




 , Xuejingzi Wu 1, Jiquan Song 1
, Xuejingzi Wu 1, Jiquan Song 11 Department of Dermatology, Zhongnan Hospital of Wuhan University, 430071 Wuhan, Hubei, China
Abstract
Psoriasis is a common, chronic, and multifactorial inflammatory cutaneous disorder that involves genetic and epigenetic factors. N6-methyladenosine methylation (m6A) is the most prevalent RNA modification implicated in various diseases; however, its role in psoriasis still needs to be further explored. We aimed to explore the mechanisms underlying the effects of m6A in psoriasis pathogenesis, prompting new therapeutic targets.
Three psoriasis-related datasets, including GSE155702, GSE109248, and GSE142582, were collected. Differentially m6A methylated genes (DMGs) between psoriasis lesions of psoriasis patients and healthy skin controls were identified from the GSE155702 dataset, and corresponding Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed. Differentially expressed genes (DEGs) and the common DEGs between the two groups were screened from the GSE109248 and GSE142582 datasets; the expression and interactions of the m6A regulators were analyzed. The m6A levels of total RNAs and the protein expression levels of METTL3, WTAP, ALKBH5, FTO, and METTL14 in imiquimod (IMQ)-induced psoriasiform lesions were evaluated.
66 significantly upregulated and 381 significantly downregulated m6A peaks were identified, corresponding to 414 genes which were particularly associated with cell and tissue development processes and cell cycle related items. 271 common DEGs were identified, associating with keratinocyte differentiation, epidermis development, cytokine-cytokine receptor interaction, and fatty acid metabolic processes. 15 crucial m6A related differentially expressed genes were obtained after the intersection of the DMGs and common DEGs, including NEU2, GALNT6, MTCL1, DOC2B, CAMK2N1, SNTB1, RNF150, CGNL1, CCDC102A, MEOX2, EEF2K, OBSCN, SLC46A2, CCDC85A, and DACH1. In addition, we found that m6A methylation and these five m6A regulators were both upregulated in psoriatic lesions.
It revealed that psoriasis pathophysiological processes encompass m6A epigenetic alterations, and that m6A alterations may specifically influence cell proliferation and neural regulation, and closely associated with osteoarticular involvement and metabolic syndrome in psoriasis.
Keywords
- psoriasis
- m6A
- differentially expressed genes
- integrated analysis
Psoriasis, characterized by well-defined erythematous scaly patches, papules, and plaques, is a common chronic relapsing and immune-mediated cutaneous condition, affecting an estimated 2–3% of the general population and predominantly affecting the extensor surfaces [1, 2, 3]. Plaque psoriasis, also referred to as psoriasis vulgaris, manifests the most frequent clinical variant of psoriasis. Psoriasis is a multifactorial disease in which both environmental factors and a complex genetic background play major roles [4, 5]. It is established that a feed-forward mechanism of inflammation, including primarily interleukin 23 (IL-23)-mediated activation of the Th17 pathway contributes substantially to the pathogenesis of psoriasis [3].
Epigenetic modifications are functional changes in gene expression without DNA sequence alterations. Epigenetic mechanisms help reveal the relationships between a genetic background and the effects of the environment on the susceptibility to a certain skin disorder [6]. Recent studies have highlighted the crucial involvement of epigenetic changes in the etiology and pathophysiology of psoriasis [7, 8, 9]. N6-methyladenosine methylation (m6A) modification, an important part of RNA epigenetics, is one of the most common internal posttranscriptional chemical modifications in messenger RNAs (mRNAs), jointly catalyzed by m6A regulators [10]. Functionally, m6A regulators are characterized into three subtypes: m6A methyltransferases (“writer” proteins), m6A demethylases (“eraser” proteins), and m6A binding proteins (“reader” proteins). m6A modification can be facilitated by “writer” proteins and removed by “eraser” proteins; moreover, these m6A-modified sites can be identified by the reader proteins. Research has demonstrated that the alteration of m6A is crucial in regulating the development and progression of multiple diseases, including the development of tumors, cardiovascular disorders, type II diabetes, and multiple inflammatory diseases [11, 12, 13, 14, 15]. Research has shown dysrugulated m6A modifications in psoriasis; however, further research is needed to better understand how aberrant m6A alterations influence the etiology of this disorder [16, 17, 18].
