






1 Immunobiology Laboratory, Institute of Science, Department of Zoology, Banaras Hindu University, 221005 Varanasi, India
Abstract
The tumor microenvironment plays a critical role in modulating immune responses
associated with tumorigenesis, tumor progression, and metastasis. Dendritic cells
(DC) play a key role in preventing and progression of metastatic neoplasia by
driving and restoring dysfunctional immune systems and obliterating
immunosuppression, thus obstructing tumor evasion. In this review, we will
discuss the functions of tumor-infiltrating DC in anti-tumor resistance,
prevention of tumor recurrence, and immunosuppression. We will also describe DC
metabolism, differentiation, and plasticity, which are essential for its
function. Cancers like Lymphomas may be able to corrupt immune surveillance by
reducing natural killer cell numbers. Thus, interactions between lymphoma and DC with
reference to cytotoxicity may be an important event, likely to be mediated via
activation with interferon-
Keywords
- cancer
- dendritic cells
- cytotoxicity
- effector functions
- immunosuppression
- metastasis
- anti-cancer drugs
- combination therapy
- tumor microenvironment
- cross-priming
- regulatory T cells
Dendritic cells (DC) represent a diverse group of lymphocytes considered specialized antigen-presenting cells (APC) having critical roles in innate immunity and initiation and regulation of adaptive immune responses [1, 2]. Modulating DC functions is one of the sought-after strategies to improve cancer immunotherapy. DC is considered as professional APC and consists of a variety of cell subsets, which are either residents in organs or migrating among the non-lymphoid and lymphoid organs. Subsets of DCs differ in morphology, ontogeny, surface phenotype, functions, and key transcription factors, which are important for their functions. In a steady state condition, DCs process and present antigens on class I and II major histocompatibility complexes (MHC). DC comprises three major subsets, which include plasmacytoid (pDC), type-1, and type-2 conventional DC (cDC1 and cDC2), respectively [3]. Other DC subtypes are inflammatory monocyte-derived DCs (MODCs) and Langerhans cells (LCs). DC is also present in the vicinity of tumors, where they are exposed to tumor-associated antigens (TAAs) and then migrate to draining lymph nodes in order to present these antigens to lymphocytes (CD8+ or CD4+ T cells) [4, 5, 6]. Cross-presentation of antigens by cDC1s contributes to the priming of tumor-associated antigens (TAA) specific cytotoxic T cells [7]. cDC1 also supports T helper 1 (Th1) cell polarization from naive CD4+ T cells [8, 9]. cDC2 comprises a heterogeneous population and preferentially prime naïve CD4+ T cells for Th2 or Th17 polarization [10, 11, 12]. Little is known about the role of the cDC2 subset in tumor immunity. DC efficiently cross-presents TAA in order to prime tumor antigen-responsive CD8+ T cells for controlling tumor growth [13, 14, 15, 16], and thus, it makes DC-based vaccines a significant therapeutic strategy to potentiate effector and memory anti-tumor CD8+ T cell responses for cancer immunotherapy [4, 17, 18, 19]. Despite being a rare population, several studies have documented the heterogeneous nature of DCs with functional flexibility [2].
Among the professional APCs, DCs have potential in terms of migration and
priming of T cells compared with monocytes/macrophages and B lymphocytes. DC
regularly surveys peripheral tissues for antigens, which are captured and
processed, followed by migration to lymphoid organs and deliver the antigens to T
lymphocytes [20, 21, 22]. The existence of danger signals from pathogens, etc.,
induces maturation of the DCs, which leads to the T cell activation and
polarization. Immature DCs, unlike their mature counterparts, induce tolerance
[23]. Semi-mature DCs stimulated by cytokine interleukin-10 (IL-10) or
transforming growth factor-beta (TGF-
Multiple strategies have been explored by targeting DC functions in cancer immunotherapies. There are approximately four approaches, which include protein and nucleic acid-based vaccines; endogenous DCs targeting antigens; tumor antigens loaded and matured ex vivo generated DCs; and reprogrammed endogenous DC using biomaterial-based platforms for in situ recruitment to tumor [24, 25]. Clinical trials performed with DC-based anti-cancer vaccines commonly depend on the use of ex vivo generated and differentiated DCs from leukapheresis enriched from CD14+ monocytes following culture in granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 [26]. Although promising with good safety and ingenuity, the therapeutic efficacy of cell therapy is only limited to less than 20% of the patients [26, 27]. The occurrence of immune suppression imposes an increasing burden of tumor antigens plus regulatory factors in mid to late-stage cancer, which may limit the efficacy of monocyte-derived DC [28, 29]. The underperformance of monocyte-derived DC involves a lack of migration from the injection site to the lymph nodes and an inability to induce strong tumor-specific cytotoxic T lymphocyte (CTL) responses [30, 31, 32, 33, 34]. The effector functions of DC include direct cytotoxicity against the tumor cells, which influence immunity and tolerance in neoplastic conditions. In this review, we will describe how different DC subsets induce and influence tumoricidal activity, including cytotoxicity and growth inhibition, and discuss their implications for established cancer treatments (chemotherapy) and novel immunotherapeutic strategies.
