






 , Zhiyu Zeng 1,2,3,*
, Zhiyu Zeng 1,2,3,*1 Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, 530021 Nanning, Guangxi, China
2 Guangxi Key Laboratory of Precision Medicine in Cardio-cerebrovascular Diseases Control and Prevention, 530021 Nanning, Guangxi, China
3 Guangxi Clinical Research Center for Cardio-Cerebrovascular Diseases, 530021 Nanning, Guangxi, China
†These authors contributed equally.
Abstract
Background: Rheumatic heart disease (RHD) is an autoimmune disease caused by recurrent infections of Group A streptococcus (GAS), ultimately leading to inflammation and the fibrosis of heart valves. Recent studies have highlighted the crucial role of C-C chemokine receptor type 2-positive (CCR2+) macrophages in autoimmune diseases and tissue fibrosis. However, the specific involvement of CCR2+ macrophages in RHD remains unclear. Methods: This study established an RHD rat model using inactivated GAS and complete Freund’s adjuvant, demonstrating a correlation between CCR2+ macrophages and fibrosis in the mitral valves of these rats. Results: Intraperitoneal injection of the CCR2 antagonist Rs-504393 significantly reduced macrophage infiltration, inflammation, and fibrosis in valve tissues of RHD rats compared to the solvent-treated group . Existing evidence suggests that C-C motif chemokine ligand 2 (CCL2) acts as the primary recruiting factor for CCR2+ cells. To validate this, human monocytic leukemia cells (THP-1) were cultured in vitro to assess the impact of recombinant CCL2 protein on macrophages. CCL2 exhibited pro-inflammatory effects similar to lipopolysaccharide (LPS), promoting M1 polarization in macrophages. Moreover, the combined effect of LPS and CCL2 was more potent than either alone. Knocking down CCR2 expression in THP-1 cells using small interfering RNA suppressed the pro-inflammatory response and M1 polarization induced by CCL2. Conclusions: The findings from this study indicate that CCR2+ macrophages are pivotal in the valvular remodeling process of RHD. Targeting the CCL2/CCR2 signaling pathway may therefore represent a promising therapeutic strategy to alleviate valve fibrosis in RHD.
Keywords
- rheumatic heart disease
- macrophage
- fibrosis
- CCR2 antagonist
Rheumatic heart disease (RHD) is a leading cause of cardiac mortality in adolescents and remains a significant global public health challenge [1]. Acute rheumatic fever (ARF) typically manifests approximately three weeks after a pharyngeal infection with Group A streptococcus (GAS), with RHD representing its most severe complication [2]. Pathological changes in RHD arise from recurrent asymptomatic ARF episodes, leading to cumulative chronic damage to the heart valves. This process ultimately results in regurgitation and/or stenosis of the mitral and/or aortic valves, precipitating complications [3]. However, understanding of the drivers and specific biological processes underlying inflammation and fibrosis development in RHD valves remains limited.
A substantial body of research on RHD supports the perspective that immune cells and the cytokines they produce act as mediators of valve damage [4, 5]. Studies on immune cells have shown widespread infiltration of CD4+ T cells in RHD valves. These CD4+ T cells not only recognize the pathogenic M protein of streptococci but also target self-antigens in cardiac tissues, resulting in secretion of various cytokines [6, 7]. Recent experimental evidence has uncovered the crucial role of macrophages in regulating inflammation and fibrosis within RHD valves [8, 9]. Notably, depleting macrophages alleviates valve inflammation, highlighting their potential role in valve myocarditis [10]. The C-C chemokine receptor type 2 (CCR2) receptor is primarily expressed on the surface of monocytes and macrophages and is considered to play a critical role in guiding macrophage infiltration into sites of inflammation [11]. The interaction between the CCR2 and C-C motif chemokine ligand 2 (CCL2) receptors has been extensively validated, emphasizing their significant involvement in the pathogenesis of various autoimmune diseases such as rheumatoid arthritis, osteoarthritis, lupus nephritis, and scleroderma [12, 13, 14, 15]. Furthermore, these receptors are closely associated with processes such as cardiac fibrosis, liver fibrosis, renal fibrosis, and cystic fibrosis [16, 17, 18, 19, 20]. Therefore, investigating the potential role of the CCL2/CCR2 axis in mediating macrophage recruitment and activation is crucial for understanding the remodeling of RHD valves.
This study investigated several pivotal questions: (1) Are CCR2+ macrophages present in the valves of patients with RHD? (2) Is CCL2 expressed in the valve cells of RHD patients? The objective was to use an animal model to determine whether activation of the CCL2/CCR2 axis during RHD facilitates the recruitment of macrophages. (3) Does blocking the CCL2/CCR2 signaling pathway result in inhibition of macrophage recruitment and infiltration, and subsequently mitigate inflammation and fibrosis in RHD valves? (4) The role of the CCL2 protein in macrophages was also investigated in vitro. The study confirmed the pivotal role of CCR2+ macrophages in the fibrosis process in RHD valves. This finding underscoresthe potential effectiveness of inhibiting the CCL2/CCR2 signaling pathway in macrophages as a therapeutic strategy for reducing fibrosis in the valves of patients with RHD.
Ethical approval for the study was obtained from the Medical Ethics Committee of the First Affiliated Hospital of Guangxi Medical University, and informed consent forms were signed by all patients and their families. Heart valves were obtained from three female patients aged 45–55 years diagnosed with RHD, who had undergone mitral valve replacement at the First Affiliated Hospital of Guangxi Medical University. Immediately after dissection, the valves were placed in pre-cooled phosphate-buffered saline, cleansed of surface blood, and then fixed in 4% paraformaldehyde (P1110, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China).
