







1 Department of Neurology, The Second Affiliated Hospital of Nanchang University, 330008 Nanchang, Jiangxi, China
2 Department of Neurology, Jiujiang First People's Hospital, 332000 Jiujiang, Jiangxi, China
†These authors contributed equally.
Abstract
Background: Alzheimer’s disease (AD) is a neurodegenerative disease
that remains a serious global health issue. Ferroptosis has been recognized as a
vital driver of pathological progression of AD. However, the detailed regulatory
mechanisms of ferroptosis during AD progression remain unclear. This study aimed
to explore the regulatory role and mechanism of methyltransferase like 14
(METTL14) in ferroptosis in AD models. Methods: Serum samples were
collected from 18 AD patients and 18 healthy volunteers to evaluate clinical
correlation. Scopolamine-treated mice and A
Graphical Abstract
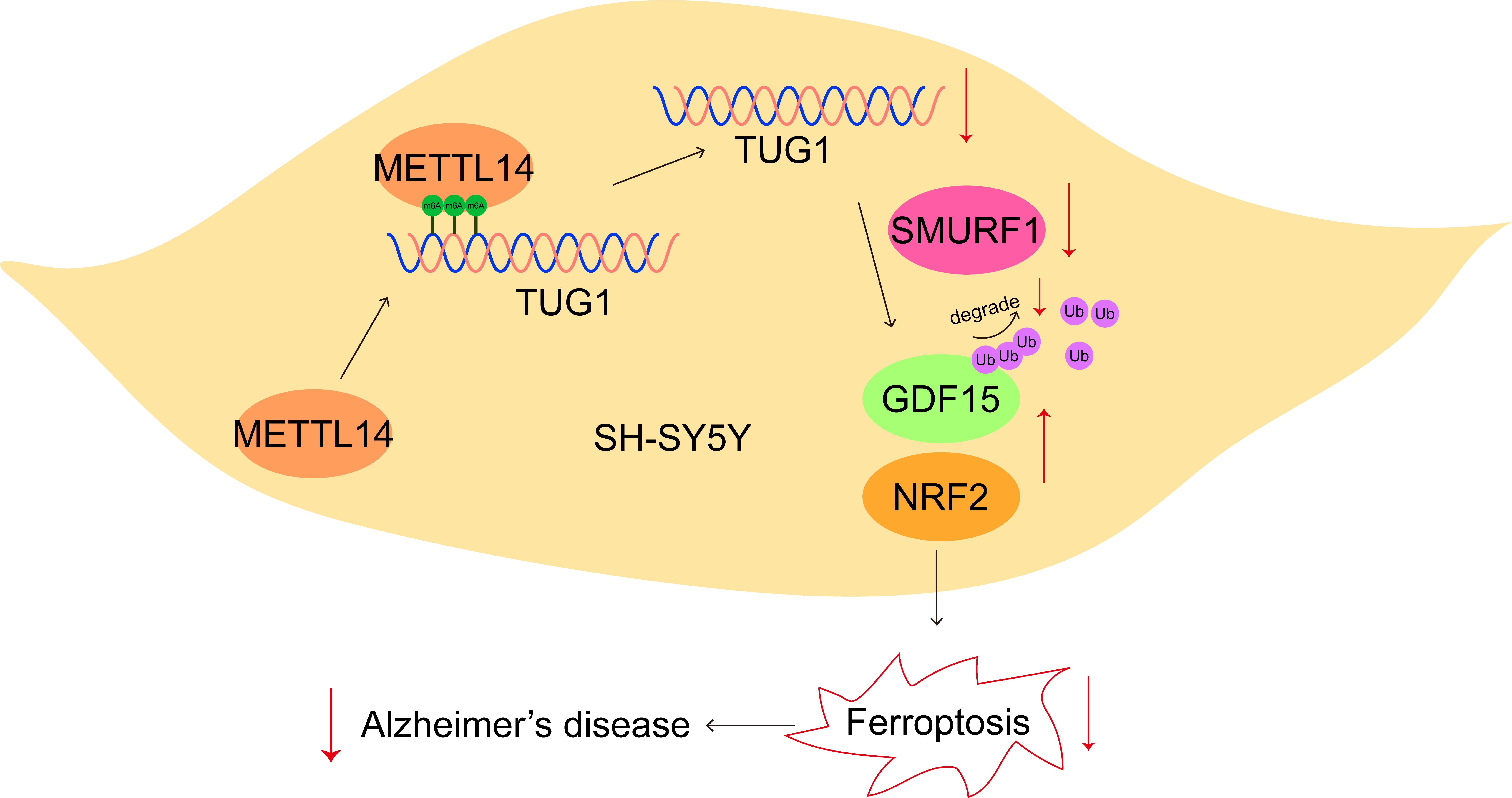
Keywords
- Alzheimer's disease
- ferroptosis
- METTL14
- m6A modification
- GDF15
As a neurodegenerative disease, Alzheimer’s disease (AD) remains the major reason for dementia, which accounts for about 60–80% of all dementia cases [1]. As reported, there will be 152 million AD patients by the year 2050 [2], suggesting that AD may be a severe health issue. Currently, the pathogenesis of AD has not been fully understood and there is still lack of effective therapeutic drugs. Ferroptosis is a novel type of cell death that is caused by iron-dependent lipid peroxidation [3]. It has been recognized that ferroptosis induction is involved in the pathogenesis of AD [4]. For instance, Ayton et al. [5] reported that ferroptosis accelerated the development of AD via increasing brain iron burden. Therefore, inhibition of ferroptosis might be an effective therapeutic strategy for AD patients.
N6-methyladenosine (m6A) is a posttranscriptional RNA modification in eukaryotic cells. Mounting evidence has indicated that m6A modification can influence the occurrence and development of a series of diseases, including AD [6]. M6A modification can be dynamically regulated by a series of methyltransferases, such as methyltransferase like 14 (METTL14) [7]. Dysregulation of METTL14 occurred in the nervous system, which affected AD progression [8]. Besides, a previous study reported that METTL14 delayed hepatocellular carcinoma development via affecting hypoxia-regulated ferroptosis by m6A modulation of solute carrier family 7 member 11 (SLC7A11) [9], suggesting that METTL14-mediated m6A might affect pathological progression of AD via ferroptosis regulation. Long non-coding RNAs (lncRNAs) are a kind of long transcripts containing more than 200 nucleotides, which have been found to be abnormally expressed in AD [10]. The metabolism and biological functions can be influenced by m6A modification [11]. METTL14-mediated m6A modification of lncRNA MSTRG.292666.16 could drive the development of non-small cell lung cancer [12]. Another study showed that METTL14-mediated m6A modification increased lncRNA tumor necrosis factor and heterogeneous nuclear ribonucleoprotein L-related immunoregulatory long non-coding RNA (THRIL) stability, which aggravated acute lung injury via autophagy induction [13]. Notably, sequence-based RNA adenosine methylation site predictor (SRAMP) predicted that lncRNA taurine upregulated gene 1 (TUG1) possessed several m6A modification sites. LncRNA TUG1 was found to be up-regulated in patients with vascular cognitive impairment, and TUG1 silencing was involved in the beneficial effect of aerobic exercise on cognitive disorder and neuronal apoptosis [14]. Zheng et al. [15] reported that m6A modification of TUG1 mediated by METTL14 accelerated diabetic kidney disease progression. Another study documented that m6A modification of Tug1 was involved in diabetes-induced testicular injury [16]. So far, whether METTL14-mediated m6A modification of TUG1 can affect AD progression remains elusive.