In the present study, we attempted to perform an integrative analysis of microarray and RNA-seq data, laying the foundation to comprehend the potential mechanisms of psoriasis pathogenesis as well as pathophysiology and to identify the regulatory mechanisms of m6A RNA methylation in psoriasis.
The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), a public database, contains a large number of high-throughput sequencing and microarray datasets submitted by research institutes worldwide. We collected psoriasis-related datasets from the GEO database. Three datasets (GSE155702, GSE109248 and GSE142582) were downloaded, whose characteristics, including accession number, platform, number of samples, and publication year, were summarized in Table 1.
| Accession number | Platform | Data type | Number of samples | Country | Years | |
| Psoriasis lesions | Healthy controls | |||||
| GSE155702 | GPL20301 | m6A methylation data | 4 | 4 | China | 2020 |
| GSE109248 | GPL10558 | RNA expression data | 17 | 14 | USA | 2018 |
| GSE142582 | GPL20301 | RNA expression data | 5 | 5 | China | 2019 |
m6A, N6-methyladenosine methylation.
For GSE155702 and GSE142582, respectively, differentially m6A methylated
genes (DMGs) and differentially expressed genes (DEGs) between
psoriasis lesions of psoriasis patients and
healthy skin controls were identified through the R package ‘edgeR’ (version
3.28.0). The DEGs between the two groups for GSE109248 were identified using the
‘limma’ package (version 3.40.2).
DMGs and DEGs were defined using cut-off
criteria of p
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
analyses were conducted for DMGs and the common DEGs using the ‘cluster profiler’
package (version 3.15.4) in R software.
p
We assembled a list of 20 m6A regulators with available RNA expression data
from the GSE109248 dataset, from which the expression matrix of these regulators
was derived. The “limma” package (version 3.40.2) was utilized to identify
differentially expressed m6A regulators between psoriasis patients and
normal controls, and it was deemed statistically significant when p
Experimental protocols with animals were conducted
following the ARRIVE guidelines and approved by
the Ethics Committee of Experimental Animal Welfare of Zhongnan Hospital of Wuhan
University. Balb/C female mice (8–10 weeks of age, purchased from Huafukang
(Beijing, China)) were housed in conventional laboratory settings and allowed
free access to food and water for 1 week prior to experimentation. The mice were
randomly assigned into Control and imiquimod (IMQ) group, and the mice in all groups were