Group A Streptococcus (GAS, American Type Culture Collection) was inoculated
into Brain Heart Infusion Powder (Guangdong Huankai Microbial Technology Co.,
Ltd., Guangdong, China) and then incubated at 37 °C for 24 h using a
rotary mixer (Fuma, Shanghai, China) at 110 r/min. Bacterial fluids were
collected and fixed in 4% paraformaldehyde for 12 h, then washed five times with
saline before being finally diluted to 10
All animal experimental procedures adhered to ethical guidelines for animal care and use, approved by the First Affiliated Medical Ethics Committee of Guangxi Medical University Affiliated Hospital (Approval Number: 2023-E751-01). Twenty-four 10-week-old female Lewis rats were obtained from Beijing Viton Lihua Laboratory Animal Technology Co., Ltd. (Beijing, China) and were randomly assigned to three groups: Control, RHD, or RHD+Rs-504393 groups, each comprising eight rats. After one week of acclimatization and feeding in the Guangxi Medical University Laboratory Animal Center (SPF grade), the rats received an initial injection of 0.2 mL of antigen B (bacterial emulsion: complete Fuchs’ adjuvant = 1:1) into the footpad under 2% concentration of isoflurane. Following a week of rest, 0.5 mL of antigen B was injected subcutaneously into the abdominal wall, repeated once a week for four consecutive weeks. Additionally, 0.5 mL of antigen A (inactivated GSA) was injected subcutaneously into the abdominal wall after a one-week rest period following the completion of the antigen B injections [21, 22]. The Rs-504393 treatment group received intraperitoneal injections of 5 mg/kg of the CCR2 antagonist (Rs-504393, BD218736, Shanghai Bide Pharmaceutical Technology Co., Ltd., Shanghai, China) concurrently with the first subcutaneous injection of antigen B into the abdominal wall. This treatment was administered once daily for eight weeks. In the control group, complete Fuchs’ adjuvant was injected instead of antigen B, while physiological saline was administered in place of antigen A, in equivalent amounts and at the same sites. Upon completion of all the interventions, 1 ml of blood was collected from the tail vein. Subsequently, all rats were euthanized using intraperitoneal administration of pentobarbital sodium at a concentration of 150 mg/kg to collect the heart valves.
The collected valve tissues were fixed in 4% paraformaldehyde for 24 h,
followed by embedding in paraffin, sectioning into 4 µm thick slices, and
mounting onto slides. Hematoxylin and eosin (H&E) staining was employed to
evaluate inflammation, while Sirius red staining was used to assess valve
fibrosis. Imaging of the H&E-stained samples was conducted using a BX43 light
microscope (Olympus Corporation, Tokyo, Japan) at a magnification of
Total RNA was extracted from both valve tissues and cells using
TRIzol® reagent (15596026CN, Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), following the manufacturer’s guidelines. The cDNA first-strand
synthesis kit (YFXM0009, Nanjing Yifeixue Biotechnology Co., Ltd., Nanjing,
China) was then employed to reverse-transcribe the total RNA into cDNA, according
to the manufacturer’s protocol. Subsequently, real time-quantitative polymerase
chain reaction (RT-qPCR) was performed using 2
| Gene (rat) | (5 | |
| Tnf- |
Forward | GCGTGTTCATCCGTTCTCTAC |
| Reverse | GTCTCGTGTGTTTCTGAGCAT | |
| Il-6 | Forward | CCGGAGAGGAGACTTCACAGAGGA |
| Reverse | AGCCTCCGACTTGTGAAGTGGTATA | |
| Tgf- |
Forward | GACCGCAACAACGCAATCTATGAC |
| Reverse | CTGGCACTGCTTCCCGAATGTC | |
| Ccl2 | Forward | CTCACCTGCTGCTACTCATTCACTG |
| Reverse | CTTCTTTGGGACACCTGCTGCTG | |
| Ccr2 | Forward | GCCACCACACCGTATGACTATGATG |
| Reverse | AGCAGGAAGAGCAGGTCAGAGATG | |
| Cd68 | Forward | CTCTTGCTGCCTCTCATCATTGG |
| Reverse | GCTGGTAGGTTGATTGTCGTCTC | |
| Col1a1 | Forward | TGTTGGTCCTGCTGGCAAGAATG |
| Reverse | GTCACCTTGTTCGCCTGTCTCAC | |
| Col3a1 | Forward | AGTCGGAGGAATGGGTGGCTATC |
| Reverse | CAGGAGATCCAGGATGTCCAGAGG | |
| Forward | GGAGATTACTGCCCTGGCTCCTA | |
| Reverse | GACTCATCGTACTCCTGCTTGCTG | |
| Gene (humam) | (5 | |
| CCL2 | Forward | ACCAGCAGCAAGTGTCCCAAAG |
| Reverse | TTTGCTTGTCCAGGTGGTCCATG | |
| CCR2 | Forward | CCAACGAGAGCGGTGAAGAAGTC |
| Reverse | CGAGTAGAGCGGAGGCAGGAG | |
| CD86 | Forward | TGCTCATCTATACACGGTTACC |
| Reverse | TGCATAACACCATCATACTCGA | |
| TNF- |
Forward | CCTCATCTACTCCCAGGTCCTCTTC |
| Reverse | TCTGGTAGGAGACGGCGATGC | |
| IL-6 | Forward | GCCTTCGGTCCAGTTGCCTTC |
| Reverse | GTTCTGAAGAGGTGAGTGGCTGTC | |
| GAPDH | Forward | TGACATCAAGAAGGTGGTGAAGCAG |
| Reverse | GTGTCGCTGTTGAAGTCAGAGGAG |
RT-qPCR, real time-quantitative polymerase chain reaction; TNF-
Valve tissues or cells were lysed in RIPA buffer (R0010, Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China), and individual samples (30 mg of
protein) were then prepared for electrophoresis on 7.5–15% sodium dodecyl
sulfate (SDS)/polyacrylamide gel electrophoresis (PAGE) gels, followed by
transfer to polyvinylidene fluoride (PVDF) membranes (ISEQ00005, EMD Millipore,
Darmstadt, Germany). The membranes were blocked with 5% skimmed milk in TBST
(T1082, Beijing Solarbio Science & Technology Co., Ltd, Beijing, China) for 1 h
at room temperature, washed three times, and subsequently incubated at 4
°C overnight with the following specific primary antibodies: anti-CCR2
(1:1000, ab223366, Abcam, Cambridge, UK), anti-CCR2 (1:1000, 16153-1-AP,
Proteintech, Wuhan, China), anti-CCL2 (1:2000, ab25124, Abcam),
anti-
Tissue samples were rapidly frozen in liquid nitrogen and processed using the
indirect immunofluorescence technique. Frozen sections were stained overnight at
4 °C in a humidified room with primary antibodies targeting anti-CD68 (1:2000,
GB113109, Servicebio, Wuhan, China), anti-CCR2 (1:200, GB11326, Servicebio),
anti-CCL2 (1:200, ab25124, Abcam, Cambridge, UK), anti-CD45 (1:300, GB115428,
Servicebio), anti-CD31 (1:2000, ab182981, Abcam), and anti-s100a4 (1:2000,
GB11397, Servicebio). Following elution, the sections underwent sequential
reactions with fluorescent secondary antibodies (1:1000, GB23303, Servicebio) for
50 min, shielded from the light. After a final rinsing step, DAPI dye was briefly
applied briefly to the tissue sections, which were then dehydrated, sealed, and
examined under light microscopy. For cellular immunofluorescence, cells were
initially cultured in 48-well plates until the intervention concluded. Samples
were washed twice with PBS, fixed in 4% paraformaldehyde for 10 min,
permeabilized with 0.1% PBST (PBS containing 0.1% Triton X-100 [9002-93-1,
Beijing Solarbio Science & Technology Co., Ltd., Beijing, China]), and blocked
with 1% bovine serum albumin (BSA, GC305006, Servicebio). Subsequently, cells
were treated with anti-CCR2 and anti-CCL2 antibodies, incubated overnight at 4
°C, washed, and exposed to fluorescent secondary antibodies in the dark
for 30 min. After additional washing, 4
Human monocytic leukemia cells were sourced from Wuhan Punosai Life Science and Technology Co., Ltd. All cell lines underwent validation by STR profiling and tested negative for mycoplasma contamination. Cells were cultured in a humidified incubator at 37 °C with 5% CO2. Initially, THP-1 cells were induced into M0 adherent morphology by treating them with 100 ng/mL phorbol 12-myristate 13-acetate (PMA, P8139, Sigma-Aldrich, Darmstadt, Germany) for 48 hours. The cells were then divided into four groups for different interventions: NC group, LPS group (100 ng/mL), CCL2 group (100 ng/mL), and LPS+CCL2 group, with LPS (100 ng/mL) serving as the positive control. Finally, cell lysates analyzed using RT-qPCR and Western blotting to assess the expression of CCR2 and CCL2, supplemented by immunofluorescence.