Growth differentiation factor 15 (GDF15) is a neurotrophic regulator that is essential for neuron survival [17]. A recent study found that up-regulation of GDF15 might be a stress response to neuronal injury in AD patients [18]. Xiong et al. [19] reported that circLPAR1 contributed to AD development via GDF-15 mRNA decay. In addition, GDF15 knockdown has been demonstrated to promote erastin-induced ferroptosis [20]. Whether TUG1 can affect AD development via regulation of GDF15 has not been clarified. The ubiquitin-proteasome system is responsible for protein degradation [21]. It has been proved that lncRNAs can regulate protein degradation with the assistance of E3 ubiquitin ligases. For instance, lncRNA LINC02878 (BREA2) restrained E3 ubiquitin ligase WW domain-containing protein 2 (WWP2)-induced ubiquitination of Notch1 to drive breast cancer metastasis [22]. Besides, LINC00941 could facilitate pancreatic cancer progression via inhibiting Annexin A2 (ANXA2) ubiquitination and degradation by preventing binding to E3 ubiquitin-protein ligase NEDD4-like (NEDD4L) [23]. Ubibrowser database predicted that Smad ubiquitination regulatory factor 1 (SMURF1) might be an E3 ubiquitin ligase of GDF15. Moreover, the potential interaction between TUG1 and SMURF1 was predicted by RNA-Protein Interaction Prediction (RPISeq) database. A previous study showed that Smurf1 facilitated AD progression via promoting lipopolysaccharide (LPS)-induced neuroinflammation [24]. Therefore, we speculated that TUG1 might facilitate GDF15 protein degradation via promoting SMURF1-mediated ubiquitination.
Based on the above background, we hypothesized that METTL14-mediated m6A modification reduced TUG1 stability to repress ferroptosis by inhibiting SMURF1-mediated GDF15 ubiquitination and subsequent inactivation of GDF15/NRF2 pathway, thereby improving cognitive impairment of AD mice. Our findings elucidate the complicated pathogenesis of AD and identify METTL14 as a novel therapeutic target for AD.
Serum samples were collected from 18 AD patients and 18 healthy volunteers at The Second Affiliated Hospital of Nanchang University. The AD patients with diabetes, cancer, or other severe diseases were excluded. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Second Affiliated Hospital of Nanchang University (No:202392). Written informed consent was collected from all participators.
SH-SY5Y cell line was purchased from American Type Culture Collection (ATCC,
Gaithersburg, MD, USA) and cultured in dulbecco’s modified eagle medium (DMEM,
11965092, Thermo Fisher, Waltham, MA, USA) containing 10% fetal bovine serum
(10099158, Thermo Fisher) at 37 °C with 5% CO2. SH-SY5Y cells were
authenticated by Short Tandem Repeat (STR) analysis and tested for mycoplasma
contamination using the PCR assay (Result revealed that SH-SY5Y cell cell line is tested negative for mycoplasma). An
in vitro AD model was established by stimulation with the oligomer of
amyloid beta (ERMDA482IFCC, A
Overexpression plasmid for METTL14 (OE-METTL14) and its vector pcDNA3.1, short hairpin RNA for METTL14 (sh-METTL14, sequences: CCATGTACTTACAAGCCGATA), sh-TUG1 (sequences: GGAACGGGCGTGCGGTCGATC), sh-SMURF1 (sequences: GCCCAGAGATACGAAAGAGAT), sh-GDF15 (sequences: CCCTCAGAGTTGCACTCCGAA), and their negative control shRNA (sh-NC, sequences: GTTCTCCGAACGTGTCACGT) were purchased from RiboBio (Guangzhou, China). SH-SY5Y cells in logarithmic growth phase were seeded into 6-well plates. SH-SY5Y cells with 70% confluence under culture condition were transfected with these segments using Lipofectamine 2000 (11668500, Thermo Fisher).
Total RNA was extracted from serum samples, SH-SY5Y cells, or hippocampus tissues using the Total RNA Extraction Kit (R1200, Solarbio, Beijing, China). Subsequently, the iscript cDNA synthesis kit (1708890, Bio-Rad, Hercules, CA, USA) was used for cDNA synthesis. RT-qPCR was performed using the Trans Start Tip Green qPCR Super Mix (AQ141-01, TransGen, Beijing, China). Gene expression levels normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were calculated by the 2-ΔΔCt method. The primer sequences are presented in Table 1.
| Name | Sequence (5 |
Length |
| Human METTL14 F | AGGGGTTGGACCTTGGAAGA | 20 |
| Human METTL14 R | GAAGTCCCCGTCTGTGCTAC | 20 |
| Human TUG1 F | ACGACTGAGCAAGCACTACC | 20 |
| Human TUG1 R | CTCAGCAATCAGGAGGCACA | 20 |
| Human SMURF1 F | GTCCAGAAGCTGAAAGTCCTCAGA | 24 |
| Human SMURF1 R | CACGGAATTTCACCATCAGCC | 21 |
| Human GDF15 F | TCAGATGCTCCTGGTGTTGC | 20 |
| Human GDF15 R | CTGGTTAGCAGGTCCTCGTAG | 21 |
| Mouse Mettl14 F | TCTGGAAAACTGCCTTTGGAT | 21 |
| Mouse Mettl14 R | AAATGCTGGACCTGGGATGAT | 21 |
| Mouse Tug1 F | GAGACACGACTCACCAAGCA | 20 |
| Mouse Tug1 R | GAAGGTCATTGGCAGGTCCA | 20 |
| Mouse Gdf15 F | TTTGGGGGGTGATGATGC | 18 |
| Mouse Gdf15 R | GCGACTTTCTGGGGAAACC | 19 |
| Mouse Il-1 |
GCCACCTTTTGACAGTGATGAG | 22 |
| Mouse Il-1 |
AAGGTCCACGGGAAAGACAC | 20 |
| Mouse Il-6 F | TGCAAGAGACTTCCATCCAG | 20 |
| Mouse Il-6 R | TCCACGATTTCCCAGAGAAC | 20 |
| Mouse Tnf- |
CCGATGGGTTGTACCTTGTC | 20 |
| Mouse Tnf- |
TGGAAGACTCCTCCCAGGTA | 20 |
| Mouse Gapdh F | AGCCCAAGATGCCCTTCAGT | 20 |
| Mouse Gapdh R | CCGTGTTCCTACCCCCAATG | 20 |
| Human GAPDH F | CCAGGTGGTCTCCTCTGA | 18 |
| Human GAPDH R | GCTGTAGCCAAATCGTTGT | 19 |
qPCR, Real-time Quantitative PCR; METTL14, mechanism of methyltransferase like
14; TUG1, taurine upregulated gene 1; GDF15, growth differentiation
factor 15; IL-1
Protein samples were isolated using the Radio Immunoprecipitation Assay (RIPA) reagent (P0013B, Beyotime, Haimen, China) and protein concentration was quantified using the Bicinchoninic Acid (BCA) Protein Assay Kit (PC0020, Solarbio). Then, the protein samples were loaded onto sodium dodecyl sulfate polyacrylamide gel electrophoresis. After blotting onto the polyvinylidene fluoride membranes and blocking with 5% non-fat milk, the membranes were incubated with primary antibodies against glutathione peroxidase 4 (GPX4, 1:500, bs-3884R, Bioss, Beijing, China), solute carri1er family 7 member 11 (SLC7A11, 1:1000, ab307601, Abcam, Cambridge, UK), SMURF1 (1:500, bs-9391R, Bioss), GDF15 (1:1000, sc-515675, Santa Cruz, TX, USA), NRF2 (1:1000, ab62352, Abcam), heme oxygenase-1 (HO-1, 1:2000, ab52947, Abcam), glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:5000, bsm-52262R, Bioss) overnight at 4 °C, followed by incubation with the secondary antibody Goat Anti-Rabbit IgG Fc and F(ab)/F(ab’)2 (H&L) (1:2000, ab205718, Abcam) for 1 h. The bands were developed with efficient chemiluminescence (ECL) solution (WBULP-100ML, Millipore, Burlington, MA, USA). The band intensity of the Control group was set to 1 for normalized statistics.
