
 , Monica Rienzo 2, Amelia Casamassimi 3, Patrizia Gazzerro 4, Ciro Abbondanza 3
, Monica Rienzo 2, Amelia Casamassimi 3, Patrizia Gazzerro 4, Ciro Abbondanza 31 Department of Medicine and Health Sciences “V. Tiberio”, University of Molise, 86100 Campobasso, Italy
2 Department of Environmental, Biological, and Pharmaceutical Sciences and Technologies, University of Campania “Luigi Vanvitelli”, 81100 Caserta, Italy
3 Department of Precision Medicine, University of Campania “Luigi Vanvitelli”, 80138 Naples, Italy
4 Department of Pharmacy, University of Salerno, 84084 Fisciano, Italy
Keywords
- PRDM2/RIZ
- transcriptome
- colorectal cancer
- EGF
- TCGA
Colorectal cancer (CRC) is the third most deadly and fourth most diagnosed cancer worldwide. Despite the advancement in diagnosis and therapy, CRC still shows a poor prognosis with a low survival rate [1]. Thus, we need to integrate our understanding of CRC biology, from genes to signal transduction and metabolism pathways.
The PRDM2 [PRDI-BF1 (Positive Regulatory Domain I-binding factor 1)] protein
shares a PR domain, endowed with H3K9 lysine methyltransferase (KMTs) activity,
followed by zinc finger domains [2]. Two main proteins, PRDM2/RIZ1 (PR+
product) and PRDM2/RIZ2 (PR– product), have been described
(Fig. 1A, Ref. [2, 3, 4, 5]). Normal tissues express PRDM2/RIZ1 and
PRDM2/RIZ2 transcripts with an equimolar ratio [2] whose alteration
impacts on cancer [2, 6]. In breast cancer MCF-7 cells 17-
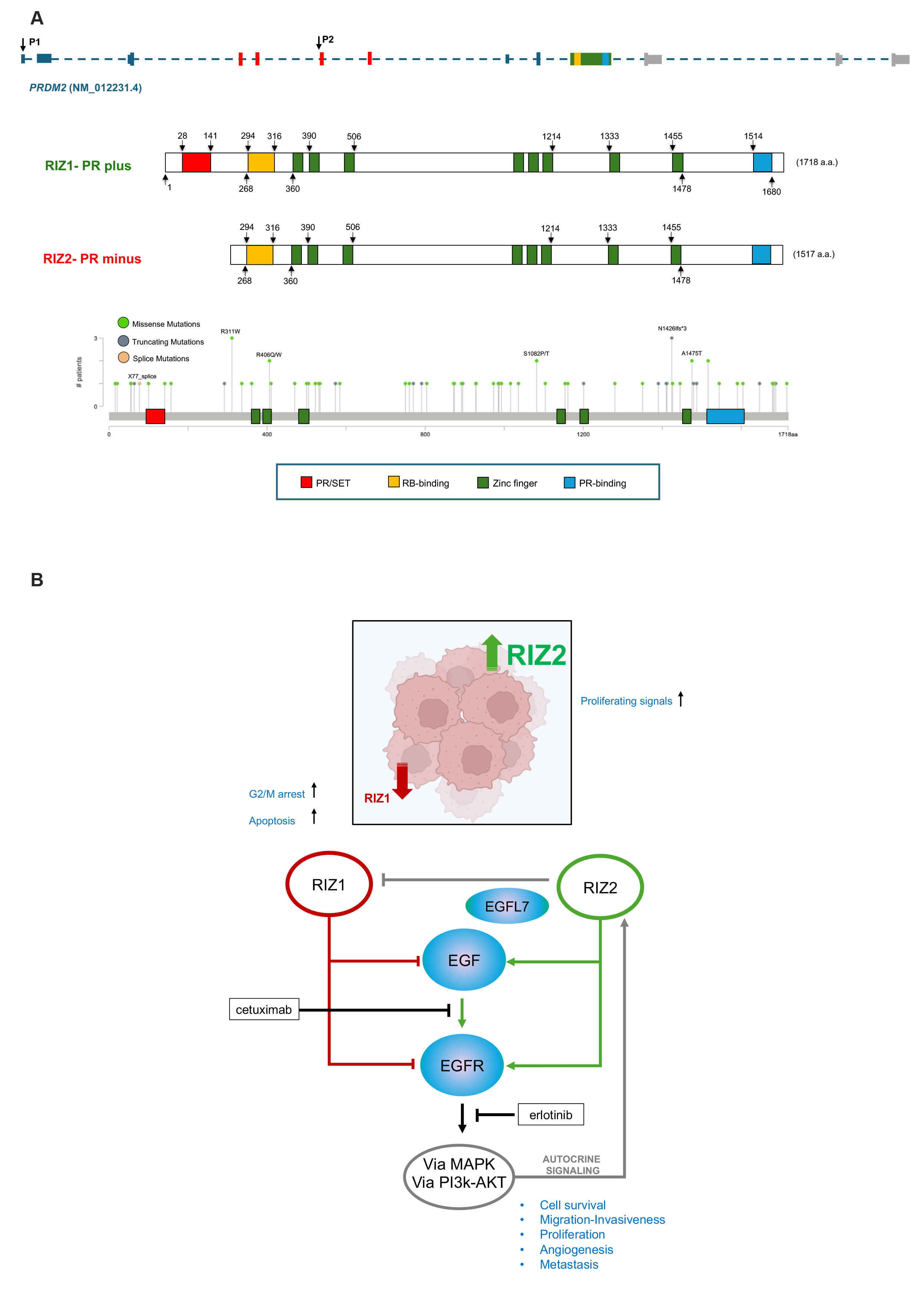 Fig. 1.
Fig. 1.
PRDM2 structure and function. (A) PRDM2 gene structure with indication of exons and promoter 1 (P1) and 2 (P2), protein structures and somatic PRDM2 gene mutations in gastrointestinal cancers as reported by cBioPortal [https://www.cbioportal.org]. (B) PRDM2/RIZ1 downregulation induces G2/M arrest with or without apoptosis depending on genetic background; PRDM2/RIZ2 overexpression increases