


 , Valeria A. Korolenya 1,3, Maxim L. Filipenko 1
, Valeria A. Korolenya 1,3, Maxim L. Filipenko 11 Laboratory of Pharmacogenomics, Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences (ICBFM SB RAS), 630090 Novosibirsk, Russia
2 Department of Fundamental Medicine, V. Zelman Institute for Medicine and Psychology, Novosibirsk State University (NSU), 630090 Novosibirsk, Russia
3 Department of Natural Sciences, Novosibirsk State University (NSU), 630090 Novosibirsk, Russia
Abstract
Making a correct genetically based diagnosis in patients with diseases associated with mitochondrial dysfunction can be challenging both genetically and clinically, as can further management of such patients on the basis of molecular-genetic data assessing the state of their mitochondria. In this opinion article, we propose a novel approach (which may result in a clinical protocol) to the use of a precise molecular-genetic tool in order to monitor the state of mitochondria (which reflects their function) during treatment of certain conditions, by means of not only signs and symptoms but also the molecular-genetic basis of the current condition. This is an example of application of personalized genomic medicine at the intersection of a person’s mitochondrial genome information and clinical care. Advantages of the proposed approach are its relatively low cost (compared to various types of sequencing), an ability to use samples with a low input amount of genetic material, and rapidness. When this approach receives positive outside reviews and gets an approval of experts in the field (in terms of the standards), it may then be picked up by other developers and introduced into clinical practice.
Keywords
- mitochondrial DNA
- mtDNA
- mtDNA copy number
- ND4-region mtDNA deletions
- mtDNA heteroplasmy
- treatment/recovery progress monitoring
Mitochondria are organelles of a eukaryotic cell that specialize in producing energy in the form of ADP and ATP via oxidative phosphorylation (OXPHOS). These partially autonomous little “power plants” containing their own circular DNA in the mitochondrial matrix replicate independently of the cell’s division cycle while using gene products from both the mitochondrial and nuclear genome. Mitochondrial DNA (mtDNA) encodes 37 genes, predominantly involved in OXPHOS [1]. In total, ~3000 genes take part in physiological processes within mitochondria, and most of these genes are contained in nuclear DNA (nDNA) [2]. mtDNA somatic mutations occur frequently because of the oxidative environment, rapid replication, and inefficient mtDNA repair [3]. The higher frequency of mutations in mtDNA compared to nDNA (10-fold) may be associated with the lack of introns, lack of protection by histone proteins, and a limited repair system [4]. The polyploid nature of the mitochondrial genome allows for a state termed heteroplasmy, when a mutation coexists with the wild-type counterpart in various proportions; and only above a certain threshold level of mutant mtDNA can biochemical defects be observed and lead to a disease manifestation [5]. Eukaryotic cells of different tissues contain different numbers of mitochondria, which are determined by cellular energy needs and can change adaptively. In addition, changes in the number of mitochondria are associated with some pathological processes [4]. Remarkably, because of uncontrolled production of reactive oxygen species, mitochondrial dysfunction is involved in the pathogenesis of numerous cardiovascular diseases such as myocardial infarction, hypertension, atherosclerosis, and varicose veins [6, 7].
There is a known group of disorders—mitochondrial diseases—associated with hereditary or somatic mutations in mtDNA or nDNA, thereby leading to disturbances of energy functions of mitochondria; these disorders affect approximately one in 5000 people worldwide [8]. Primary mitochondrial diseases (PMDs) can be caused by germline mutations in mtDNA and/or nDNA genes encoding either OXPHOS structural proteins or mitochondrial proteins of the complex machinery needed for the OXPHOS process to operate optimally [9]. In contrast, secondary mitochondrial dysfunction (SMD; when mitochondria do not work properly because of another disease or aberration) can either occur due to mutations in other genes not related to OXPHOS or can be induced by environmental factors (that exert oxidative stress). Accordingly, PMD can only be inherited, in contrast to SMD, which can be either inherited or acquired [9]. Nevertheless, we cannot rule out a possibility that PMD and SMD may affect a person at the same time; and even by neoteric testing, it is challenging to differentiate between the two.