To evaluate SH-SY5Y cell viability, 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) solution (G4100, Promega, Madison, WI, USA) was added into each well of cells that were seeded in 96-well plates at a density of 3000 cells per well. After incubation at 37 °C for 4 h, the formed formazan crystals were dissolved by dimethyl sulfoxide solution (DMSO, D8418, Sigma-Aldrich). The absorbance was measured at 490 nm on a Microplate Reader (VA000010C, Thermo Fisher).
The Iron Assay Kit (ab83366, Abcam) was used to determine Fe2+ and total iron levels. In brief, SH-SY5Y cells were lysed by Iron Assay Buffer. Then, cell lysates were incubated with the Assay Buffer for Fe2+ detection or the Iron Reducer for total iron detection at 37 °C for 30 min, followed by incubation with the Iron Probe for 60 min at 37 °C. The results were measured at 593 nm on a Microplate Reader (VA000010C, Thermo Fisher).
The ROS level in SH-SY5Y cells was measured using the ROS Detection Assay (ab287839, Abcam). The cells were seeded into 96-well plates and incubated with the ROS/Superoxide Detection Solution at 37 °C for 1 h away from light. The results were detected using a fluorescence microplate reader ((excitation) Ex = 488 nm, emission (Em) = 520 nm, VA000010C, Thermo Fisher).
The concentrations of interleukin-1beta (IL-1
Total RNA m6A level was detected using the m6A RNA methylation quantification kit (P-9008-48, EpiGentek, Farmingdale, NJ, USA). Briefly, total RNA was isolated from SH-SY5Y cells and incubated with the capture antibody and detection antibody. After incubation, color developing solution was added into cells. Finally, the optical density at 450 nm was measured and the relative total RNA m6A level was calculated.
To determined TUG1 stability, SH-SY5Y cells were treated with 5 µg/mL actinomycin D (SBR00013, Sigma-Aldrich) for 3, 6, 9 h. Then, TUG1 level was assessed by RT-qPCR as described above.
The m6A level of TUG1 was assessed by MeRIP using the riboMeRIPTM m6A Transcriptome Profiling Kit (C11051-1, RiboBio). Briefly, total RNA was fragmentated and incubated with anti-m6A antibody (ab208577, Abcam) or anti-IgG (ab18448, Abcam) conjugated in magnetic beads A/G at 4 °C for 2 h. After elution, the RNA was collected from the precipitates and detected by RT-qPCR.
RIP assay was performed using the Magna RIP kit (17-701, Millipore). SH-SY5Y cells were lysed in the RIP lysis buffer and then coimmunoprecipitated in Protein A/G MagBeads that were pre-coated with anti-SMURF1 antibody (H00057154-M01, Thermo Fisher) or isotype control IgG (ab18448, Abcam). The specific binding RNAs to the antibody-protein complex were isolated and the level of TUG1 was analyzed by RT-qPCR.
RNA pull-down assay was performed using the Magnetic RNA-Protein Pull-Down Kit (20164, Thermo Fisher). Briefly, the protein samples of SH-SY5Y cells were incubated with the Biotin-labeled TUG1 probe that was pre-coupled in streptavidin beads at 4 °C for 2 h. After washing for five times, the RNA-protein complexes were retrieved from the beads and subjected to Western blotting.
The cell samples were lysed in Radio Immunoprecipitation Assay (RIPA) Lysis Buffer (P0013B, Beyotime) containing various protease inhibitors. After centrifugation at 12,000 g for 10 min, cell lysates were immune-precipitated with protein G Plus-Agarose pre-bound with anti-GDF15 antibody (5 µg, sc-515675, Santa Cruz). After immunoprecipitation overnight, the immune-precipitates were eluted and subjected to Western blotting with GAPDH as loading control. For ubiquitination assay, SH-SY5Y cells were treated with 10 µM MG132 for 8 h, and the cell lysates were immunoprecipitated by GDF15 antibody (sc-515675, Santa Cruz), and GDF15 ubiquitination was analyzed by Western blotting with ubiquitin antibody (ab140601, Abcam).
C57BL/6J mice (aged 6–8 weeks, weighing 20–25 g) were purchased from Slac Jingda Laboratory Animal Co., Ltd. (Hunan, China). AD was induced by scopolamine according to a previous study [25]. A total of 32 mice were divided into four experimental groups (n = 8 per group): control, AD model, AD + vector, and AD + OE-Mettl14. The mice in AD groups were intraperitoneally injected with 2 mg/kg scopolamine (Sigma-Aldrich) for 7 consecutive days. The control mice were intraperitoneally injected with equal volume of saline vehicle. The mice in AD + vector and AD + OE-Mettl14 groups were intrahippocampally injected with Adeno-associated virus type 2 (AAV2) -vector or AAV2-OE-Mettl14 (1010 pfu/mouse, Genechem, Shanghai, China) before the injection with scopolamine. At 24 h after the final scopolamine injection, all mice were subjected to behavioral test and then euthanized by cervical dislocation. The hippocampal tissues were collected for further examination. All experimental protocols were approved by the Animal Care and Use Committee of the Second Affiliated Hospital of Nanchang University (No:2023095).
The learning and memory capacities of mice were analyzed by MWM assay using a
steel pool (120 cm diameter
The pathological changes in hippocampal tissues were determined by HE staining. The hippocampal tissues were fixed in 4% paraformaldehyde, embedded in paraffins, and sectioned into 5 µm slices. The slices were subjected to HE staining using the Hematoxylin and Eosin Staining Kit (C0105S, Beyotime). The stained slices were observed under a light microscope (BX51M, Olympus, Japan). The number of damaged neurons was quantified as previously described [26].
