects and
resistance mechanisms in targeting the Warburg effect for hepatic fibrosis
treatment in order to more fully assess the therapeutic potential of this
therapy. Future researchers should establish correlations between key channels of
signaling implicated in hepatic fibrosis, such as the Warburg effect,
Wnt/
HF, hepatic fibrosis; HSCs, hepatic stellate cells; KCs, kupffer cells; HK, hexokinase; PFK, phosphofructose kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PGK, phosphoglycerate kinase; GLUT, glucose transporter; PK, pyruvate kinase; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; PEP, phosphoenolpyruvate; PA, pyruvate; PDH, pyruvate dehydrogenase; acetyl CoA, acetyl coenzyme A; OXPHOS, oxidative phosphorylation; MCT, monocarboxylic acid transporter; ECM, extracellular matrix; VEGF, vascular endothelial growth factor; EMT, pithelial to mesenchymal; TGF-
XYS initiated the study and contributed to review design. MP and HYL wrote the first draft of the manuscript and drafted Figures and Tables. MP performed literature searches. XYS reviewed manuscript for inclusion, and reviewed and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by the National Natural Science Foundation of China Youth Science Foundation Project (81704073); General Projects of Key R & D Program of Science and Technology Department of Shaanxi Province (2022SF-225).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.