Origin and differentiation of DCs from blood or splenic monocytes and
macrophages constituted the major focal areas of studying DC biology. Monocytes
are reported to differentiate in vitro into DCs upon stimulation with
GM-CSF, tumor necrosis factor-
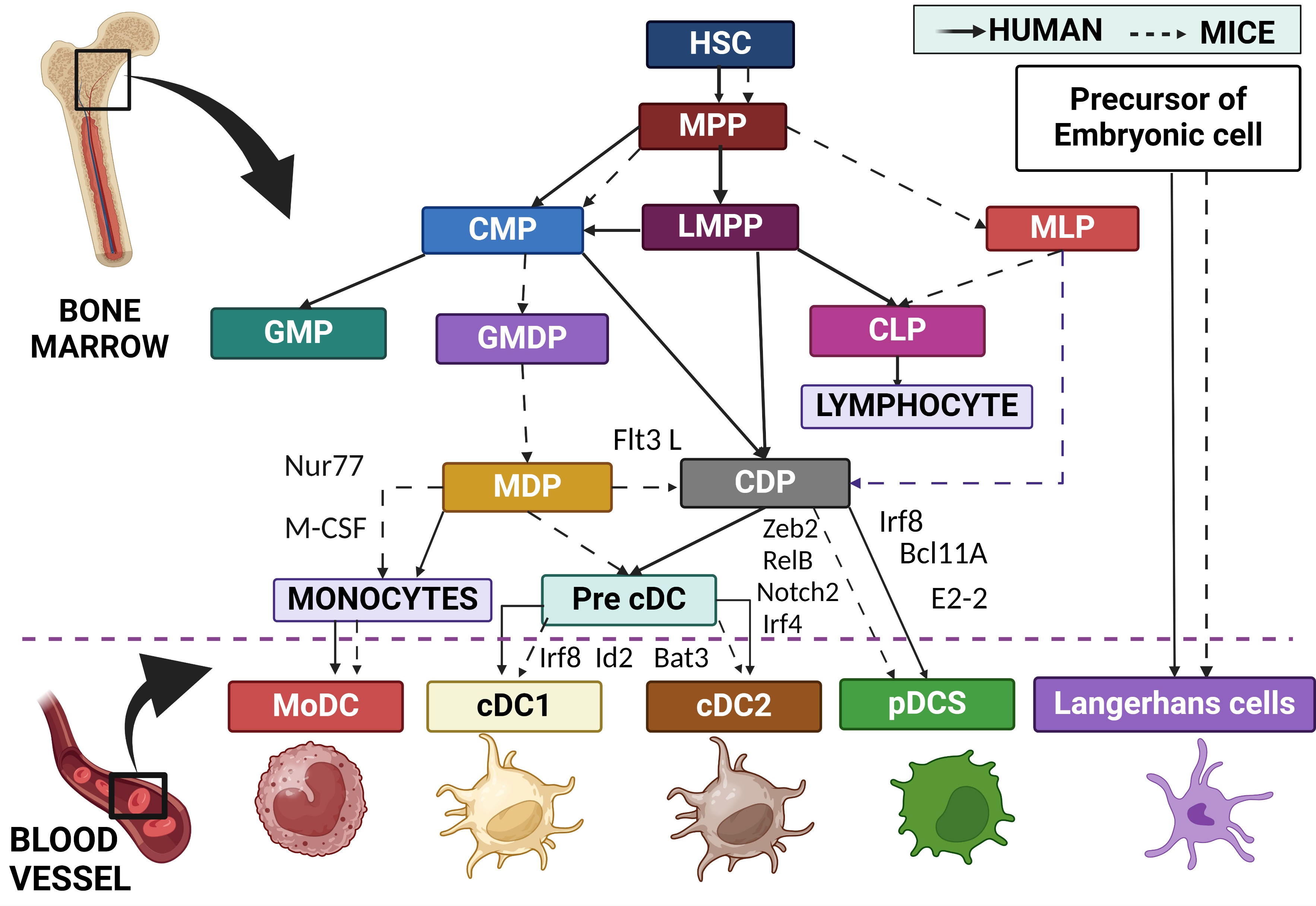 Fig. 1.
Fig. 1.
Origin and development of DC subtypes in mice and humans as indicated with acronyms depicted. DC, Dendritic cells; HSC, Hematopoietic stem cell; MPP, Multipotent progenitor; CMP, Common myeloid progenitor; LMPP, Lympho-myeloid primed progenitor; MLP, Multipotent lymphoid progenitor; GMP, Granulocyte and monocyte progenitor; GMDP, Granulocyte, monocyte DC Progenitor; CLP, Common lymphoid progenitor; MDP, Macrophage and DC progenitor; CDP, Common or conventional DC progenitor; Pre-cDC, pre-Conventional DC; Nurr77, Nuclear receptor NR4A1; M-CSF, Macrophage colony-stimulating factor; Irf8, Interferon regulatory factor 8; Id2, Inhibitor of DNA Binding 2; Zeb2, Zinc finger E-box-binding homeobox 2; Irf4, Interferon regulatory factor 4; Notch 2, Neurogenic locus notch homolog protein 2; Bcl11A, Transcription factor B-cell lymphoma/leukemia 11A. Created with BioRender.com.
Exposure to antigen matures DCs and upregulates the expression of MHC and co-stimulatory molecules (CD80/86), cytokines, and chemokines receptors, including CCR7 [51]. Mature DCs migrate to regional lymphoid tissues via CCR7 and activate naïve T lymphocytes and enable them to differentiate into effector T cells, including T helper (Th) 1 cells, Th2 cells, Th17 cells, T follicular helper (TFH) cells, regulatory T (Treg) cells, and CD8+ CTLs, resulting in T-cell responses [51, 52]. Experimental evidence has revealed that DCs are heterogeneous, and there are different DC subsets that are specialized in priming different types of effector T lymphocytes and skew effector response accordingly [53]. Four major subsets of DC are recognized, namely, conventional DCs (cDCs), plasmacytoid DCs (pDCs), monocyte-derived DCs (MODCs), and Langerhans cells (LCs). cDCs are further subdivided into cDC1s and cDC2s. Analogous subsets have been identified in humans and mice [4, 53, 54]. The phenotypic and functional characteristics of these DC subsets are summarized in Table 1 (Ref. [55, 56, 57]).