The THP-1 cells were seeded at a density of 1
The concentration of CCL2 in rat serum and levels of Interleukin-12A (IL-12A),
IL-6, and IL-1
The number of animals used in the in vivo experiments was denoted as
“n”. For the in vitro investigations, “n” represented the number of
experiments conducted. Statistical analyses were performed using GraphPad Prism
9.5 (GraphPad Software, San Diego, CA, USA) and SPSS 27 (IBM-SPSS Statistics,
Chicago, IL, USA). Data were expressed as mean
The co-expression of CD68, CCR2, and COL1A revealed by immunofluorescence is shown in (Supplementary Fig. 2A). CCL2 was also expressed in leukocytes marked with CD45, endothelial cells marked with CD31, and fibroblasts marked with s100a4 (Supplementary Fig. 2B). These findings provide evidence for the presence of CCR2+ macrophages in the valves of RHD patients, notably in close proximity to fibrin. The expression of CCL2 on endothelial cells and fibroblasts suggests a potential mechanism for recruiting CCR2+ macrophages from the bloodstream to the valve site.
Key observations included a notable increase in the infiltration of inflammatory
cells within RHD rat valves, as evidenced by HE stains (Fig. 1A), and
exacerbation of valve fibrosis in the RHD group, observed in Sirius red polarized
light images (Fig. 1B). Remarkably, these adverse changes were significantly
mitigated following administration of Rs-504393. Analysis via RT-qPCR (Fig. 2A)
and western blot (Fig. 2C) in RHD rats revealed heightened activation of the
CCL2/CCR2 axis, elevated levels of pro-inflammatory and pro-fibrotic cytokines
(TNF-
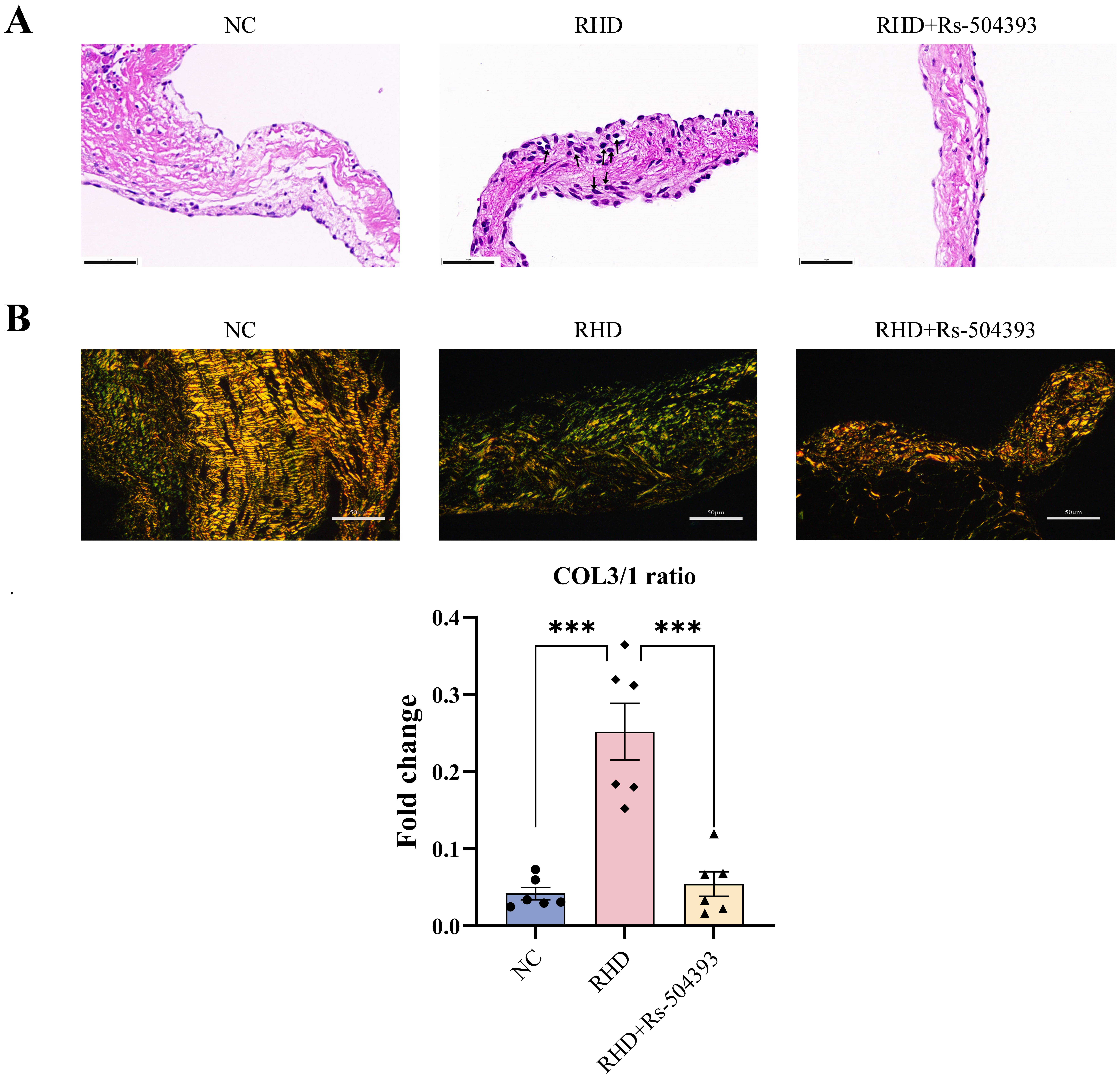 Fig. 1.