All data from three independent experiments are expressed as mean
First, RT-qPCR validated a lower expression of METTL14 in the serum samples of
AD patients in comparison with healthy volunteers (Fig. 1A). Besides, SH-SY5Y
cells were stimulated with A
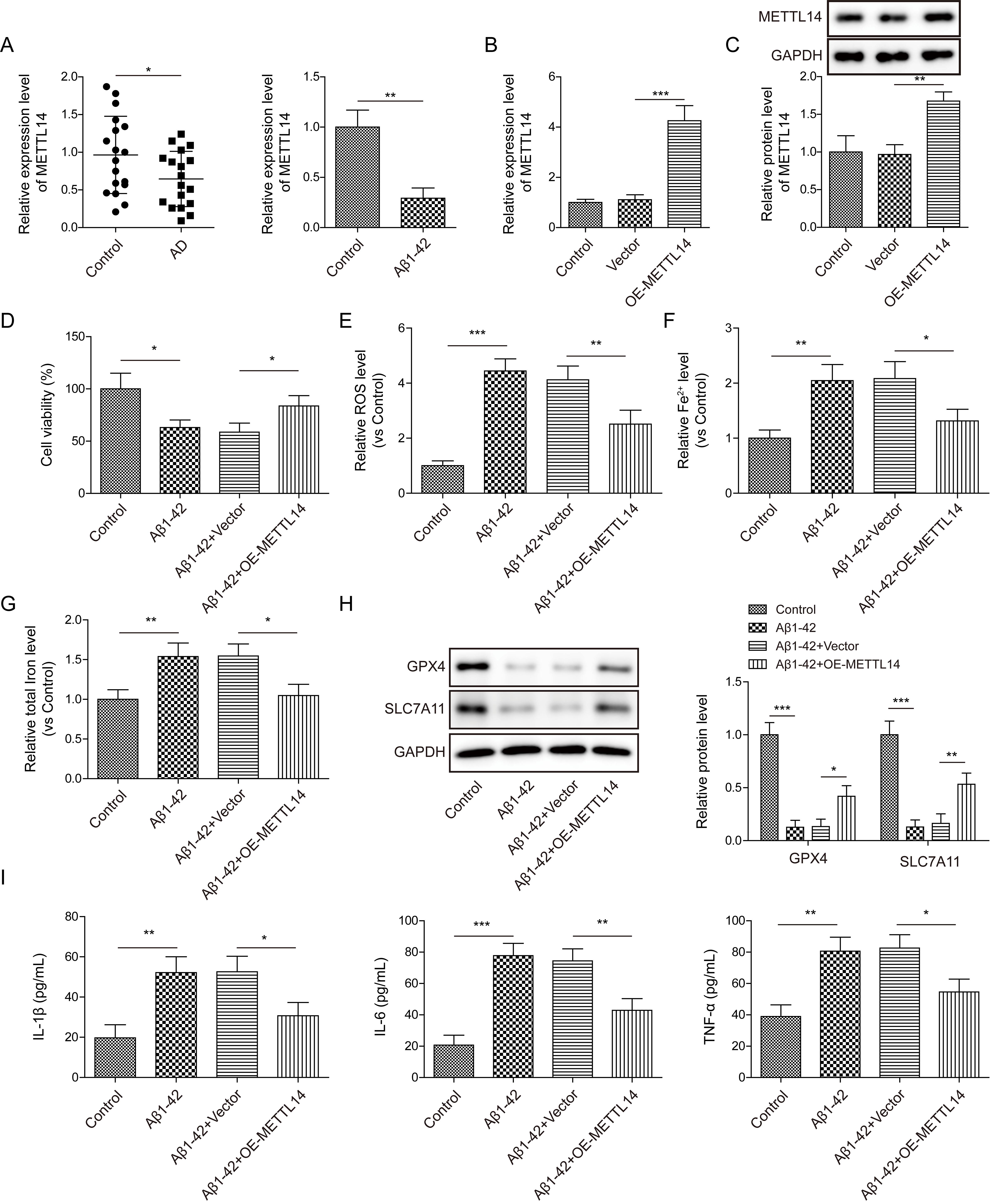 Fig. 1.
Fig. 1.
METTL14 overexpression inhibited A
To further explore the downstream regulatory mechanism of METTL14 in AD progression, TUG1 was focused on. The silencing efficiency of sh-METTL14 in SH-SY5Y cells was validated by RT-qPCR and Western blotting (Fig. 2A,B). Notably, TUG1 expression was increased in METTL14-silenced cells, but reduced in METTL14-overexpressed cells (Fig. 2C). We also found that total RNA m6A level was declined after METTL14 knockdown, while elevated after METTL14 overexpression (Fig. 2D). Of note, SRAMP database predicted several m6A modification sites on TUG1. Moreover, METTL14 overexpression significantly enhanced the m6A level of TUG1, while METTL14 knockdown resulted in the opposite result (Fig. 2E). In addition, the stability of TUG1 RNA in actinomycin D-treated SH-SY5Y cells was decreased in METTL14-overexpressed cells, but increased in METTL14-depleted cells (Fig. 2F). There was no significant difference between control and sh-NC/vector group. These results revealed that METTL14 facilitated the decay of TUG1 RNA via m6A modification.