| Dendritic Cell Subsets | Conventional type 1 dendritic cells (cDC1) | cDC2 | Plasmacytoid Dendritic Cells (pDC) | Monocyte derived dendritic cells (MoDC) | Langerhans Cells (LC) | References |
| Surface marker for Mice | CD11c | CD11c | B220 | Fc |
CD11c | [55, 56, 57] |
| CD205 | MHCII | PDCA1 | CD14 | NHCII | ||
| CD207 (Langerin) | CD172 |
Siglec-H | FcεRI | CD207 | ||
| CD24 | CD11b | Ly6C | CD11b | CD326 | ||
| CD8 |
TLR1/TLR6 | TLR7/TLR9 | CD172 (SIRP |
CD11b | ||
| CD103 (Tissue) | CD301b (Tissue) | CCR9 | CD206 | CX3CR1 | ||
| Clec9A | CD317 | CD88 | ||||
| MHCII | Bst2 | MerTK | ||||
| TLR3/TLR8 | MHCII | Ly6C | ||||
| XCR1 | CD11clow | CD64 | ||||
| CD11c | ||||||
| MHCII | ||||||
| CD209 | ||||||
| CCR2 | ||||||
| Surface Marker for Human | CD141 (BDCA3) | CD11c | CD123 | CD11c | CD11c low | [55, 56, 57] |
| CD11c | human leukocyte antigen DR (HLA-DR) | CD303 (BDCA2) | HLA-DR | HLA-DR | ||
| HLA-DR | CD172 |
CD304 (BDCA4) | CD1c | CD207 | ||
| XCR1 | CD1c(BDCA1) | HLA-DR | CD14 | CD326 | ||
| Clec9A | CD11b | B220 | CD206 | CD11b | ||
| SIRP |
Bst2 | CD11b | CD172 |
|||
| CD301a | CXCR3 | CD64 | CX3CR1 | |||
| IRF4 | CD209 | CD1a | ||||
| CCR2 | CD1c | |||||
| Fc |
||||||
| CD14 | ||||||
| FcεRI | ||||||
| CD1a/CD1c | ||||||
| CD172 |
||||||
| Significant Characteristics | IL 12 initiation, The cytotoxic responses, and Cross-presentation | T helper 1 (Th1), Th2 and Th17 reactions. Assists CD4 anti-tumor immunity. | IFN- Interferon secretion. | Functions of regulation in a stable state. Modulate the immune system against certain experimental tumor model | Sustain skin homeostasis via mediating tolerance. Cancer vaccinations. Elicit anti-tumor immunity | [55, 56, 57] |
| Antiviral immunity. | ||||||
| Direct apoptosis of tumor cells | ||||||
| Enhanced chemotaxis towards CCL21. | ||||||
| Initiate IL10+, IL22+, IL4+ | ||||||
| T cell polarization. | ||||||
| Enhanced MHC-II expression |
Fc
Both cDC1s and cDC2s are formed from the common DC precursors; however, they
possess different functions. cDC1s play an important role in antigen-specific
immune responses against intracellular pathogens and induce CD8+ T cell
responses via MHC class I [4, 53, 54, 58]. Batf3 deficiency in mice causes the
elimination of CD103+ cDC1s in the intestine, lung, mesenteric lymph nodes,
skin, and skin-draining lymph nodes and thus reduces CD8+ CTL responses
[59, 60]. cDC1s are responsible for the cross-presentation of extracellular
antigens to CD8+ T lymphocytes besides other DC subsets [61, 62, 63].
CD8
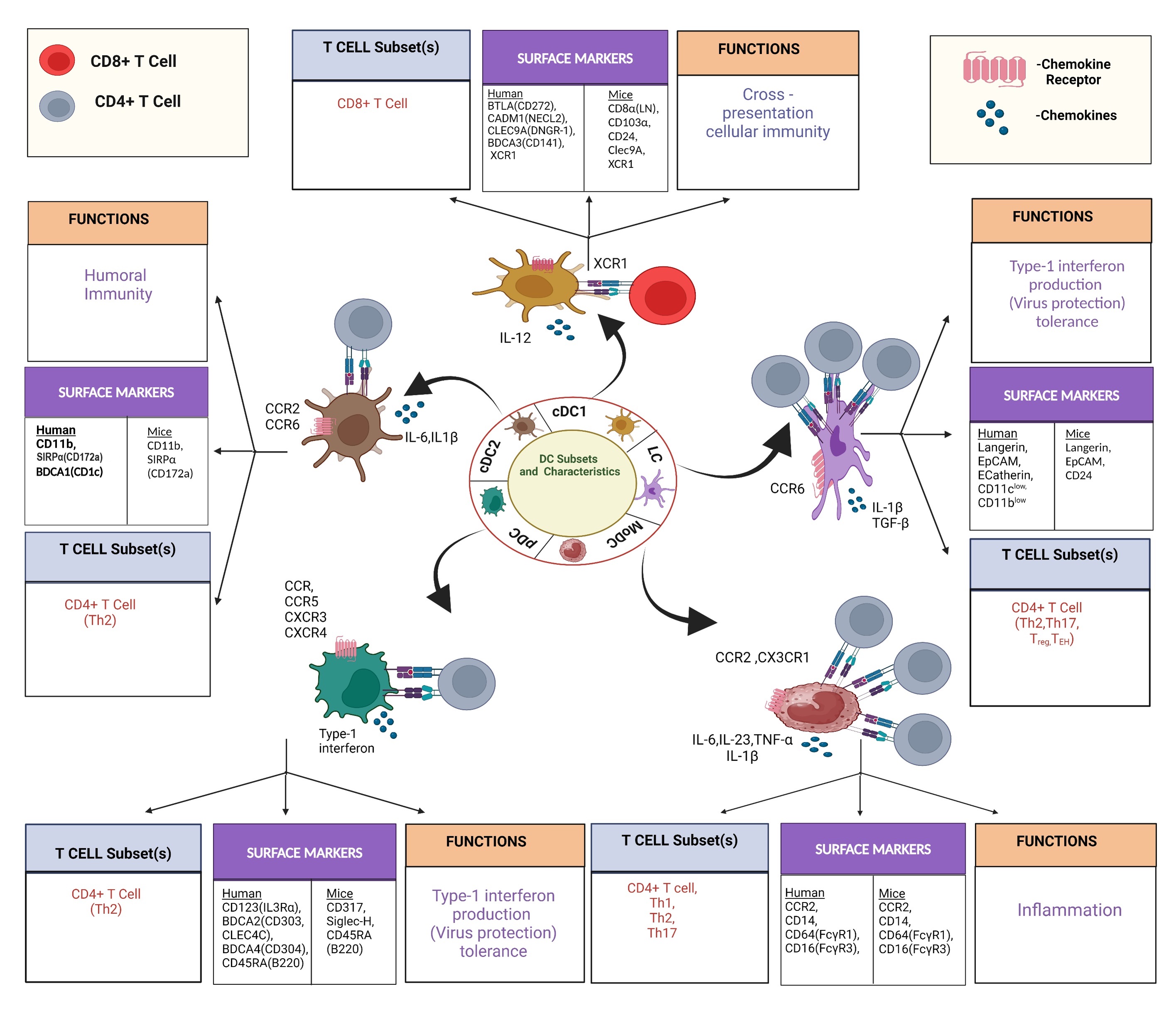 Fig. 2.
Fig. 2.
DC subtypes with cellular interaction involving DC and other
immune cells with cytokine and chemokine involvement in DC functions.