Fig. 1.
Pathological staining in each rat group. (A) Inflammatory
changes in the valves of the NC group, RHD group, and RHD+Rs-504393 treatment
group were observed through HE staining, with images captured at the original
magnification of
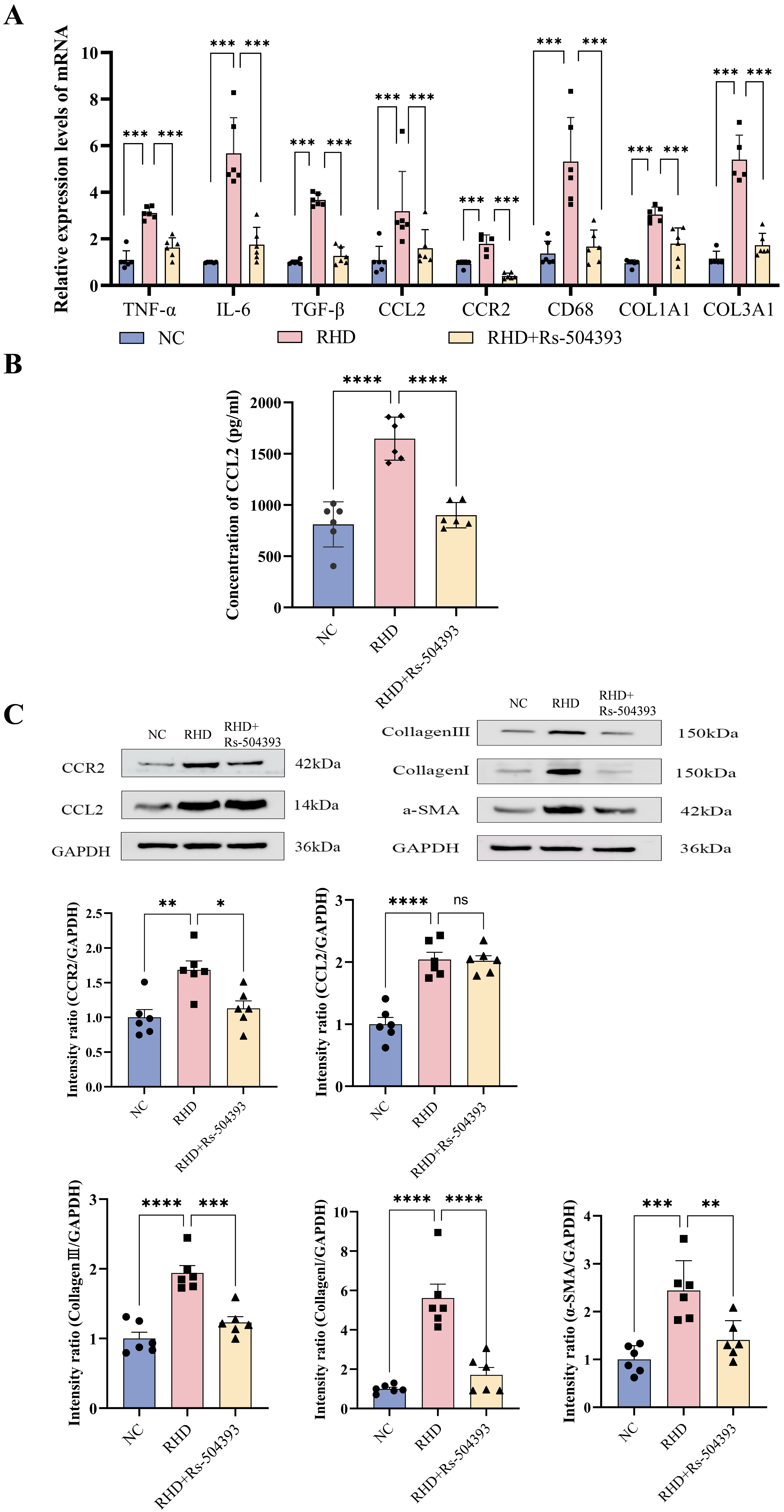 Fig. 2.
Fig. 2.
Changes in the expression levels of pro-inflammatory cytokines,
fibrosis markers, and macrophage markers in valve tissues after Rs-504393
treatment. (A) RT-qPCR was employed to detect mRNA levels of
TNF-
Protein blot analysis (Fig. 2C), ELISA (Fig. 2B), RT-qPCR (Fig. 2A) and immunohistochemistry (Fig. 3) conducted on the valves of RHD rats showed a significant upregulation in CCL2 expression and a notable increase in macrophage numbers compared to controls. Treatment with Rs-504393 resulted in a marked reduction in the total count of CD68-labeled macrophages, specifically decreasing both CCR2+ and M1-type macrophages. Statistical analysis of the immunohistochemistry showed a minimal change in the number of M2-labeled macrophages, potentially attributable to the acute nature of this experimental model. These findings collectively suggest that Rs-504393 treatment attenuates inflammation and fibrosis in rheumatic rat valves by reducing the infiltration of CCR2+ macrophages.