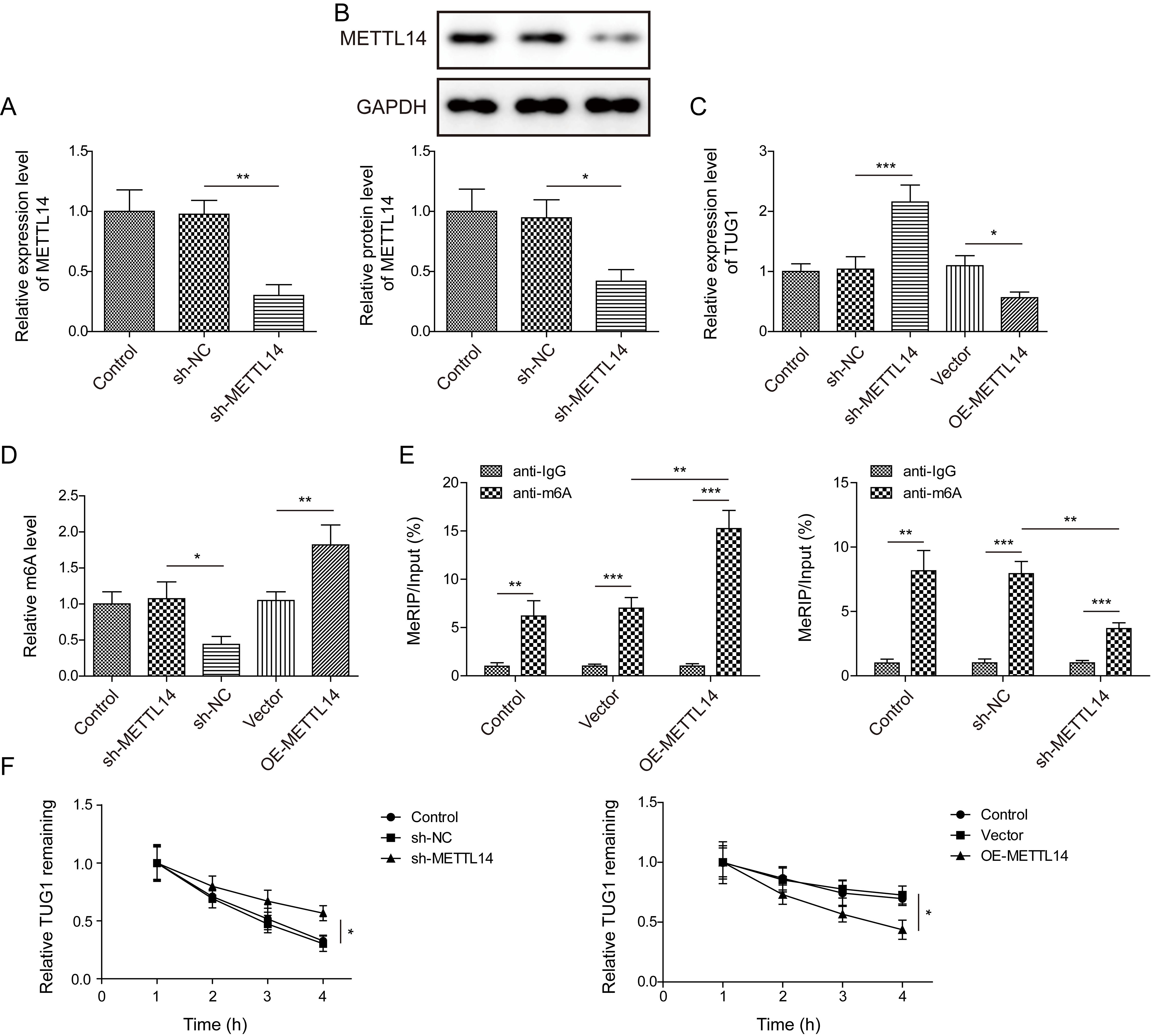 Fig. 2.
Fig. 2.
METTL14 reduced the stability of TUG1 via m6A
modification. Expression of METTL14 was detected by RT-qPCR (A) and Western
blotting (B). (C) TUG1 expression after transfection was evaluated by
RT-qPCR (n = 3). (D) Total RNA m6A level was assessed by the m6A RNA methylation
quantification kit (n = 3). (E) MeRIP was adopted to determine m6A level of
TUG1 (n = 3). (F) After exposure to actinomycin D, the remaining
TUG1 expression was determined by RT-qPCR (n = 3). * p
Given that TUG1 was a downstream target of METTL14, we further
investigated the regulatory role of TUG1 in AD progression. As compared
with normal volunteers, TUG1 expression was higher in the serum samples
of AD patients, as well as in A
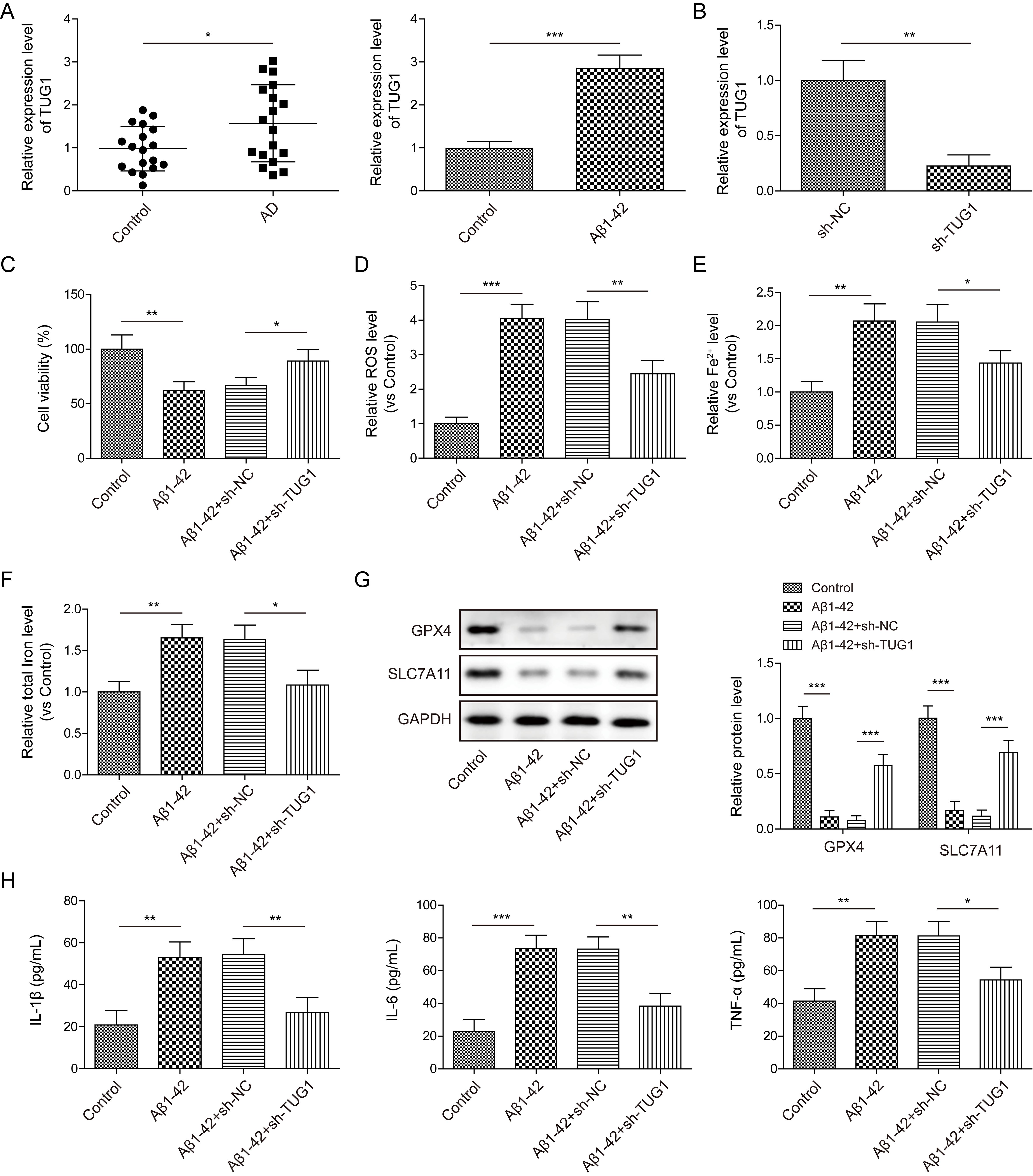 Fig. 3.
Fig. 3.