proliferating signals; in CRC PRDM2/RIZ1 and PRDM2/RIZ2 show an opposite role: PRDM2/RIZ1 negatively controls EGF signal transduction while PRDM2/RIZ2 positively controls autocrine signaling of EGF, by stimulating its secretion and EGFR signal transduction via MAPK and PI3K-AKT. PRDM2, PRDI-BF1 (Positive Regulatory Domain I binding factor 1); RIZ1, retinoblastoma protein interacting zinc finger 1; CRC, colorectal cancer; EGFR, epidermal growth factor receptor; PI3k, phosphatidylinositol 3-kinase; AKT, serine/threonine-protein kinase; MAPK, mitogen-activated protein kinase; PR, PRDF1-RIZ; SET, Su(var)3-9, Enhancer of zeste and Trithorax; RB, Retinoblastoma.
Our analysis of PRDM2 transcripts in TCGA-COAD (colon adenocarcinoma) datasets on GEPIA2 revealed a higher expression of RIZ2 vs. RIZ1 transcript and a frequent PRDM2/RIZ1 methylation in CRC (Fig. 1A) [7]. PRDM2/RIZ1 is commonly mutated in microsatellite instability (MSI) positive CRC and its expression abrogated growth and prompted apoptosis of MSI positive -HCT116 CRC xenograft tumors [8].
In addition, RIZ2 was overexpressed in tumors, particularly in those with Microsatellite stability (MSS). Interestingly, frameshift mutations of microsatellite repeats in the PRDM2 C-terminal coding region were often found in MSI positive CRC. Particularly, 1- or 2-bp deletions occur in two PRDM2 poly-adenosine regions [8, 9, 10]. Mostly, these mutations lead to the PR-binding motif removal, which is critical for PR functions [10, 11, 12]. However, the significance in tumorigenesis of these truncated PRDM2 proteins is still unknown. Furthermore, the restoration of PRDM2 wild-type sequence repaired H3K9me2 activity and impaired CRC cell growth [2].
Interestingly, in a cohort of sessile serrated lesions (SSLs) PRDM2promoter hypermethylation, reducing mainly the PR+ product (PRDM2/RIZ1) expression, could be an early event in dysplasia progression to CRC [13].