Mitochondrial disorders differ in their genetic etiology and clinical
manifestation [10] and contribute to the development of age-, stage-, and
stress-related diseases by altering complex cellular and physiological functions
[11]. Pathological variants in the mitochondrial genome induce many syndromes and
disorders [10], and a deviation of the mtDNA copy number beyond a certain
threshold may result in mitochondrial dysfunction followed by cellular, tissue,
and eventually organ impairment. For instance, Grady et al. [12] have
demonstrated that in case of pathogenic mutation m.3243A
Among frequent mtDNA mutations, the most common large deletion, 4977 bp in length, is of particular interest; it accounts for up to 30% of all mtDNA deletions and causes many syndromes [13]. Deletions are the most functionally significant mutations because they can lead to the loss of an entire protein and to serious disturbances in the energy function of the cell. Detection of deletions is one of possible screening methods for diagnosing mitochondrial abnormalities. The mtDNA copy number is considered one of markers of mitochondrial functioning; it depends on the number of mitochondrial genomes per mitochondrion and on the number of mitochondria per cell [14]. Thus, clinically significant events in the mitochondrial genome are a mutation in an mtDNA gene and a change in the number of mitochondria per cell (which correlates with changes of the mtDNA copy number).
The genetic approach has apparently surpassed biochemical and histological approaches in popularity thereby transforming diagnostics of diseases associated with mitochondrial dysfunction. Therefore, it is essential to tackle this important topic, which is actually congruent with the concept of personalized medicine. In this opinion article, we propose a novel molecular-genetic approach (described in detail in section 3 “The Proposed Approach”) to the monitoring of mitochondrial function during treatment of patients with certain diseases affecting mitochondria’s state and consequently their function. This approach involves a precise quantitative polymerase chain reaction (qPCR)-based method for monitoring a current mitochondrial state in the course of treatment of certain conditions so that physicians could rely not only on signs and symptoms but also on the molecular-genetic basis of that condition. It is our aim to draw the reader’s attention to this advanced approach which—when introduced into clinical practice—may improve diagnostic accuracy and clinical management of affected patients.
The advent of mitochondrial medicine helps to deal with a range of mitochondria-related diseases (including vascular diseases) at the organelle level, and molecular-genetic technologies may bring about advancements in molecular-level monitoring of dynamics of mitochondrial improvement during treatment.
Current treatments (including physical therapies) of mitochondrial diseases are mostly symptomatic and supportive, often involving a cocktail of antioxidants, vitamins, amino acids, and other bioactive compounds and auxiliary factors in combination with antipsychotic/antiepileptic drugs that have not been standardized; the treatments of mitochondrial diseases have a limited evidence base for clinical efficacy and actually have had limited success [15, 16, 17]. All this leads to suboptimal control of disease progression [15], even though new treatments are being developed, and the number of clinical trials in mitochondrial disorders is increasing, as is awareness of mitochondrial involvement in more common diseases [16]. Another problem with clinical guidelines is lack of high-quality evidence and screening for drug-induced mitochondrial toxicity [18]. Thus, proper monitoring of mitochondrial function for diagnosis, continuous treatment response analysis, and safety testing in patients may bring considerable benefits. In this regard, finding quantifiable disease-specific biomarkers is necessary for rational treatment strategies [19].
It is not always easy to accurately diagnose mitochondrial diseases because of the absence of pathognomonic tests. Tests of oxidative metabolism or respiratory chain enzymatic activities may confirm a diagnosis but are not sensitive enough, and the methods used are not consistent among laboratories [4]. Quite a few strategies for clinical diagnosis of mitochondrial diseases have been outlined [20]. For diagnostic purposes, different types of tissues (such as blood, the buccal epithelium, or muscles) [21] are being employed to detect mitochondrial diseases. Using muscle biopsies and metabolic markers to quantify mitochondrial proteins has turned out to be nonspecific or low specific. For instance, although fibroblast growth factor 21 (FGF-21) has been considered a biomarker of muscle-manifesting mitochondrial deficiencies [22], after some medical treatments, it may be upregulated, and its serum concentrations will no longer reflect a muscle treatment response [4]. A more comprehensive analysis should not be conducted within a muscle biopsy sample alone but should include a clinical examination accompanied by tests for serological biomarkers and by a genetic analysis [4]. Considering the advancements in genetic tests, Gharti et al. [4] have suggested that mtDNA mutation and heteroplasmy as genotypic endpoints (especially when several endpoints are examined simultaneously) are promising indicators of mitochondrial diseases and may be the most helpful for diagnostics.