Participation of major T cell subsets and the functions plus expression of
surface markers of individual DC subtypes is presented. IL, cytokine interleukin; CCR, Chemokine Receptor; TGF, transforming
growth factor-beta; Th 1, T helper 1;
BTLA, B and T cell lymphocyte attenuator; CADM, Cell Adhesion Molecule; NECL, Nectin like protein; SIRP
cDC2s sdubset are more abundant in blood and lymphoid tissue and are
heterogeneous in their phenotype and functions. Murine cDC2s express high CD11
band signal regulatory protein alpha SIRP
Plasmacytoid DC (pDC) is derived from common DC precursors and also from common lymphoid progenitors [81]. pDCs express lymphocyte marker B220, but not the myeloid markers CD11b and CD33 [23]. PDCs rapidly induce antiviral immune response and produce significant amounts of type I and III IFNs via TLR-mediated activation in viral infection. They are also poor in antigen-presenting capacity to I T lymphocytes [82]. pDCs in mice can be identified as CD11c intermediate, Ly6C+, B220+, Bst2 (PDCA1, CD317)+, and Siglec H+ cells, and the human counterpart express human leukocyte antigen DR (HLA-DR), B220, CD303 (BDCA2), CD304 (BDCA4), CD123 (IL-3R) without CD11c [82, 83, 84]. pDCs also develop from common lymphoid progenitors (CLP) and are differentiated from IL-7R+ lymphoid progenitors, which also differentiate into B cells [85, 86]. IL-7R+ lymphoid progenitors constitute the majority of murine mature BM and splenic pDCs [86] (Fig. 2).
pDCs cooperate with cDCs to induce optimal cross-priming and CD8+ T CD8 T
cell immunity under different settings [87, 88], indicating that pDCs transfer
antigens to cDCs and provide an explanation for the observed synergy [89]. pDCs
are found to be tolerogenic and perform suppressive functions as they accumulate
in multiple types of cancers like head and neck, ovarian, breast, and melanoma
with poor prognosis [82, 90, 91, 92]. pDCs induced the expansion of T regulatory cells
(Tregs) through inducible T cell co-stimulatory ligands (ICOSL) [93]. Human and
murine pDCs kill cancer cells directly through tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) and/or granzyme B-dependent mechanisms [94, 95].
pDCs may also induce anti-tumor immunity against human colon cancer [96];
activation of pDCs by TLR ligands induces anti-tumor immunity in multiple
clinical trials, indicating the role of pDCs for cancer immunotherapy [97, 98, 99, 100].
OX40+ pDC induces potent anti-tumor CD8+ T cell function in synergy with
cDC1s subset [101]. pDCs are also reported to be associated with poor prognosis
in breast and ovarian cancer by promoting expansion and activation of Treg cells
via ICOSL expression [102, 103]. pDCs activated by TLR7 ligands in mouse models of
breast cancer exhibit anti-tumor properties and suppress tumor growth and
proliferation in vivo [104]. TLR7 ligand-activated CD8
Circulating blood monocytes under the inflammatory settings differentiated into inflammatory DC populations and are designated as monocyte-derived DC (MODCs) [105]. MODCs are a highly heterogeneous group of cells and share markers with monocytes, macrophages, and cDC2s. Unlike pDC and cDC, MODCs differentiated from Ly6Chi monocytes in inflammatory sites through the CCR2/CCL2 axis, independent of Flt3L [18, 106]. Human MODCs express CD1c+CD11c+HLA-DR+CD14intCD206+ while murine MODCs hasCD11b, CD11c, MHCII, CCR2, and CD209 with an intermediate level of Ly6C and CD64. For in vivo development, MODCs need macrophage colony-stimulating factor (M-CSF) but not FLT3L or GM-CSF and are dependent on IRF4 for differentiation [107]. MODCs are also generated ex vivo from human and mouse blood or bone marrow monocytes in the presence of GM-CSF and/or IL-4 for use in pre-clinical and clinical studies. MODCs play critical roles in anti-tumor immunity [108]. MODCs are inflammatory DC differentiated from monocytes during inflammation and infection, which are recruited in the inflamed zones for the removal of inflammation and then differentiated into macrophages [109]. A subtype of MODCs also includes TNF/iNOS-producing DCs (Tip-DCs) [110]. MODCs share functional similarities with both cDC1s and cDC2s, which include the expression of co-stimulatory molecules and cytokines. Like cDC1 and cDC2, MODC has the ability to present antigens to CD4+ and CD8+ T cells. MODCs promote the differentiation of CD4+ T cells into Th1, Th2, and Th17 cells, as well as their potential to differentiate CD8+ T cells into CTLs [111, 112, 113, 114]. MODC-based DC vaccines have been developed against various cancers, including clinical trials with mixed results [115] (Fig. 2).
LCs are DC subsets present in the skin that have the efficient migratory
potential to drain lymph nodes. They share a common ontogeny with macrophages but
function as DC. Developmentally, the origin of LC is traced to embryonic
macrophage lineage precursors and not to common DC precursors [116]. LCs can also
be formed under inflammatory conditions from circulating monocytes [117]. LCs
share similarities in phenotype and function with cDCs as well as with tissue
macrophages. Like tissue macrophages, LC undergoes self-renewal in the skin
[118, 119]. LCs express langerin/CD207, a C-type lectin that forms the Birbeck
granules, and epithelial cell adhesion molecule (EpCAM) [120, 121] serves as the
first line of immunological defense [122]. LCs present antigens to both CD4+
and CD8+ T lymphocytes and are dispensable for cross-presentation for
induction of CD8+ CTL responses [65]. LCs also efficiently induce Th2, Th17,
and T follicular helper (TFH) responses [123, 124, 125]. In LC-deficient mice, there
are impaired Th2 responses and antibody production, while TH1 responses remain
unaltered [126]. LCs also induce the differentiation of Treg cells [127]. LCs
require transforming growth factor-
DCs in the Tumor Microenvironment (TME) interact with other immune and stromal cells and lymphoid organs,
resulting in induction or inhibition of DC functions and anti-tumor immunity
based on the type of cells encountered. Damage-associated molecular patterns
(DAMPs), predominantly intracellular proteins released from apoptotic dying
cells, induce the generation of cytokines and activation of T lymphocytes [132].
Tumor cells evade IR by following strategies like preventing the infiltration of
cDC1 cells in the TME and thus increasing its chance to survive [72]. In patients
with ovarian and breast cancer, pDC function becomes aberrant with poor
production of type I interferon accompanied by Treg differentiation [133, 134].