A
Next, the underlying molecular mechanism of TUG1 in AD was explored. As predicted by RPISeq database, TUG1 might interact with SMURF1 protein (interaction score, 0.7; SVM 0.99). RNA pull-down assay suggested that SMURF1 protein could be immunoprecipitated by TUG1 probe (Fig. 4A). Furthermore, RIP assay demonstrated that the enrichment of TUG1 was elevated after immunoprecipitation with anti-SMURF1 antibody (Fig. 4B). These results proved that there was a direct interaction between TUG1 and SMURF1. In addition, ubibrowser predicted that SMURF1 was a potential E3 ubiquitin ligase of GDF15. Co-IP assay further verified that SMURF1 could directly interplay with GDF15 protein (Fig. 4C). Transfection with sh-SMURF1 evidently reduced SMURF1 mRNA and protein levels in SH-SY5Y cells (Fig. 4D,E). Furthermore, SMURF1 silencing lowered the ubiquitin level of GDF15 and consequently enhanced GDF15 protein level in SH-SY5Y cells (Fig. 4F). Besides, silencing of TUG1 or SMURF1 reduced SMURF1 protein level, but enhanced GDF15, NRF2, and HO-1 protein levels in SH-SY5Y cells, which were reinforced by co-transfection with sh-TUG1 and sh-SMURF1 (Fig. 4G). Collectively, TUG1 depletion activated GDF15/NRF2 pathway via repressing SMURF1-mediated ubiquitination of GDF15.
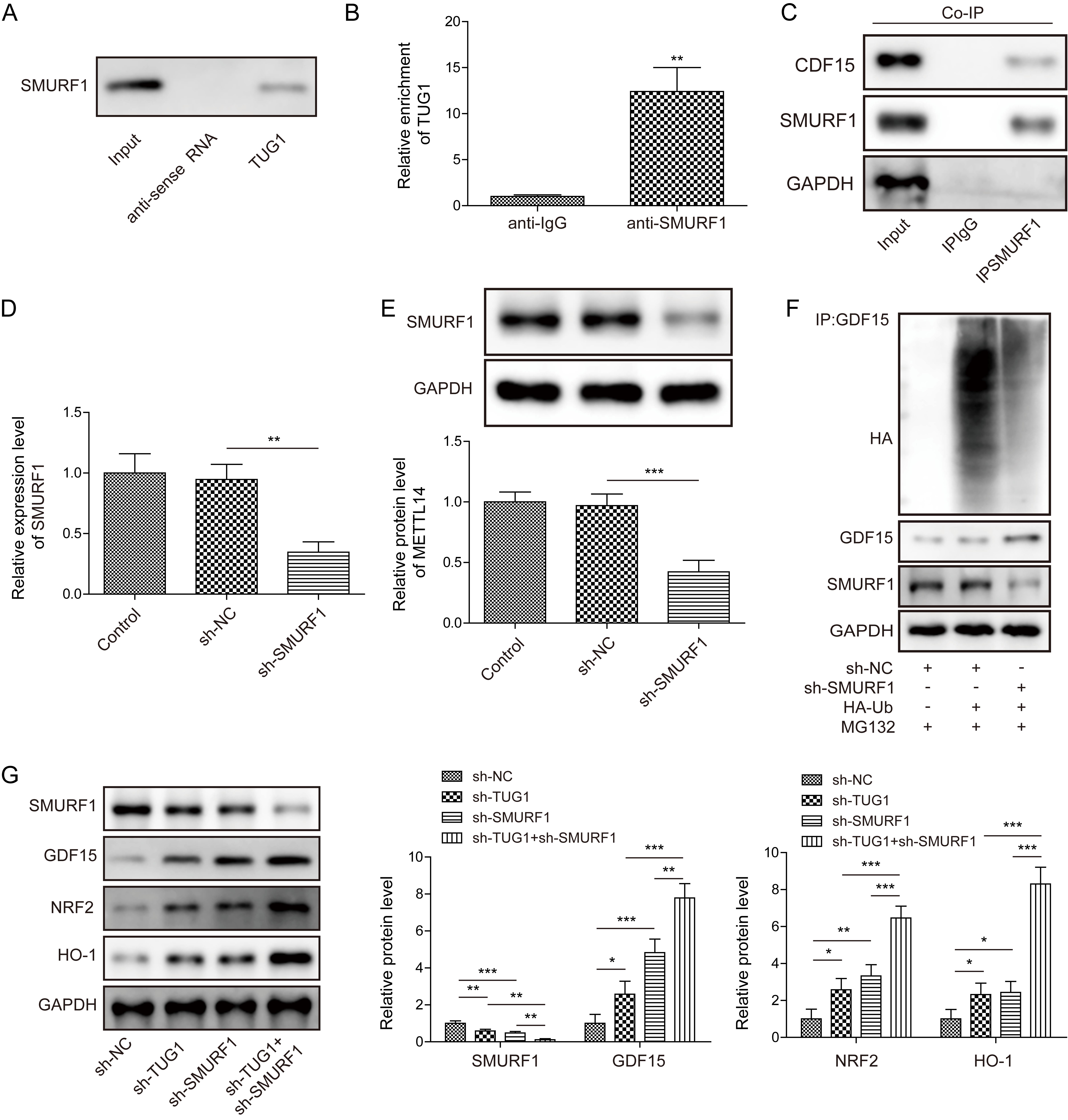 Fig. 4.
Fig. 4.