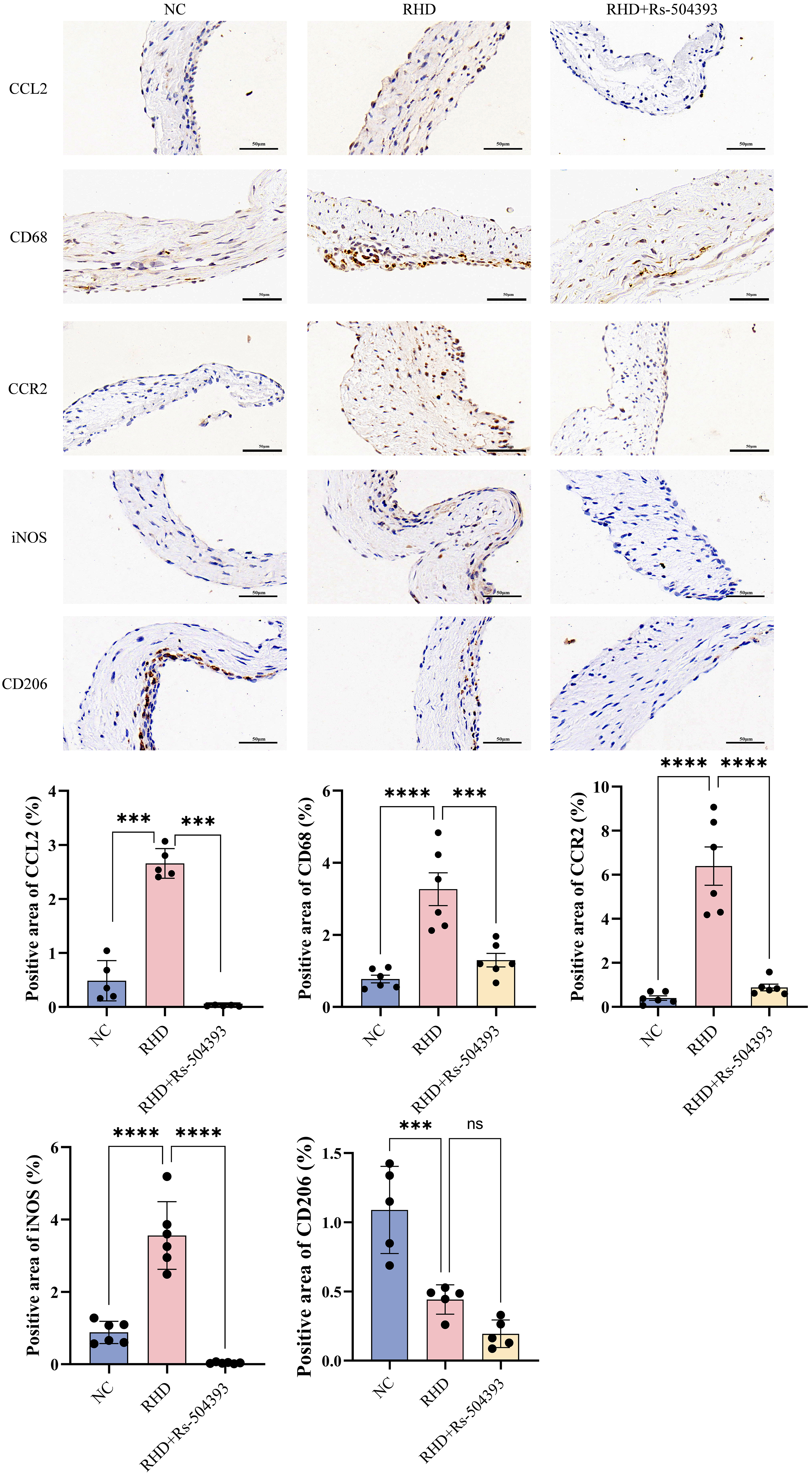 Fig. 3.
Fig. 3.
Immunohistochemistry reveals infiltration of different
macrophage subtypes in each group. Upper Panel: Immunohistochemical expression
of CCL2, CD68, CCR2, iNOS, and CD206 in the NC group, RHD group, and
RHD+Rs-504393 treatment group. Images were captured at the original magnification
of
Various concentrations of CCL2 were applied to intervene on M0 macrophages for
24h, and the optimal concentration that maximized CCR2 expression was determined
to be 100 ng/mL based on the CCK8 assay (Fig. 4A) and RT-qPCR experiment (Fig. 4B). RT-qPCR (Fig. 4C) and flow cytometry (Fig. 4D) demonstrated elevated levels
of inflammatory factors (CCL2, IL-6, TNF-
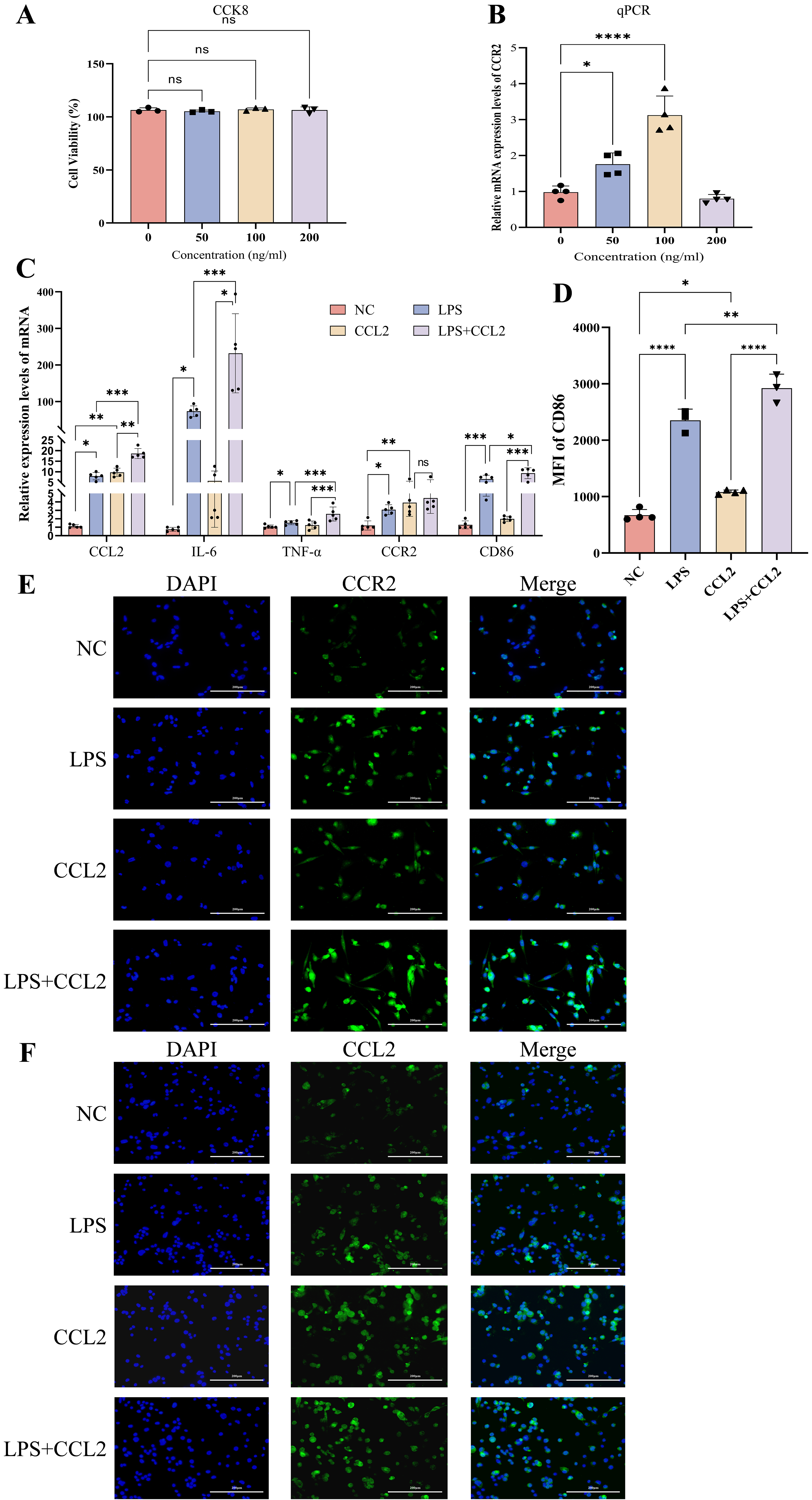 Fig. 4.