Augmentation in infiltration, expansion, and activation of cDC1 control the
tumors and response to immunotherapies via stimulation with NK cells or
intratumoral delivery of XCL1 and sFlt3L, encoded in recombinant Semliki forest
virus-derived vectors [72, 135]. In a recent clinical study, PDL-1 expression was
found to be significantly augmented in DCs located in TME and also in
circulation. PD-L1 blocking relieved B7.1 receptors, which allow it to interact
with CD28 and enhance the priming of T lymphocytes [136]. Anti-PD-1 immunotherapy
depends on DC-derived IL-12 in association with IFN-secreting T cells [137]. DCs
are also essential for the reactivation of circulating T memory lymphocytes
[138]. Wingless-related integration site (WNT)/
Prolong physical interactions of Tregs with DCs, an engagement which is
significantly intense compared with DC-CD8+ T cell association in the TME,
results in upregulation of the immunosuppressive Indoleamine 2,3 dioxygenase (IDO) and reduces the
co-stimulatory molecules on DC [152]. DCs also interact with NK cells, resulting
in the generation of cytokines and chemokines, including XCL1, which allow the
recruitment of cDC1 to the TME [72]. DC-derived cytokines like IL-12,
IL-15/IL-15R
Transcriptomic analysis demonstrated high frequencies of different DC subtypes in the TME compared to non-neoplastic tissue [161, 162]. Several factors are responsible for determining the variability of DC phenotype and its function, which include tumor immunophenotype (hot versus cold), TME characterization, tumor stage, age, gender, histology, treatment history, and schedule. Qualitative and quantitative features specific to each DC subtype have been observed in breast cancer subtypes (“cold” versus “hot” tumors) [149]. The TME harbors ontogenically distinct DC populations with resistant effects on tumor malignancies [163]. Tumor-infiltrating DC states are conserved across solid human cancers as studied by meta-analysis of eight currently available scRNA-seq datasets revealing five different DCs regardless of tissue origin, genetic signatures of cancer cells, or composition of the TME [164]. Transcriptomic analysis in colorectal, lung, ovary, and breast cancers has shown infiltration of alternative cDC2 subtype with a Langerhans-like phenotype (CD1C, CD1A, and CD207), indicating a pan-cancer blueprint of heterogeneous TME [162]. Here, we document the recent status of each subset in relation to cancer.
cDC1 (CD45+CD141+CD8
Enhanced IFN-
CD1c+CD14–cDC2s infiltrate tumors and trigger CD4+ T cell
responses at draining lymph nodes [77, 163]. Together with pDC, cDC2 constitutes
35% of cellular constituents in melanoma. In the TME, cDC2s show higher
expression of CD1A, CD1B, CD1E, CD207, a”d FC”R1A and IRF8 [163, 177, 178]. cDC2
is associated with a positive prognosis and infiltrates abundantly in head and
neck and non-small cell lung cancer (NSCLC) cancer patients with better clinical outcomes [77, 177]. Treg
depletion positively correlates with enhanced cDC2 percentage, aids in the
induction of CD4+ T cell responses, and protects against tumors [77]. cDC2
also induces CTL responses, indicating their potential in DC-based vaccination
besides their role in Th17 activation and secretion of inflammatory cytokines,
including IL-1
In TME, a cell population has been identified with high expression of
TLR9, IL3RA, CLEC4A, GZMB, LILRA4,
IRF7, and TCF4 genes [177, 178]. Following activation with
TLR7/9, pDCs participate in IR via antigen presentation; however, it is weaker
than the cDC subsets [183]. pDCs derived from IFN-
A unique DC type, characterized by the expression of maturation markers (MHC-II,
IL-12, CD40, CD86, PD-L1 and 2) and immunoregulatory markers like CD200, CD274,
and PD-L1 has been documented in human and murine non-small lung cancers [143].
This DC has been named DC3 [164], LAMP3+ DC [195], CCR7+ DC [162], or
BATF3+ DC [167]. High mregDCs are observed inside the tumor lesions and are
positively associated with improved survival in patients with NSCLC and
colorectal cancer [80, 167]. mregDCs in tumors stimulate anti-tumor CD8+CTLs
or NK cells by IL-12, produced upon sensing IFN-
MODCs represent HLA-DR+CD11c+CD14+ in the TME derived from
differentiation from the monocytes [196]. CCL2 neutralization or inhibition of
colony-stimulating factor-1 receptor (CSF-1R) downscale monocyte infiltration in
the lymph node or tumor lesions, reducing the recruitment of tumor-responsive T
lymphocytes and downregulates anti-tumor immune responses [196]. APCs, like
monocytes, are important for the PD1 blockade and act as a therapeutic target for
binary or combination therapy [197]. MODCs in the TME are associated with the
production of IL-15, which promotes Th1 responses [198]. MODC exerts tumoricidal
effect via iNO production, serves as APC, and performs effector functions via the
production of TNF-
DCs are regarded as the sentinels and messengers of the immune system and are universally considered professional APCs, playing a fundamental role in anti-tumor immunity. DCs have the unique ability to acquire, process, and present tumor-derived antigens to T cells, as well as the potential to drive the differentiation of naïve T cells into activated tumor-specific effector cells. DC also engages in crosstalk in NK-T cells, anti-tumoral immunity, and B cell functions. In addition to innate and adaptive immune functions, DCs also functioned as direct cytotoxic effectors, including growth inhibition against various types of tumors. This DC function is less conventional and may be controversial since it goes against the documented origin and functions of DC in addition to induction, regulation, and mechanisms of tumoricidal functions. In recent times, these unconventional functions and another face of DC, in addition to their capability of antigen presentation, have become the subject of intensive research [203] (Fig. 3).