shaved on their backs (average area of 2
MeRIP (Methylated RNA Immunoprecipitation) sequencing data of psoriatic lesions in GSE155702 was analyzed. Chromosome location analysis showed that variant m6A peaks were found in all chromosomes. The number of m6A peaks differed among individual genes. Besides, the majority of genes had one or two m6A peaks (Fig. 1). We further evaluated the actual m6A modification between psoriasis and control. m6A dot blot assays suggested that m6A modification was increased in psoriasiform dermatitis compared with the control (Fig. 1D).
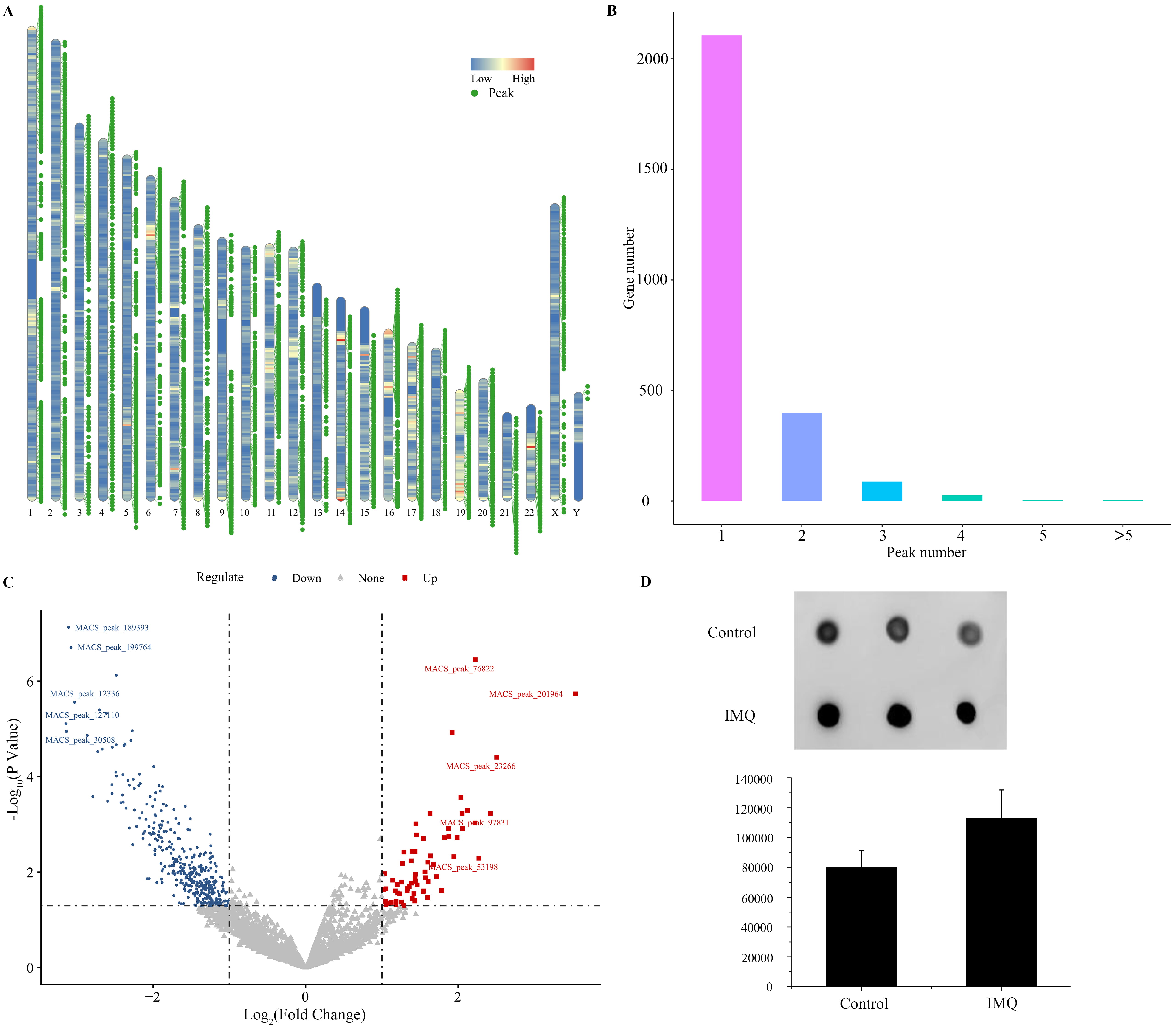 Fig. 1.
Fig. 1.
Overview of m6A methylation in psoriasis. (A) The chromosome location of transcripts having m6A peaks. (B) Number of m6A methylated genes with different m6A peak number. (C) Volcano plot showing differentially methylated RNAs between psoriasis and healthy controls. (D) The m6A levels of skin samples from control mice and imiquimod (IMQ)-induced mice were tested by m6A dot blot assay.
We identified 381 significantly downregulated m6A peaks and 66 significantly upregulated m6A peaks by comparing the m6A modification differences between psoriasis patients and normal controls, corresponding to 414 differentially m6A methylated genes (DMGs) in psoriasis. It suggested that, compared to hypermethylated m6A peaks, there were more hypomethylated m6A peaks in psoriatic skin. To explore the biological relevance of m6A in psoriasis, GO and KEGG analyses of these DMGs were conducted. GO analysis revealed that the hypomethylated DMGs in psoriasis were mainly enriched in cell and tissue development processes (e.g., connective tissue development, positive regulation of neuron projection development, somitogenesis, positive regulation of cell projection organization, axonogenesis). Additionally, hypomethylated DMGs in psoriasis were linked to the Wnt signaling pathway (Fig. 2B). Hypermethylated DMGs in psoriasis were linked to cell cycle-associated items (like G2/M transition of mitotic cell cycle, cell cycle G2/M phase transition, regulation of G2/M transition of mitotic cell cycle, regulation of cell cycle G2/M phase transition), implying that these hypermethylated DMGs regulate the proliferation/differentiation of keratinocytes (Fig. 2A). KEGG analysis found that hypomethylated DMGs in psoriasis were mainly associated with axon guidance, endocytosis, and the Wnt signaling pathway, while hypermethylated DMGs were mainly associated with the mRNA surveillance pathway and the NF-kappa B signaling pathway.