Fig. 4.
Expression of cytokines and M1 phenotype after intervention with
recombinant CCL2 (100 ng/mL) and LPS (100 ng/mL) in THP-1 cells. (A) CCK8 assay
assessing the impact on cell viability when THP-1 cells in the M0 state were
intervened with different concentrations of recombinant CCL2 for 24 hours, n = 4.
(B) RT-qPCR measuring the expression levels of CCR2 in M0 state THP-1
cells intervened with different concentrations of recombinant CCL2, n = 4. (C)
RT-qPCR detecting the mRNA levels of CCL2, IL-6,
TNF-
RT-qPCR analysis (Fig. 5A) showed that the mRNA expression levels of signature
indicators of M1-type macrophages (including IL-6,
TNF-
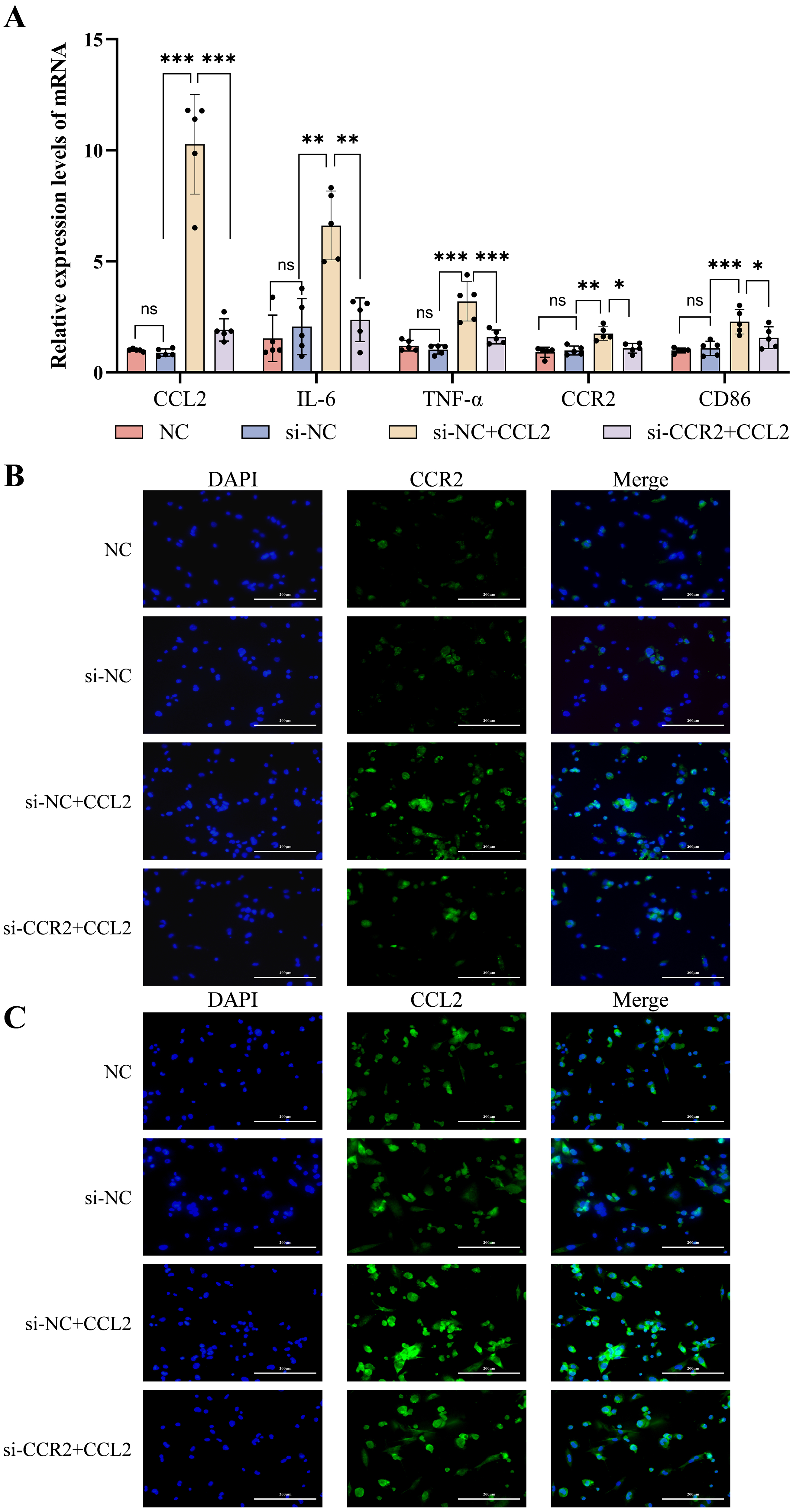 Fig. 5.