Recently, we demonstrated that RIZ2 overexpression increased cell viability, growth, colony formation, migration, and organoid formation in DLD1 cells [14]. Additionally, PRDM2/RIZ2 overexpression modifies transcriptome via epidermal growth factor (EGF) pathway and induces EGF secretion, suggesting that RIZ2 is involved in the EGF autocrine loop regulation of DLD1 cell behavior that fosters tumorigenesis (Fig. 1B) [14]. Similarly, in chronic myelogenous leukemia blastic crisis cell lines CML-BC PRDM2/RIZ1 is involved in the autocrine regulation of the insulin-like growth factor-1 (IGF-1) signaling [15]. Our finding concerning the RIZ2-induced increase in EGF secretion represents a very intriguing aspect that could be responsible for CRC pathogenesis and progression [14]. As mentioned before, the PRDM2/RIZ gene alterations observed in cancers induce PRDM2/RIZ1 silencing often without modifying PRDM2/RIZ2 expression. Thus, the imbalance in RIZ1/RIZ2 expression level is relevant for neoplastic transformation [2, 4, 5]. Several reports, indeed, revealed that PRDM2/RIZ1 overexpression induces blockade of cell growth and prompts apoptosis in several cancer cell lines, including those of colorectal origin, while PRDM2/RIZ1 silencing increased cell division [4, 5, 14] (Fig. 1B). The only evidence to date about PRDM2/RIZ2 function is that reported by our research team [6, 14]. The PRDM2/RIZ2 overexpression increased cell viability and growth- and the cell cycle transition from G2 to M phase. RIZ2, in fact, controls the expression of several mitosis genes, as Cyclin B and anaphase-promoting complex/cyclosome (APC/C) [6]. These findings highlighted the putative PRDM2/RIZ2 tumor-promoting function in CRC and its ability to impact pathways, such as EGF/EGFR signaling, involved in tumor growth, microenvironment remodeling, stemness and chemosensitivity. However, the molecular mechanisms subtending our evidence need to be clarified. Moreover, the behavior of CRC cell lines with high PRDM2/RIZ2 endogenous levels and the possible rescue of cell phenotype by PRDM2/RIZ2 downregulation are currently unknown. In addition, further investigations are required to unveil the interactome of RIZ1and RIZ2and to identify key PRDM2-related pathways in CRC. The PRDM2 interacting partners could be easier to target than PRDM2 proteins, considering the difficulties to study PRDM2/RIZ1 and PRDM2/RIZ2 because of their huge sequence overlap, or for which selective inhibitors are already available [6]. The discovery of PRDM2 interactome and compounds directly targeting PRDM2/RIZ1 intrinsic catalytic activity or the signaling transduction pathways differentially regulating and/or involving RIZ1-RIZ2 could identify therapeutic strategies targeting on PRDM2-interacting elements or on their specific domains, currently undruggable.
Additional attempts are warranted to transfer our knowledge to the clinical practice and many relevant aspects remain to be elucidated in CRC. Although we recognize the difficulties of achieving these ambitious goals, we believe that they represent a challenge that should not be overlooked in this era. These points are of key weightiness in CRC, where little is known about the PRDM2 protein functions and the identification of new targets for pharmacological intervention is a clinical priority.
CIMP, CpG island methylator phenotype; CIN, chromosomal instability; COAD, colon adenocarcinoma; CRC, colorectal cancer; EGF, epidermal growth factor; KMT, lysine methyltransferase; MSI, microsatellite instability; MSS, microsatellite stability; PRDM, PR/SET domain family; PRDM2, PRDI-BF1 (Positive Regulatory Domain I-binding factor 1); RIZ, retinoblastoma protein interacting zinc finger; SSL, sessile serrated lesion; TCGA, the cancer genome atlas.
Conceptualization, AC, CA, EDZ, MR, and PG; investigation, EDZ, AC and MR; visualization MR; writing—original draft preparation, AC, EDZ, MR; writing—review and editing, AC, MR and EDZ; supervision, PG and CA. All authors have read and agreed to the published version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Given Dr. Erika Di Zazzo is the Editorial Board member of the journal, Erika Di Zazzo had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Haseeb Ahmad Khan.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.