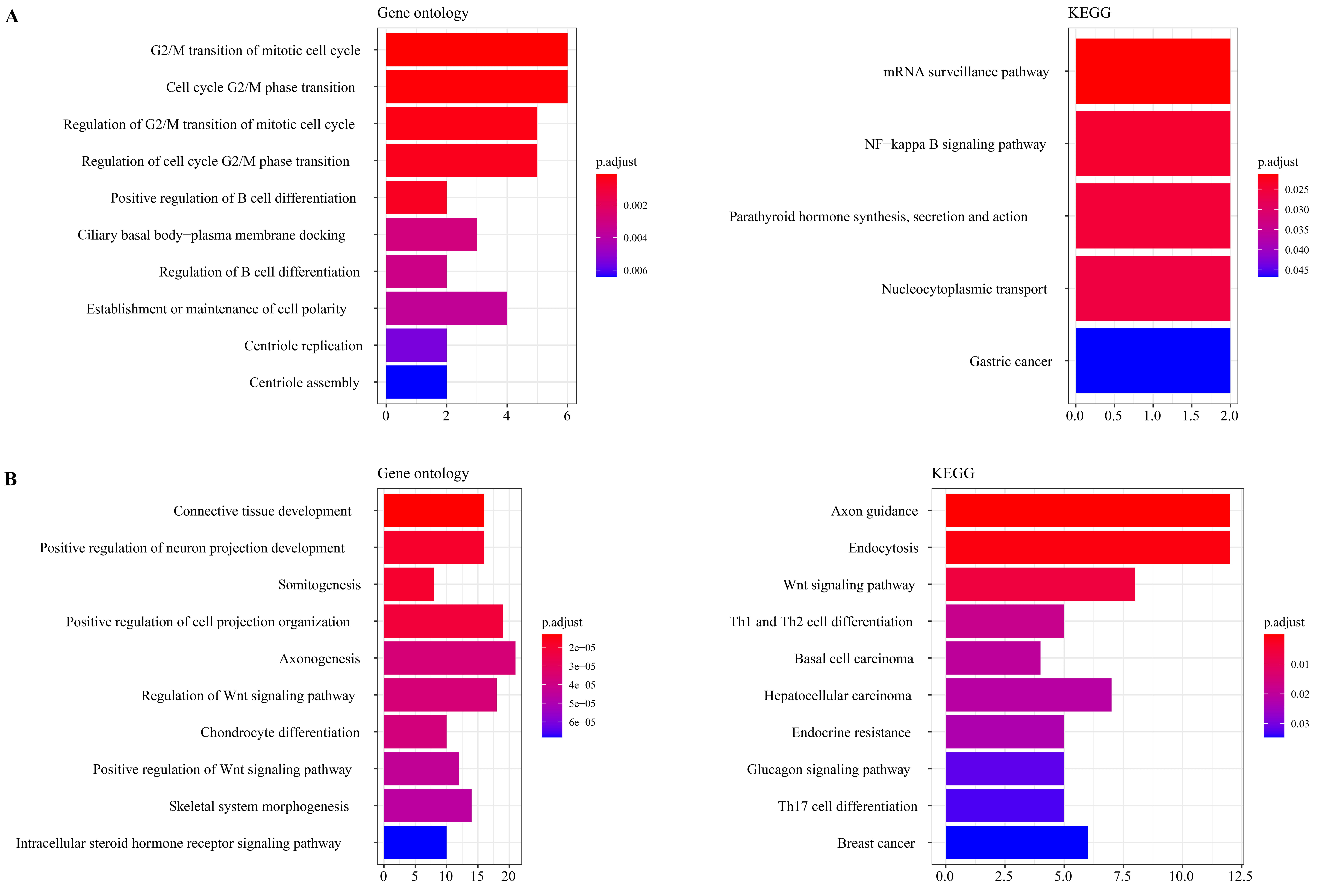 Fig. 2.
Fig. 2.
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of genes containing altered m6A peaks. (A) The top 10 significantly enriched gene ontology terms and top 5 significantly enriched pathways for the hypermethylated genes in psoriasis verses controls. (B) The top 10 significantly enriched gene ontology terms and pathways for the hypomethylated genes in psoriasis verses controls.