TUG1 knockdown activated GDF15/NRF2 pathway through
interaction with SMURF1. The direct interaction between TUG1 and SMURF1
was validated by RNA pull down (A) and RIP assays (B) (n = 3). (C) The
interaction between SMURF1 and GDF15 proteins was validated by Co-IP (n = 3). (D,E) RT-qPCR and Western blotting analysis of SMURF1 mRNA level (n = 3). (F) The
ubiquitin level of GDF15 was assessed by Co-IP (n = 3). (G) Western blotting
determination of SMURF1, GDF15, NRF2, and HO-1 protein levels (n = 3). *
p
Given that GDF15 was modulated by TUG1 in SH-SY5Y cells, we further
investigated the involvement of GDF15 in the influence of TUG1 on AD
progression. For this purpose, A
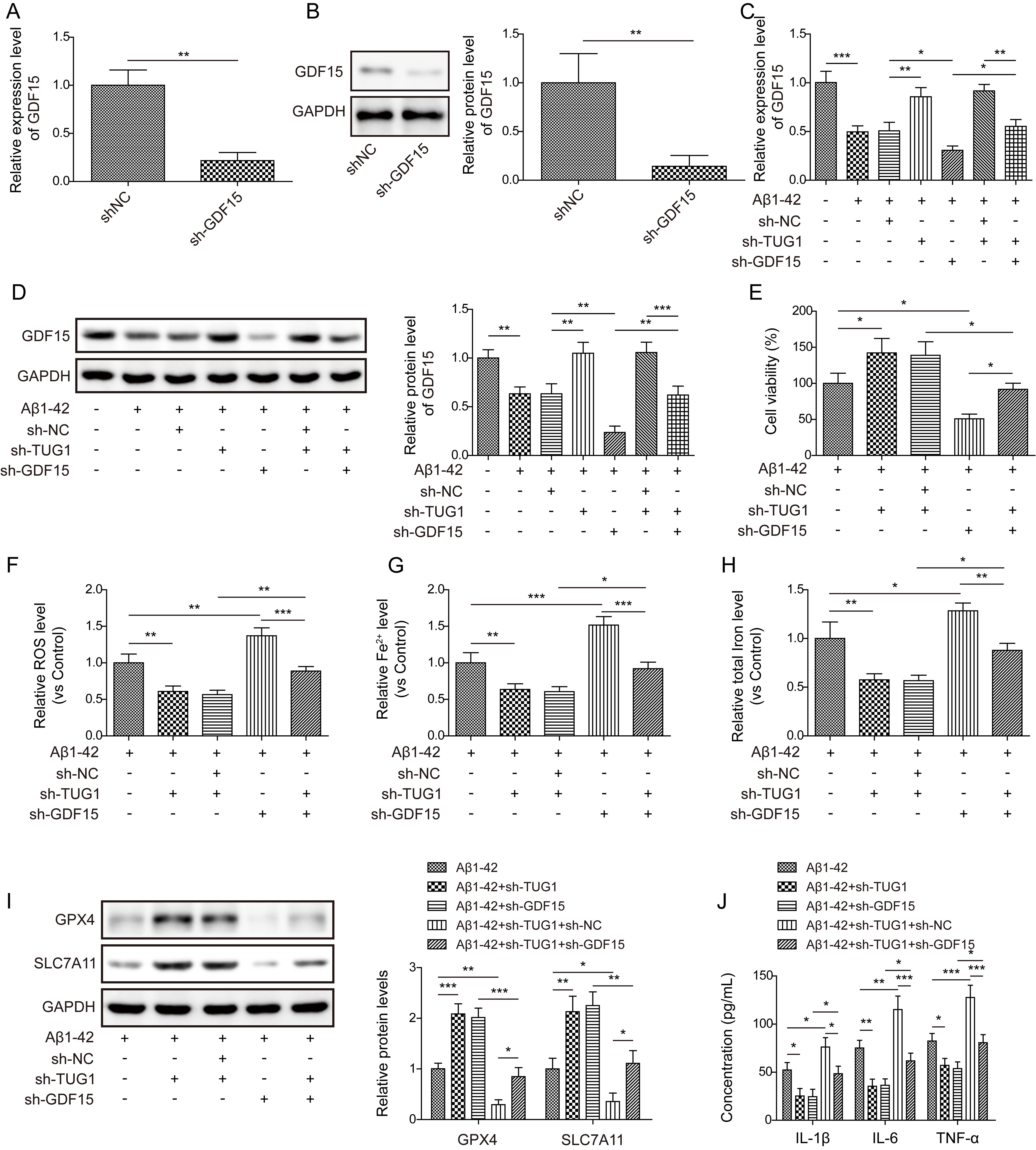 Fig. 5.
Fig. 5.
TUG1 depletion repressed ferroptosis via activating
GDF15. GDF15 expression level was detected by RT-qPCR (A) and
Western blotting (B) (n = 3). (C,D) RT-qPCR and Western blotting analysis of
GDF15 expression in cell from various groups. (E) Cell viability was determined
by MTT assay (n = 3). Levels of ROS (F), Fe2+ (G), and total iron (H) in
cells received multiple treatments were assessed (n = 3). (I) Protein levels of
GPX4 and SLC7A11 was measured by Western blotting (n = 3). (J) IL-1
Finally, the protective effect of Mettl14 was validated in AD mice in
vivo. As evaluated by MWM, AD mice exhibited obvious memory impairment, however;
Mettl14 overexpression effectively improved behavioral deficits of AD mice (Fig. 6A–D). HE staining indicated intact structure of hippocampal neurons, and no
obvious necrosis was observed in WT mice. However, abnormal shape, unordered
arrangement, and apoptosis were found in hippocampal neurons of AD mice (Fig. 6E). These pathological changes in hippocampus were prominently relieved by
Mettl14 overexpression (Fig. 6E). Moreover, the damaged neurons were indicated as
red arrows and quantified. We found that the number of damaged neurons was
strikingly increased, which was reduced after Mettl14 overexpression (Fig. 6E).
In addition, METTL14 and Gdf15 mRNA levels were down-regulated, while
Tug1 level was up-regulated in the hippocampal tissues of AD mice,
whereas Mettl14 overexpression reversed these changes (Fig. 6F). Furthermore, the
protein levels of Gdf15, Nrf2, HO-1, Gpx4 and Slc7A11 were reduced in the
hippocampal tissues of AD group, which could be counteracted after Mettl14
overexpression (Fig. 6G). RT-qPCR analysis showed that the increased
Il-1
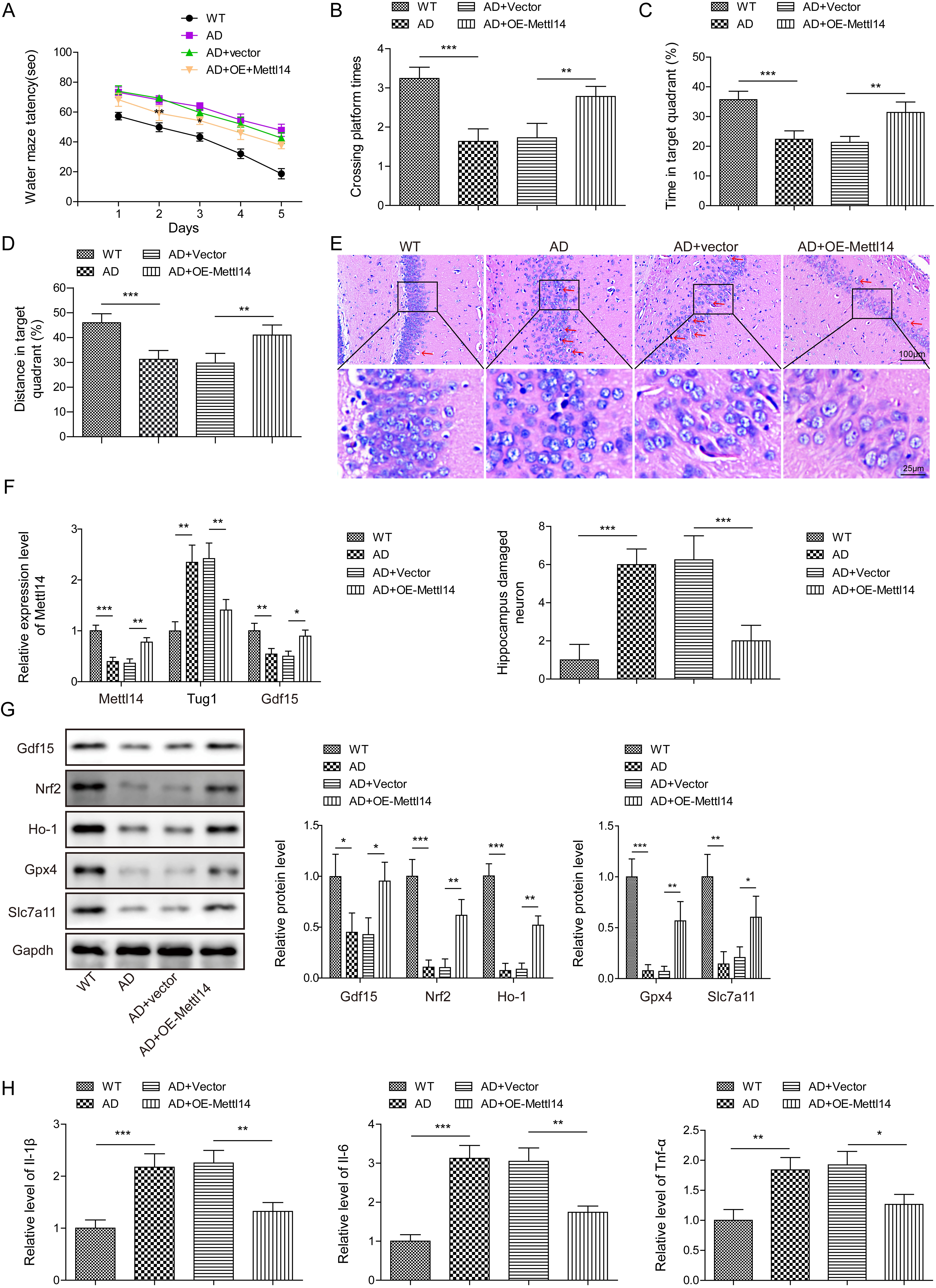 Fig. 6.