Herein, for clinicians managing patients who need accurate monitoring of treatment responses to prove convalescence, we propose a trailblazing method for monitoring the state (and consequently function) of mitochondria via assessment of the quantitative and structural characteristics of mtDNA. The invention can be used to identify structural rearrangements (the proportion of mtDNA copies carrying deletions within the common large 4977 bp deletion-prone region) and to evaluate a relative level (copy number) of mtDNA in biological samples [23]. The method includes DNA extraction from samples and simultaneous amplification of the appropriate DNA regions in triplex mode by qPCR, followed by determination of starting quantities of selected DNA fragments: “D-loop”, “ND4”, and “LINE-1”. “D-loop” is located in the control region of the mitochondrial genome where large deletions are rare; “ND4” lies within the major arc of the mitochondrial genome where large deletions are common, including the “common” 4977 bp deletion [13]. The product of the MT-ND4 gene is a part of mitochondrial respiratory chain complex I and is involved in its assembly; MT-ND4 participates in the implementation of a proton gradient used by complex V to produce most of cellular ATP [24]. Deletions in MT-ND4 and other mitochondrial genes (such as neighboring MT-ATP6 and MT-ATP8) within the large 4977 bp deletion region result in a spectrum of clinical phenotypes related to an impairment of ATP synthesis [25]. LINE-1 stands for long interspersed nuclear element 1, which is used here for normalizing the mitochondrial genome data to the nuclear genome’s DNA. In the method, in each patient’s sample, mtDNA fragments “displacement loop (D-loop)” and “ND4” and nDNA fragment “LINE-1” are simultaneously amplified and detected via fluorescein amidite (FAM), hexachloro-fluorescein (HEX), and rhodamine (ROX) fluorescence channels, respectively; starting amounts of these DNA fragments are determined, then the relative amount of mtDNA is calculated using the [D-loop]/[LINE-1] ratio, after which the proportion of mtDNA copies containing deletions in total mtDNA is determined according to the formula: 1 – [ND4]/[D-loop] [23]. There is a feature related to primers for “D-loop” (which was chosen because of the rarity of large deletions in this region [13]), which are used for amplification and subsequent quantification of mtDNA. The D-loop in the control region of mtDNA regulates replication and transcription of mitochondrial genes and is considered an important noncoding yet highly polymorphic region in mtDNA; D-loop includes two hypervariable segments: hypervariable segments I and II (HVI and HVII) [26]. Although our fragment “D-loop” spanning mtDNA positions 263–338 only partially overlaps with one of those hypervariable regions, namely HVII, any mutations within