Differentially expressed m6A regulators were detected in dataset GSE109248, containing 20 m6A regulators, including 8 m6A methylation transferases (METTL15, METTL14, METTL3, WTAP, ZC3H13, RBMX, RBM15, and RBM15B), 2 demethylases (FTO and ALKBH5), and 10 m6A binding proteins (YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, HNRNPA2B1, HNRNPC, IGF2BP1, IGF2BP2, and IGF2BP3). It showed that the expression levels of METTL15, METTL14, WTAP, and FTO were higher in psoriatic lesions, while the expression of IGF2BP1 was significantly downregulated in psoriatic lesions (Fig. 3A). Moreover, the changes of METTL3, WTAP, ALKBH5, FTO, and METTL14 expression in psoriasis were validated by Western blot. We found that these five m6A regulators were both upregulated in psoriatic lesions (Fig. 3B). Considering the similarity of their biological functions, we analyzed the correlation and interaction among the 20 m6A regulators. The STRING database analysis suggested that HNRNPA2B1 was the “hub” gene of these m6A regulators, as it interacted with 18 of them (Fig. 3C). The protein-protein interaction (PPI) networks showed that these m6A regulators interacted with each other frequently. Correlation analysis showed that HNRNPA2B1 was significantly correlated to these five differentially expressed m6A regulators in psoriasis. HNRNPA2B1 and IGF2BP1 had a positive correlation, whereas there was an inverse correlation between HNRNPA2B1 and METTL15, METTL14, and FTO in psoriasis (Fig. 3D).
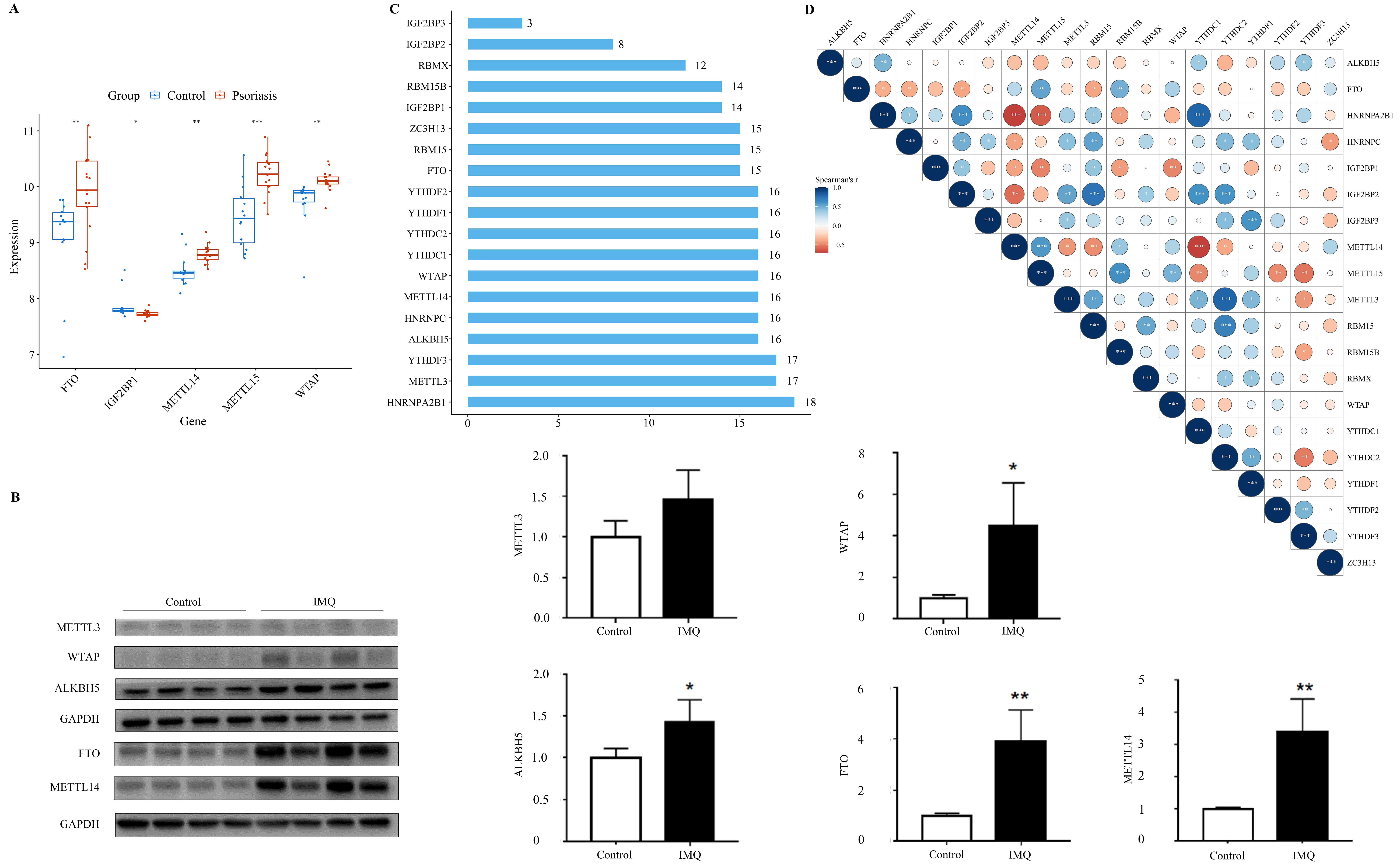 Fig. 3.