Fig. 5.
Expression of inflammatory cytokines and M1 phenotype molecules
after knockdown of CCR2 in THP-1 cells using siRNA, followed by
intervention with CCL2. (A) RT-qPCR detecting the mRNA levels of CCL2,
IL-6, TNF-
The ELISA results from cell supernatants showed elevated levels of inflammatory
factors such as IL-12A, IL-6, and IL-1
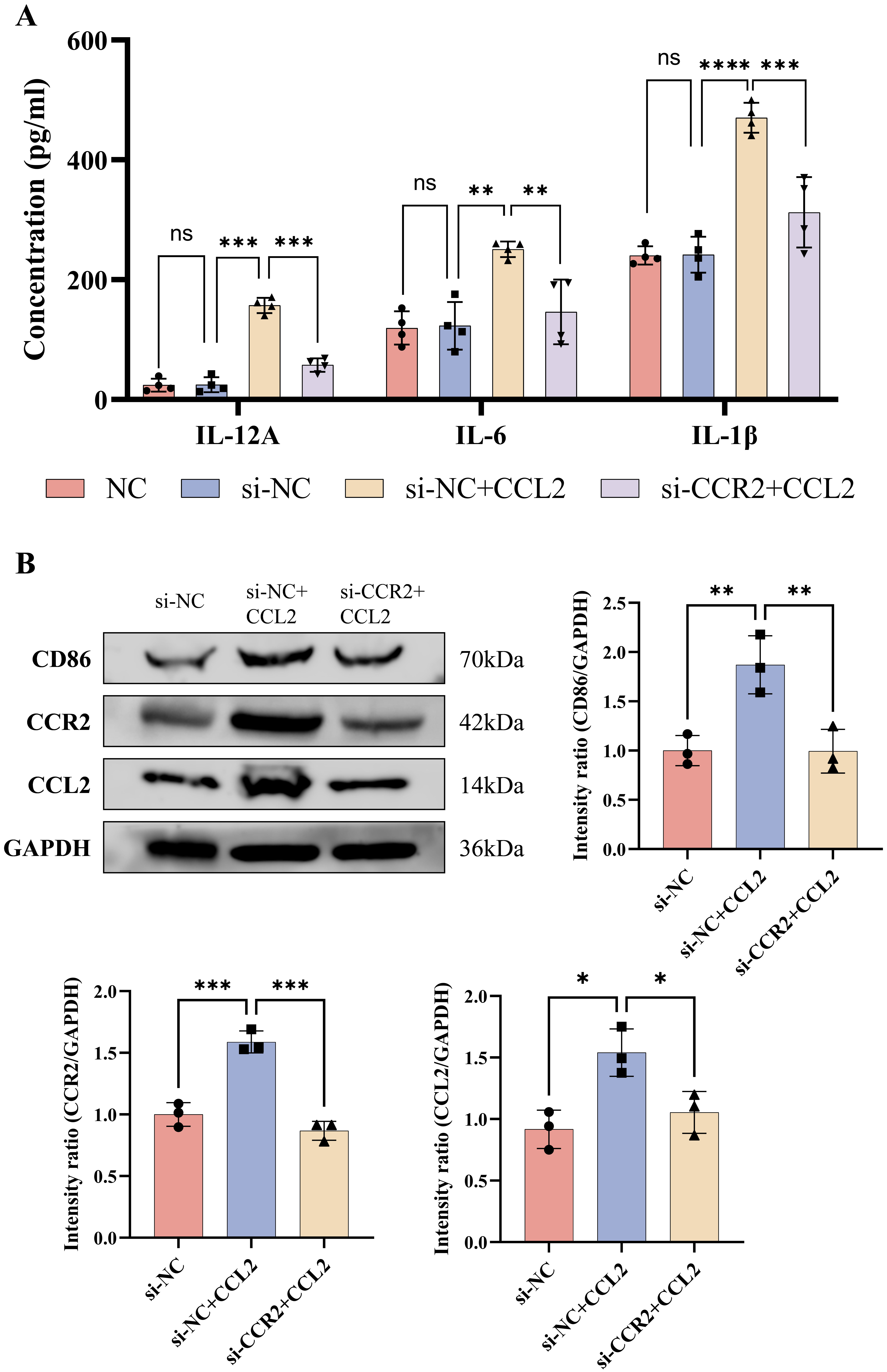 Fig. 6.
Fig. 6.
Silencing CCR2 in THP-1 cells followed by treatment
with CCL2 was conducted to assess the levels of inflammatory cytokines in the
cell supernatants and the expression of CCL2, CCR2, and CD86 in THP-1 cells. (A)
ELISA was used to measure the levels of IL-12A, IL-6, and IL-1
This study investigated macrophage recruitment and polarization both in vivo and in vitro to assess the impact of inhibiting the CCL2/CCR2 axis on the progression of RHD. We observed an expanded macrophage population in the valves of Lewis rats infected with GAS for 8 weeks, characterized by a significant increase in the proportion of the M1 phenotype and only a minimal change in the M2 phenotype. Elevated levels of the monocyte chemotactic factors and their chemokine receptors were identified as primary contributors to the increased macrophage presence in the valves. Additionally, we demonstrated a correlation between macrophage infiltration in the valves of RHD rats and the development of fibrosis. Blocking the CCL2/CCR2 axis with the CCR2-specific antagonist Rs-504393 resulted in a substantial reduction in macrophage numbers in RHD rats valves, along with a significant decrease in valve inflammation and fibrosis. In vitro experiments simulating CCL2 action on macrophages revealed that the CCL2 protein promotes polarization towards the pro-inflammatory M1 type. However, this effect was attenuated when CCR2 was silenced in macrophages using small interfering RNA. Together, these findings suggest that macrophage recruitment and polarization play a crucial role in RHD progression, highlighting them as potential targets for treating valve fibrosis associated with RHD.