this locus will not drastically affect assay results (in terms of “D-loop” quantification). This is because we compare the copy number/nondel rate within one person, and therefore we actually ignore interindividual differences. It should be noted that the molecular-genetic tool for the proposed approach is valid for monitoring dynamics of mitochondrial function improvement during treatment of diseases associated with mitochondrial dysfunction that is relevant to the mtDNA copy number and/or deletions affecting the ND4 region being analyzed (see Fig. 1 for a schematic explanation of the proposed approach).
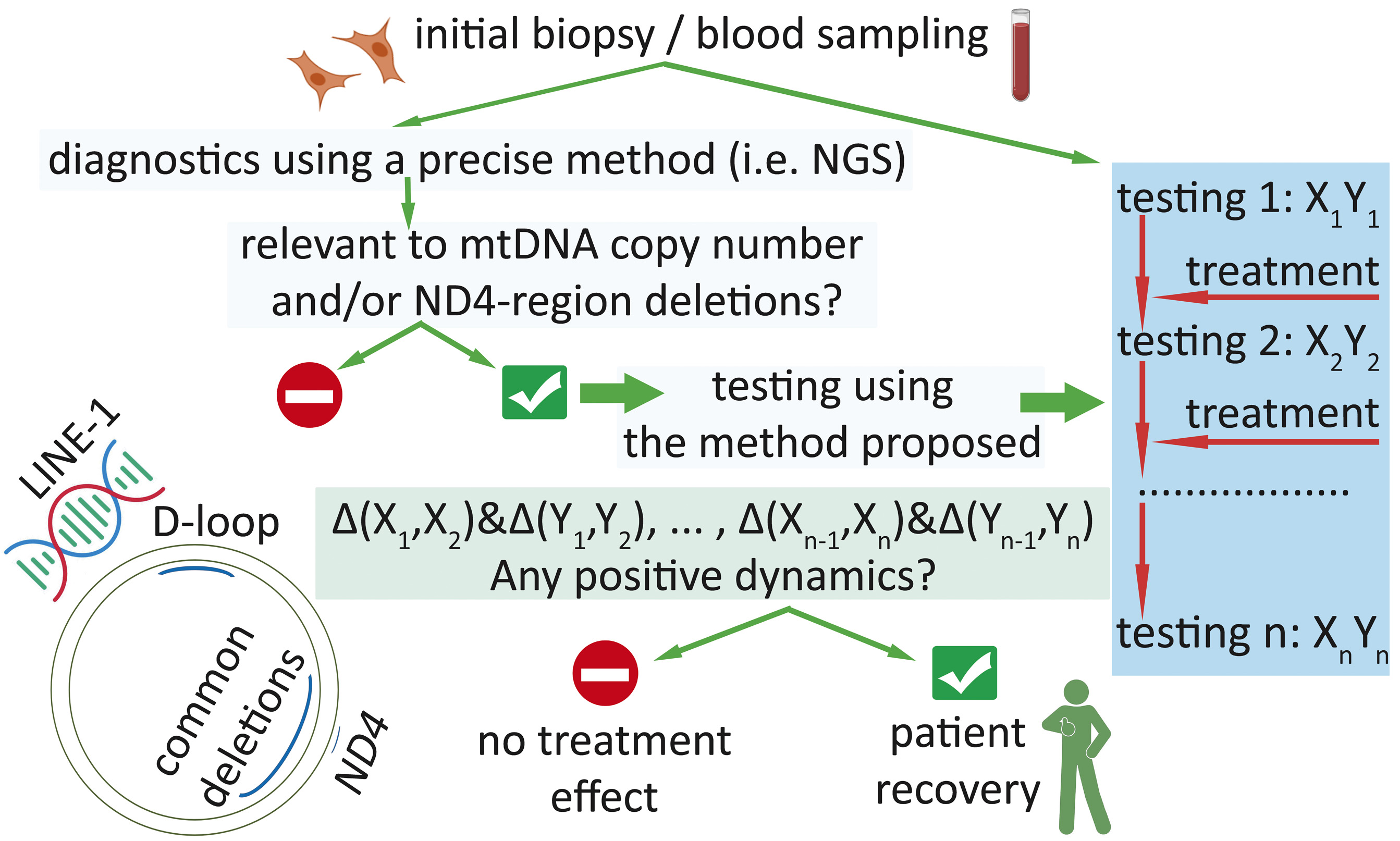 Fig. 1.
Fig. 1.
Schematic representation of the proposed approach. “D-loop”
denotes a segment of the control region of mtDNA (where large deletions are
rare); “ND4” means an mtDNA region in the MT-ND4 gene (where large
deletions are common); “LINE-1” stands for long interspersed nuclear element 1
of the nuclear genome. “Xn” denotes the mtDNA copy number (determined using the
formula [D-loop]/[LINE-1]) during the nth testing; “Yn” stands for the
mtDNA integrity level (determined via the formula [ND4]/[D-loop]) during the
nth testing; “testing 1” means initial testing (after initial biopsy
sampling) with the proposed method; as a result, “X1Y1” data are obtained;
“testing n” means the nth testing (after biopsy sampling n, during the
nth period of the treatment course) with the proposed method; as a result,
“XnYn” data are obtained. “
For diseases associated with mitochondrial dysfunction, the course of treatment is usually long and can be divided into periods during which the attending physician can modify treatment (for example, by adding/deleting/substituting medications or procedures). After each of these periods, it would be prudent to schedule another testing of the patient (by the described method) to monitor the patient’s condition. If, after each subsequent period of treatment, stable positive dynamics are observed, then the physician can conclude that the treatment is effective. Ultimately, the patient can be discharged or remain on supportive care.
The large copy number of our reference DNA fragment (LINE-1 from the human nuclear genome) reduces the contribution of DNA degradation to the assay results. Furthermore, because the amounts of LINE-1 and mtDNA are comparable and abundant within the cell, the bias within the analysis results is minimized, and we can test samples containing a low input amount of genetic material. Nonetheless, there is an open question which type of biological samples is the most appropriate for this analysis. Another important feature of this method is rapidness of the procedures, in contrast, e.g., to sequencing-based techniques, which usually require time-consuming analysis of experimental data and special skills of the personnel.