Fig. 3.
The expression and interaction of m6A regulators in
psoriasis and controls. (A) The expression levels of the twenty m6A
regulators in psoriasis and controls. (B) The protein expression levels of
methyltransferase-like 3 (METTL3), Wilms’ tumor 1-associated protein (WTAP), alkB homolog 5 (ALKBH5), fat-mass and obesity-associated protein (FTO), and methyltransferase-like 14 (METTL14) in
skin samples derived from control mice and IMQ-induced mice. (C) The interaction
counts among the twenty m6A regulators. (D) Spearman correlation analysis of
the twenty m6A regulators. *p
DEGs were identified between psoriasis patients and normal skin samples. The present study screened 2153 and 2174 DEGs in GSE109248 and GSE142582, respectively. The intersection of the Venn diagrams revealed 271 common DEGs, including 32 common upregulated genes and 239 common downregulated genes. Both GO analysis and KEGG pathway enrichment were performed to further investigate the underlying biological information of these common DEGs (Fig. 4). GO analysis showed that the upregulated DEGs, in terms of the biological process, were primarily involved in keratinocyte differentiation, epidermis development, and epidermal cell differentiation, while the downregulated DEGs were primarily involved in the fatty acid metabolic process, the fatty acid derivative metabolic process, and the long-chain fatty acid metabolic process. KEGG analysis showed that the upregulated DEGs were largely enriched in the IL-17 signaling pathway, cytokine-cytokine receptor interaction, and amoebiasis, while the downregulated genes were largely enriched in glyoxylate and dicarboxylate metabolism, ovarian steroidogenesis, and arachidonic acid metabolism.