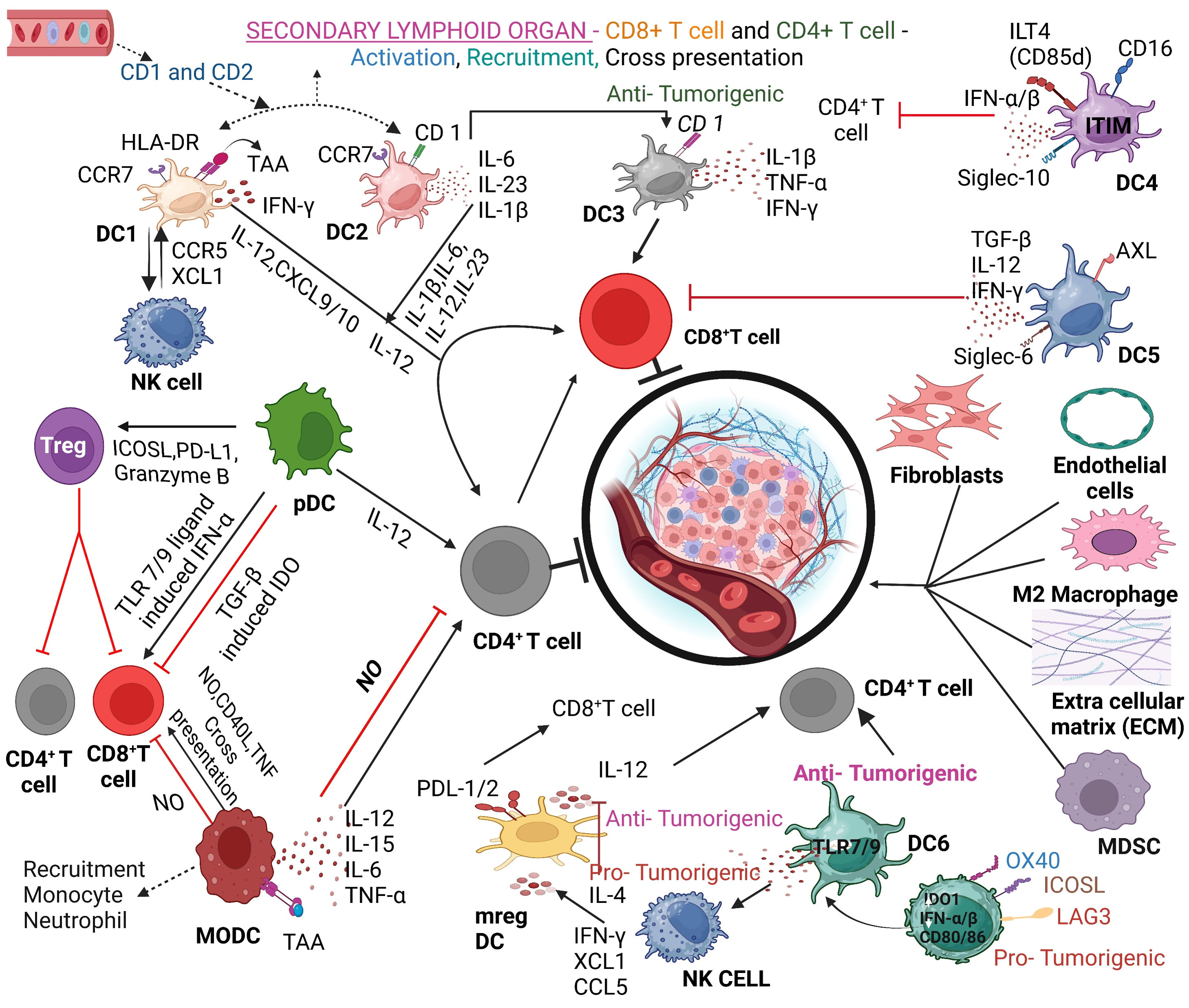 Fig. 3.
Fig. 3.
DC and tumor microenvironment involving participation of DC subtypes and cellular interactions. DC subtypes and their crosstalk with cells in the tumor microenvironment (TME) are presented in the context of cytokine and chemokine-induced intervention of various functions presented. TAA, tumor-associated antigen; NK, natural killer; MDSC, myeloid-derived suppressor cell; ITIM, immunoreceptor tyrosine-based inhibition motif; AXL, a receptor tyrosine kinase; ICOSL, Inducible T Cell Costimulator Ligand; PD-L1, programmed death-Ligand 1; TAA, tryptophan aminotransferase; ICOSL, Inducible T Cell Costimulator Ligand; LAG3, Lymphocyte activation gene 3 protein. OX40 is a type of tumor necrosis factor (TNF) receptor, also called CD134. Created with BioRender.com.
MODCs are the widely studied cell type for demonstrating the functions of human
DCs ex vivo (Table 1). Several differentiating and maturating agents,
like pattern recognition receptors (PRR), can recognize and occupy antigens in
order to trigger cytotoxic effector functions of human MODC. Cytolytic potential
in these monocytic precursor cells has been reported (Table 1). CD14+ and
CD16+ human monocytes stimulated with type I or II IFN, a ligand for TLR4
(LPS), TLR7, and TLR8 (R848), exert anti-tumor activity against a panel of tumor
cell lines [204, 205, 206] (Table 1). TRAIL was implicated in the direct tumoricidal
activity by human monocytes [204, 207, 208]. IFN-
Killer blood DC has two main subsets, mDCs, and pDCs, which can be cytotoxic. In
humans, the blood mDC subset is characterized as HLADR+CD11c+CD123
(IL-3R
Murine splenic DCs, following stimulation with recombinant IL-15 (rIL-15), express TRAIL, which induces cytotoxicity and growth inhibition against a murine lymphoma called Dalton lymphoma (DL) [228]. DC-mediated effector functions against DL tumor cells occur downstream to STAT3 since inhibition of STAT3 by cucurbitacin I (a selective Janus kinase/STAT3 inhibitor) augments the anti-tumor effects by DC-derived TRAIL. Recombinant IL-15 priming in combination with cucurbitacin I in DL tumor-bearing mice prolonged the survival, which partly restores the TRAIL expression in DCs following its downregulation in DC of untreated animals. In the case of chronic myeloid leukemia (CML), peripheral blood DC-derived TRAIL mediates anti-tumor activity via DR5 and not by DR4 as indicated by the effect of neutralizing anti-DR5 antibody, which reduces DC-mediated cytotoxicity [228] (Fig. 4A).