Fig. 6.
Mettl14 overexpression improved cognitive disorder and
suppressed ferroptosis in AD mice via TUG1/Gdf15/Nrf2 pathway. (A–D)
Morris water maze was performed to evaluate the learning and memory capacities of
mice (n = 8). (E) The pathological changes in hippocampus were observed by HE
staining (n = 8). Scale bars: 100 µm or 25 µm. (F) Expression of
Mettl14, Tug1, and Gdf15 was detected by RT-qPCR (n = 8). (G) Protein
abundance of Gdf15, Nrf2, HO-1, Gpx4, and Slc7a11 was determined by Western
blotting (n = 8). (H) The mRNA levels of Il-1
Ferroptosis is a novel type of cell death mediated by iron-dependent lipid peroxidation, which has been considered as a driver of neuronal loss in various neurodegenerative diseases, including AD [27]. Thus, inhibition of ferroptosis may be a potential treatment strategy for AD. Nevertheless, the detailed regulatory mechanisms of ferroptosis during AD development deserve to be clarified. In this study, we provided first evidence that TUG1 was down-regulated by METTL14-mediated m6A modification, which mitigated ferroptosis to delay AD progression through inhibiting GDF15 ubiquitination to activate Nrf2 pathway.
Mounting evidence has proved that m6A modification is
implicated in the pathogenesis of AD [28]. Mettl14, an m6A writer, has been
reported to be down-regulated in AD brain [29]. A recent study found that METTL14
deficiency reduced the number of oligodendrocyte and resulted in central nervous
system hypomyelination [30]. Importantly, METTL14-mediated m6A Modification of
TRAF6 was demonstrated to relieve dopaminergic neuron degeneration [31].
Recently, the regulation of ferroptosis by METTL14 has been reported. For
instance, Zhuang et al. [32] suggested that METTL14 facilitated
doxorubicin-induced ferroptosis in cardiomyocytes via modulation of
KCNQ1OT1/miR-7-5p pathway. METTL14 exerted anti-tumor effect via regulating
YT521-B homology domain family member 2 (YTHDF2)/SLC7A11/ROS axis-mediated
ferroptosis in hepatocellular carcinoma [9]. In line with the previous studies,
we found a reduction in METTL14 expression in AD patients and mice.
Overexpression of METTL14 restrained ferroptosis in A
It has been identified that M6A modification can regulate the splicing, distribution, and stability of lncRNAs. Recent studies have revealed that m6A-modulated lncRNAs participate in multiple brain diseases. For example, METTL3-mediated lnc-D63785 m6A modification contributed to anoxia/reoxygenation-induced neuronal damage [33]. Chang et al. [34] discovered that METTL3-mediated m6A modification stabilized MALAT1, which accelerated glioma progression. Furthermore, RMVar database predicted several m6A modification sites in TUG1. Moreover, RM2Target database predicated that there were 5 binding motifs between METTL14 and TUG1. Here, we demonstrated that METTL14 facilitated lncRNA TUG1 degradation via m6A modification. So far, whether TUG1 accelerated AD progression via ferroptosis induction has not been clarified. In this study, we for the first time found that knockdown of TUG1 suppressed ferroptosis in the in vitro model of AD. Therefore, our findings suggested that METTL14 led to degradation of TUG1 through m6A modification, which inhibited ferroptosis to delay AD development.
Finally, we uncovered the potential downstream mechanism through which
TUG1 promoted AD progression. Dysregulation of GDF15 has been widely
documented in the development of AD. It has been shown that GDF15 expression was
abnormally elevated in the brains of AD patients, suggesting GDF15 as a stress
mechanism to protect against AD [18]. In addition, GDF15 conferred protection
against AD by facilitating hippocampal neurogenesis and A
There are several limitations in this study. First, although the learning and memory capacities of mice was analyzed by MWM assay, the trajectory diagrams were not saved and shown. In the future study, we will present these trajectory diagrams to evaluate memory impairment more intuitively. Secondary, MeRIP-seq is a method to study the expression of m6A-modified lncRNAs [43]. However, due to lack of time, it was difficult for us to perform MeRIP-seq in the current study. Thirdly, m6A modification can be mediated by m6A-binding proteins (readers) [44]. The m6A reader for TUG1 m6A modification in AD needs to be further explored.
Taken together, our findings demonstrated that METTL14 repressed ferroptosis and neurological damage to improve cognitive impairment in AD mice via m6A modification of TUG1 to activate GDF15/Nrf2 pathway by inhibiting SMURF1-mediated GDF15 ubiquitination (Graphical abstract). Our data provide theoretical evidence that METTL14 delays the development of AD, suggesting METTL14 as a novel effective target for the treatment of AD.
AD, Alzheimer’s disease; METTL14, methyltransferase like 14; TUG1,
taurine upregulated gene 1; m6A, N6-methyladenosine; lncRNAs, long non-coding
RNAs; SMURF1, smad ubiquitylation regulatory factor-1; GDF15, growth
differentiation factor 15; ATCC, American Type Culture Collection;
A
The data sets generated and/or analyzed during the current study are not publicly available due to Further research is needed, but are available from the corresponding author on reasonable request.
XG, YS and YZ designed the research study. XL and ZC performed the research. XG and JM analyzed the data. XG and YS wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Animal and clinical experiments were approved by the Second Affiliated Hospital of Nanchang University Medical Research Ethics (No.2023095, No. 202392). All participators signed the written informed consent.
Not applicable.
This work was supported by the Science and Technology Program of Jiangxi Provincial Health Commission (Grant number: 202310029), the National Natural Science Foundation of China (Grant number: 82260262).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.