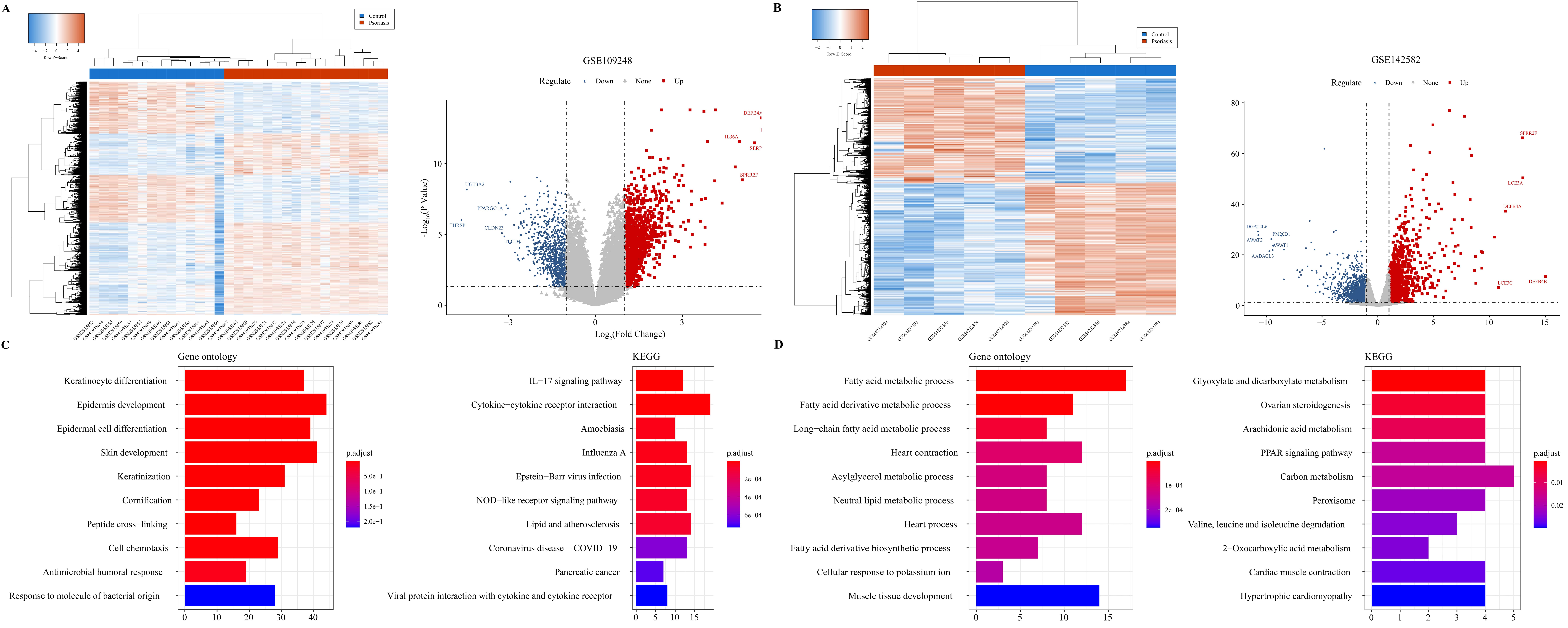 Fig. 4.
Fig. 4.
Differentially expressed genes (DEGs) identified between psoriasis and controls. (A,B) Heatmap and volcano plot showing DEGs between psoriasis patients and normal controls in GSE109248 and GSE142582. (C,D) Top ten enriched GO terms (biological processes) and KEGG pathways of the common upregulated and downregulated DEGs identified in psoriasis.
To explore whether the DEGs between patients and controls were associated with m6A modification, we overlapped m6A modified transcripts with the DEGs. By constructing a Venn diagram, we obtained the consistent differential genes between the aforementioned common DEGs and DMGs, which mostly could be the genes regulated by m6A methylation. 15 genes, namely NEU2, GALNT6, MTCL1, DOC2B, CAMK2N1, SNTB1, RNF150, CGNL1, CCDC102A, MEOX2, EEF2K, OBSCN, SLC46A2, CCDC85A, and DACH1, were identified.
Psoriasis leads to a substantial burden for individuals and society. Despite tremendous advancements in our knowledge of the pathophysiology of psoriasis, the definite molecular mechanism is still not fully elucidated. Recently, epigenetic changes have been reported to be involved in the pathogenesis of psoriasis. m6A is the most abundant modification of mRNA in eukaryotes, and its continuous and dynamic regulation at the posttranscriptional level influences mRNA localization and metabolism, participating in various physiological and pathological processes accordingly [11, 12, 14]. Psoriasis is characterized by hyperproliferation and inflammation of the epidermis; m6A modification has been found to exert a role in regulating cell proliferation-related pathways and immune function, thereby suggesting the potential role of m6A in the pathogenesis of psoriasis.