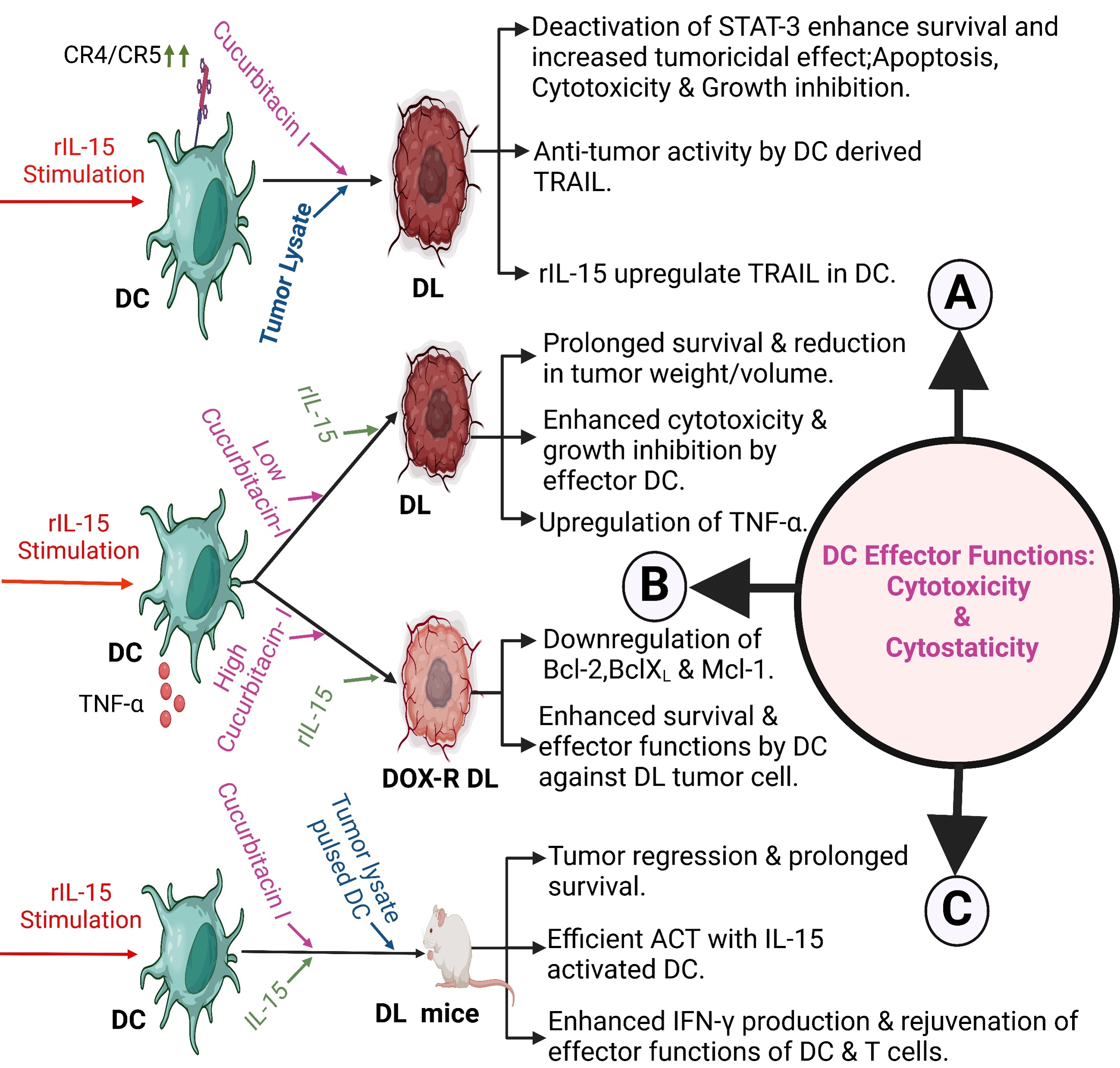 Fig. 4.
Fig. 4.
Effector functions, including cytotoxicity and cytostaticity of
splenic DC against experimental lymphoma. (A) IL-15 stimulation to splenic DC,
in addition to cucurbitacin I treatment, enhances TRAIL-mediated tumoricidal
response against DL lymphoma, inducing cytotoxicity, broad-range growth
inhibition, and tumor reduction, plus increased survival [228]. (B) Assistance by
high-dose cucurbitacin I and rIL-15 enhance cytotoxicity and cytostaticity in
doxorubicin-resistant Dalton Lymphoma by downregulating Mcl-1 Bcl-2 and BclXL [229]. (C) Enhanced cytotoxicity and growth inhibition plus restoration of
DC-mediated adaptive immunity in DL tumor-bearing mice treated with rIL-15
activated DC cucurbitacin I with additional priming with
Splenic DC-derived TNF-
rIL-15 activated killer DCs, cucurbitacin I plusrIL-15 prolongs survival and
cures mice with highly aggressive and metastatic DL lymphomas. It rebuilds
impaired DC functions and restores CD8+ T-cell-mediated immune functions in
vaccinated mice. It also increases TRAIL and TNF-
pDCs are a unique lineage of cells, and they do not express CD3, CD19, CD14,
CD16, CD56, and CD11c but do express multiple signature markers. This includes
blood dendritic cell antigen-2 (BDCA-2, CD303), dendritic cell antigen-4 (BDCA-4,
CD304), immunoglobulin-like transcript7 (ILT7), CD123 and CD4 and are restricted
to bone marrow and in peripheral blood [231]. pDCs induce indirect cytotoxicity
against cancer cells via induction of apoptosis and by anti-angiogenesis via
signaling through a common IFN-
Recent studies on clinical trials of DC-based immunotherapy reveal many novel
aspects of this therapy against cancer. In a phase I trial, in newly diagnosed
and recurrent glioblastoma (GBM) with autologous DC vaccine pulsed with lysate derived from an
allogeneic stem-like cell line, it was found to be safe and well tolerated. This
therapy increased median progression-free survival and overall lifespan for newly
diagnosed and recurrent GBM patients. Besides that, a subset of patients
developed a cytotoxic T-cell response as determined by the production of
IFN-
In acute myeloid leukemia (AML), high rates of relapses can be stabilized using
a combination of GM-CSF and Prostaglandin E1 (Kit-M), which converts myeloid
blasts into DC of leukemic origin. Stimulation with these DC ex vivo
activates anti-leukemic immune cells. This therapy induces leukemia-specific
immunoreactive cells (e.g., non-I, effector, memory, CD3+
DC-NK Cross Talk is at the center stage of immuno-surveillance, where DC
is endowed to activate the cytotoxic potential of NK cells. Cytokines like IL-12,
IL-15, IL-18, and type-I IFN are secreted by activated cDCs and pDC, which
potentiate NK cell proliferation, cytotoxic potential, and production of
IFN-
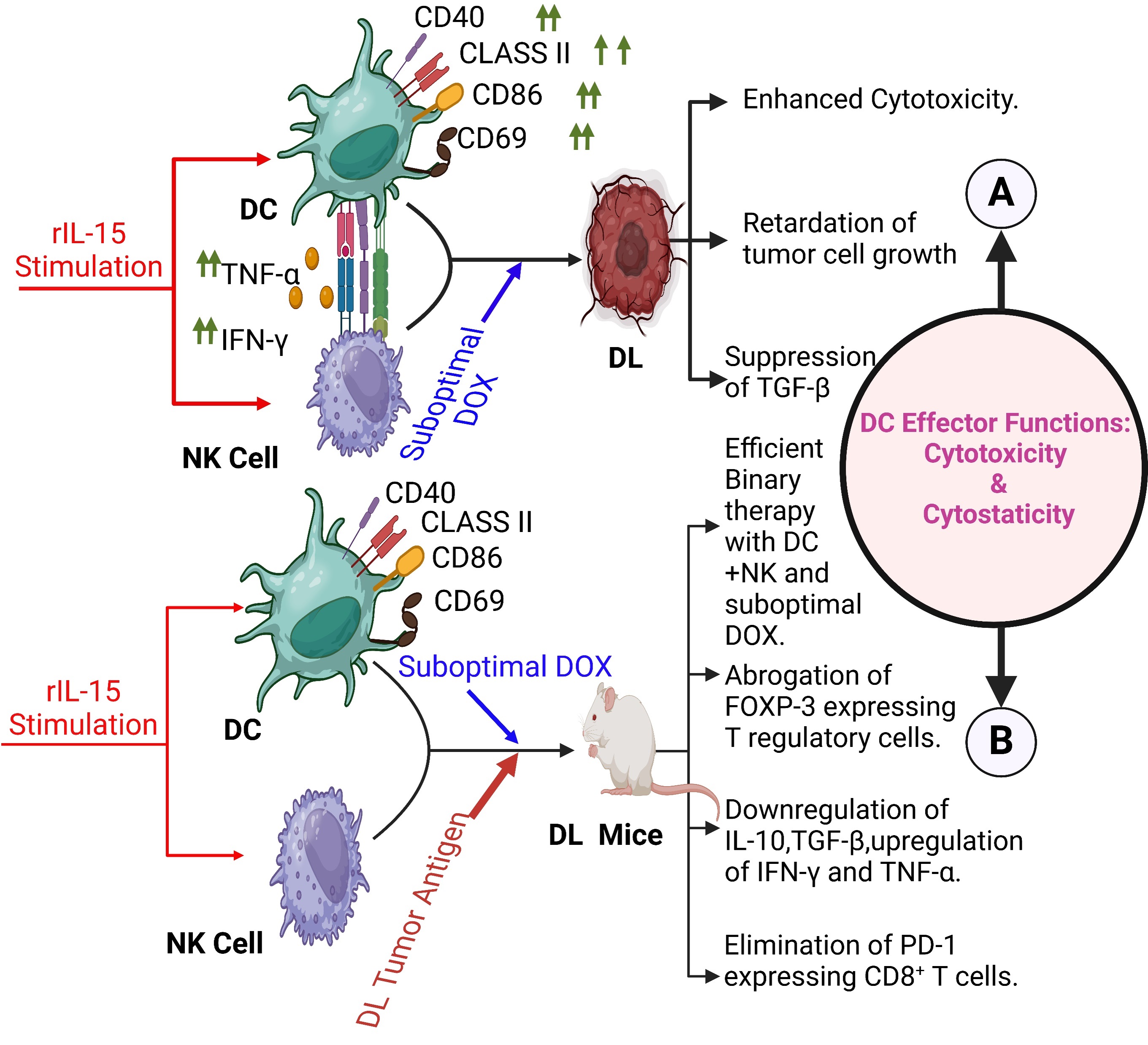 Fig. 5.
Fig. 5.