The newly developed diagnostic system has been successfully tested on DNA samples isolated from vein segments and successfully determines quantitative (in terms of mtDNA/nDNA) and structural (in terms of common deletions in the MT-ND4 gene region) characteristics of mtDNA in varicose veins [7]. It should be emphasized that in the work just cited, while using the same multiplex diagnostic system of primers and a probe, we employed the droplet digital PCR methodology instead of qPCR, for more accurate detection of mtDNA deletions (in terms of assay sensitivity and measurement precision) [7]. This method enabled us to catch tiny differences (as small as 2%) in heteroplasmy levels between vein tissue samples. The proposed method is quantitative. Many other techniques currently used for mitochondrial function assessment appear to be quite laborious, time consuming, and costly. Moreover, many of the tested biochemical parameters can be easily affected by environmental factors or by comorbidities (therefore occasionally causing false positive or false negative results [27]) and, in our opinion, ought to be considered secondary endpoints (and indirect metrics) rather than primary endpoints. Nevertheless, this mtDNA analysis is close to direct evaluation of mitochondrial function.
A question may arise: To which cells and tissues is this approach applicable? Indeed, there are differences in the mtDNA copy number/proportion of deletion-containing mtDNA copies among cell types. In our previous work, we demonstrated the validity of this method on vein samples [7]; venous walls predominantly consist of endothelial cells (t. intima), smooth muscle cells (t. media), and fibroblasts (t. adventitia). In our opinion, there is no limitation regarding the measurement of the mtDNA copy number: any tissue is suitable for extraction of mtDNA because it is present in all (even non-nucleated) cells of the body. Regarding another parameter, it has been demonstrated that common mtDNA deletions occur more frequently and more abundantly in highly energy-consuming human tissues and increase with age [28]. For instance, one deletion is detectable in blood [29], muscle fibers [30], adipose tissue [31], vastus lateralis and deltoid muscle biopsies (as well as brain and fibroblasts) [32], muscle and liver tissues but not in testes (until the age of 60 years) [28]. Accordingly, in order to monitor a disease’s status, it is important to take into account the accessibility of the sampling and to choose biological samples most appropriate for the analysis: this open question about the type of tissue remains to be resolved. Preferably, these should be samples from affected tissues or those in the immediate vicinity or from more accessible tissues (such as blood or the buccal epithelium), provided that changes in the mitochondrial parameters being evaluated are relevant to a target cell/tissue type. Therefore, the limitation of this methodology should be mentioned again: it is not applicable to all mitochondrial dysfunctions. This approach is applicable to mitochondria-related pathologies relevant to the mtDNA copy number and/or ND4 region deletions (within the common large 4977 bp deletion region).
A clinical diagnostic molecular-genetic approach is proposed for monitoring the state of mitochondria (which reflects their function) for continuous treatment response analysis in order to prove a patient’s convalescence by means of not only signs and symptoms but also the molecular-genetic basis of the current condition. The method can be used for primary mtDNA screening as well. Advantages of this diagnostic approach include its low cost (compared to various types of sequencing), an ability to test samples with a low input amount of genetic material, and speed. Even though this is a feasible option, some challenges remain to be overcome. We believe this information will help to develop evidence-based clinical care protocols for future diagnostics and theranostic management of human mitochondria-related pathologies. All in all, this eventually is intended to maintain a healthier population.
ADP, adenosine diphosphate; ATP, adenosine triphosphate; bp, base pair; FAM, fluorescein amidite; HEX, hexachloro-fluorescein; LINE, long interspersed nuclear elements; mtDNA, mitochondrial DNA; nDNA, nuclear DNA; OXPHOS, oxidative phosphorylation; PMD, primary mitochondrial diseases; qPCR, quantitative polymerase chain reaction; ROX, rhodamine; SMD, secondary mitochondrial dysfunction.
MAS and MLF designed the study. MAS performed literature data acquisition and analysis. VAK performed visualization and formal analysis. MAS wrote the manuscript. MAS and MLF provided administrative, technical, and material support. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
Not applicable.
Not applicable.
This research was funded by the Program of Fundamental Scientific Research of the Russian Federation (PFSR RF) “Fundamental Basics of Health Preservation”, grant number 121031300045-2.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.