Effective tissue repair following injury is crucial for the survival of all organisms [23]. After tissue damage, various immune cells are recruited to the site to clear cellular debris and infectious microorganisms, thereby coordinating the tissue repair response. The extent and duration of this repair response may vary, influencing the final outcome. While enhancing the inflammatory response is beneficial, it can also activate fibrotic reactions leading to excessive accumulation of collagenous connective tissue and subsequent organ dysfunction [24]. There is evidence that macrophages play a critical regulatory role at all stages of repair and fibrosis [25]. In cardiac tissue, a significant population of resident macrophages exists, with increases during injury due to infiltration of circulating monocytes [26]. Circulating monocytes also serve as a source of myofibroblasts in the injured heart, contributing to cardiac repair [27]. Recent studies in mouse hearts following transverse aortic constriction surgery have shown that macrophages derived from recruited monocytes stimulate fibrosis. In contrast, resident macrophages inhibit cardiac fibrosis, their expression of CCR2 helps categorize them as either resident (CCR2-) or circulating-derived (CCR2+) types [28]. Furthermore, in Marfan’s syndrome (MFS), CCR2 deficiency in monocytes inhibits macrophage recruitment. Studies in mice have demonstrated that this protects against the progression of mucoid valve degeneration, resulting in minimal leaflet thickening and preserved stromal integrity [29]. Numerous studies have reported that CCR2+ mesenchymal macrophages play a role in promoting pulmonary fibrosis, with an increase in CCR2-dependent monocytes and mesenchymal macrophages found in the lungs of patients with this condition [30, 31]. These mesenchymal macrophages produce factors that induce fibroblast recruitment and collagen production, highlighting their involvement in the pro-fibrotic process [32]. Importantly, studies using CCR2-deficient mouse models have shown significantly reduced pulmonary fibrosis [33, 34]. These findings are consistent with our observations in RHD rats, where an increase in CCR2 macrophages correlates with inflammation and fibrosis of the valves, both of which are significantly reduced by inhibiting the aggregation of CCR2 macrophages. This suggests an indispensable role for CCR2 macrophages in tissue repair and fibrosis.
The migration of CCR2-expressing cells from the circulation into specific tissues and organs during disease progression has been extensively studied across various conditions. CCR2 binds to several chemokines, including CCL2, CCL7, CCL8, CCL11, CCL13, CCL16, and CCL26 [35, 36], with CCL2 recognized as its primary ligand [37]. The amino-terminal region of CCL2 plays a crucial role in determining binding affinity and signaling selectivity for CCR2 [38]. In our study of rats with RHD, we observed a significant elevation of CCL2 levels in both tissue and serum, potentially contributing to increased cell numbers in the valves. Research on osteoarthritis has highlighted that the CCL2/CCR2 axis, rather than CCL5/CCR5, mediates monocyte recruitment, inflammation, and cartilage destruction [13]. In atherosclerosis, CCL2 facilitates the movement of inflammatory monocytes between the bone marrow, circulation, and atherosclerotic plaques by binding to CCR2 [35, 39]. Studies across different tumors have similarly emphasized the critical role of the CCL2/CCR2 axis in orchestrating the migration of immune cells into the tumor microenvironments [40]. CCL2 expression is widespread across various cell types, including stromal cells, smooth muscle cells, fibroblasts, leukocytes, endothelial cells, and tumor cells [41, 42, 43]. Our experiments identified CCL2 expression in leukocytes, endothelial cells, and fibroblasts within the valves of RHD patients. These findings underscore the potential of targeting the CCL2/CCR2 axis as a therapeutic strategy in numerous diseases.
The prevalence of RHD remains high in some populations in both developing and developed countries, with global estimates indicating that 40.5 million people currently suffer from RHD and 306,000 people die from the disease each year [3]. Despite this high prevalence and mortality rate, the pathogenesis of the disease remains poorly understood. Previous studies have focused on the pathogenicity of GSA, the role of various cytokines, the function of T lymphocytes during valve injury, and the involvement of macrophages in RHD, including their infiltration pathways into the valve [7, 44, 45]. Our findings may therefore shed light on these aspects and lay the groundwork for further investigations into the pathogenesis of RHD, potentially confirming the CCL2/CCR2 axis as a therapeutic target to mitigate macrophage recruitment in heart valves.
We are also concerned about the the investigation of targeted therapeutic agents against the CCL2/CCR2 signaling axis in various autoimmune diseases. A randomized controlled trial showed that treatment of rheumatoid arthritis with a human anti-CCL2 monoclonal antibody (ABN912) did not result in clinical or immunohistological improvement, possibly due to a significant increase in serum total CCL2/Monocyte Chemotactic Protein 1 (MCP-1) levels observed after ABN912 treatment [46]. In another study, patients with rheumatoid arthritis treated with an anti-CCR2 antibody (MLN1202) in a phase IIa clinical trial did not show significant clinical improvement compared to placebo [47]. Rs-504393, a selective CCR2 antagonist inhibiting ERK1/2 activation, has shown promise in early-stage osteoarthritis by reducing pain [48]. Additionally, the humanized anti-CCR2 monoclonal antibody MLN1202 has been effective in reducing brain lesions in patients with multiple sclerosis [49]. The potential therapeutic role of these drugs targeting the CCL2/CCR2 axis in treating RHD requires validation through further animal and clinical trials. We regret that we were unable to collect valvular tissue samples from RHD patients and controls for validation, and that we could not utilize a CCL2/CCR2 knockout animal model for further validation. Future studies could enhance this research by incorporating larger population samples and employing knockout animal models.
In conclusion, this study demonstrated that pharmacological blockade of the CCR2 signaling pathway with Rs-504393 significantly reduced the severity of valve inflammation and fibrosis in rats with RHD. This effect was attributed to mechanisms involving reduced macrophage accumulation in the valves.
The raw data supporting the conclusions of this article will be made available by the correspondence author (zengzhiyu@gxmu.edu.cn) on reasonable request.
ZYZ, FH and YX conceived and designed the study. LB and YL participated in the experimental design. LB, YL, CHL and ZRL conducted the experiments. LB, YL and ZYM analyzed the data. LB and YL drafted the manuscript. All authors contributed to editorial changes in the manuscript. All authors approved the final version. All authors agree to be accountable for all aspects of the work to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The protocol involving animals and patients has been approved by the Medical Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (Approval Number: 2023-E751-01), and all study participants provided written informed consent.
We thank the Department of Cardiothoracic Surgery of the First Affiliated Hospital of Guangxi Medical University for providing the valve specimens. The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
This study was supported by the National Natural Science Foundation of China (Grant No. 81960082), Guangxi Key Laboratory Base of Precision Medicine Control and Prevention of Cardiovascular and Cerebrovascular Diseases (Grant No. 17-259-85), and Guangxi Clinical Research Center of Cardiovascular and Cerebrovascular Diseases (Grant No. AD17129014).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.