We described the m6A distribution pattern in psoriasis, and it suggested
that variant m6A peaks were found in all chromosomes, with most individual
genes containing one single m6A peak. By analyzing the
m6A modification differences between psoriasis patients and healthy donor
controls, DMGs were acquired. Compared with controls, there were more
hypomethylated DMGs in psoriasis than hypermethylated ones, indicating that
psoriasis-related DMGs were preferentially hypomethylated. GO analysis revealed
that hypermethylated DMGs in psoriasis were mainly linked to cell cycle-related
terms, indicating the effects of m6A through speeding up the cell cycle of
keratinocytes. Hypomethylated DMGs in psoriasis were found to be particularly
connected to the Wnt signaling pathway. The
Wnt signaling pathway participates in regulating cell proliferation,
differentiation, migration, and polarity [19, 20]. Studies reported that the Wnt
signaling pathway or expression of some specific Wnt proteins, like Wnt5a, was
altered in psoriasis skin, which may affect the occurrence and development of
psoriasis [21, 22]. Regarding KEGG analysis, compared with healthy controls,
hypermethylated DMGs in psoriasis were mainly connected to
the mRNA surveillance pathway and the
NF-
In a comparison between the identified DMGs and common DEGs in psoriasis, 15 overlapping genes, defined as dysregulated m6A modification genes, remained, including NEU2, GALNT6, MTCL1, DOC2B, CAMK2N1, SNTB1, RNF150, CGNL1, CCDC102A, MEOX2, EEF2K, OBSCN, SLC46A2, CCDC85A, and DACH1, which could be mostly regulated by m6A modification in psoriasis. Studies have shown that GALNT6, MTCL1, CAMK2N1, EEF2K, OBSCN, and DACH1 are related to cell proliferation and growth. GALNT6, a member of the N-acetylgalactosaminyltransferases, has been shown to have oncogenic functions and has been implicated in the metastasis of multiple cancers [35]. Circular RNA MTCL1 (circMTCL1) has been found to exert oncogenic biological characteristics by promoting cell proliferative capability and invasive and migrative abilities [36]. CAMK2N1 is an endogenous suppressor of calcium/calmodulin dependent kinase II and has been described to have a cancer-inhibiting function [37]. EEF2K has been found to promote cell proliferation, survival, and aggressive tumor characteristics [38]. OBSCN has been identified as a potent tumor suppressor whose loss of function accelerates pancreatic cancer progression and metastasis [39]. DACH1 has been associated with various tumor diseases, and its reduced expression in breast cancers has been found to promote tumor progression and poor differentiation [40]. The pathomechanism underlying psoriasis is considered to involve the acceleration of cell proliferation and the rapid migration of keratinocytes from the basal layer to the granular layer. Given the hyperproliferation of psoriatic keratinocytes that partially resembles the excessive proliferation of cancer cells, we hypothesized that m6A affects cell proliferation in psoriasis by regulating the m6A modification in the mRNAs of some critical oncogenes or tumor suppressors. Of note, studies have revealed the possibility of an association between psoriasis and non-melanoma skin cancer (NMSC) [41]. Cytosolic NEU2 sialidase has a pivotal role in neuronal differentiation [42]. The neural gene SNTB1 is related to neural precursor cell proliferation [43]. DOC2B participates in neuronal activity and neurotransmitter release [44]. Consistent with what has been mentioned above, m6A could affect the pathophysiology of psoriasis through neural regulation. Additionally, psoriasis patients often suffer from moderate to severe pruritus, and the nervous system plays an important role in promoting itching [34]. Therefore, it suggested that m6A might function as a novel therapeutic target in psoriasis pruritus. CGNL1 is known to be involved in promoting angiogenesis by strengthening adherens junctions, leading to stabilization and further elongation of newly formed vascular tubules [45]. It demonstrated that m6A could be closely related to increased angiogenesis and microvascular dilation in psoriasis lesions.
Along with its cutaneous symptoms, psoriasis has been linked to a higher risk of various metabolic comorbidities [46]. Our study unravel that hypomethylated DMGs in psoriasis were particularly related to endocrine resistance and the glucagon signaling pathway, which disclosed the regulatory role of m6A in metabolic abnormalities and could help explain why psoriasis is correlated to a metabolic syndrome.
Our study indicates that psoriasis pathophysiological processes are complex molecular processes encompassing m6A epigenetic alterations, which are implicated in various critical pathogenic processes in psoriasis, including specifically influencing cell proliferation and neural regulation and closely associating with osteoarticular involvement and metabolic syndrome in psoriasis. Our research has expanded the regulatory mechanisms contributing to psoriasis.
The public datasets to support the results of this subject can be gained from GEO (https://www.ncbi.nlm.nih.gov/geo/). The raw experimental data of the present study are available from the corresponding author on reasonable request.
All authors contributed to the study conception and design. LG, XW and JS performed data analysis and validation. LG drafted the manuscript. JS and XW revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors have read and agreed to the published version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All experimental protocols with animals were conducted following the ARRIVE guidelines, and approved by the Ethics Committee of Experimental Animal Welfare of Zhongnan Hospital of Wuhan University (ID ZN2023073).
Not applicable.
This research was funded by the Fundamental Research Funds for the Central Universities (No.2042020kf0173).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.