DC-NK crosstalk in lymphoma. (A) Pharmacological intervention of suboptimal doxorubicin in DC-NK crosstalk mediated cytotoxicity and growth inhibition against DL tumor cells. Active participation of regulatory cytokine for the outcome of effector functions of dual cell intervention [259]. (B) Novel binary therapy with DC-NK combination, in addition to a suboptimal dose of doxorubicin, offers superior protection and cure response by enhancing effector functions of DC and NK cells and ablation of disease exacerbating T regulatory cells and PD-1 responsive CD8+ T cells [260]. DOX, Doxorubicin; FOXP, Forkhead box P. Created with BioRender.com.
Cytotoxicity is also considered as a form of immunogenic cell death (ICD) incorporating efficient tumor antigen cross-priming. Studies have suggested that several anti-cancer agents, including chemotherapy and physical therapeutic modalities, exert immunomodulatory activities. These affect immune cells like DCs in the TME, modify the tumor cell immunogenicity via induction of immunogenic cell death (ICD), and release and expose a series of DAMPs through a well-defined spatiotemporal scheme [261]. Immune cells like DCs express pattern recognition receptors (PRRs), which recognize these DAMP sin stress, damage, or death. DCs play an essential role in the IR, triggered when malignant cells undergo ICD, suggesting functions of ICD inducers to stimulate robust T cell response aided and assisted by the occurrence of activated DC in TME [262, 263, 264, 265, 266, 267, 268]. All these indicate great potential and influence in synergy with other therapeutic approaches aiming at boosting efficient and robust anti-tumor immunity. The ex vivo ICD induction is a relatively recent strategy to understand the tumor immunological features providing significance of the role of TAAs, relevant molecules act as DC activating signals. ICD inducers like doxorubicin or radiotherapy can be combined with immunogenic TAAs in addition to the application of adjuvants encapsulated in nanoparticles, liposomes, or immunostimulatory complex for optimal delivery in DC vaccination [269, 270]. Calreticulin (CRT), a Ca2+-binding protein in the lumen of the endoplasmic reticulum (ER), acts as a strong “eat me” signal in ICD, which facilitates phagocytosis of dying cells by DCs. CRT-CD91 interaction triggers the activation pathways of NF-kB in DCs, releasing pro-inflammatory cytokines in the extracellular milieu and causing Th17 priming [271]. In non-small cell lung cancer patients, CRT expression is linked with higher infiltration of myeloid DCs (mDCs) and effector memory T-cells and correlated with favorable clinical outcomes [272]. Heat shock protein 90 (HSP90) and CD91 interaction activate DC and enhance the presentation of TAA to CTLs [273]. The binding of extracellular High mobility group box 1 (HMGB1) to TLR4 induces high-functioning cross-presentation of neoplastic antigens by DCs [274]. Dying cell-derived ATP during ICD in the extracellular milieu acts as a “find me” signal to attract DCs besides neutrophils and monocytes via the P2X7 receptor. Mice deficient in NLRP3 or P2RX7 are unable to mount organized and proactive adaptive immune responses during ICD [263]. This data indicates that immunogenic dying apoptotic cells are vital sources of both antigens and adjuvants, contributing to DC activation followed by effector T cell stimulation [275, 276]. The process of cytotoxicity is a form of ICD cross-prime CD8+ T cells, responsible for optimal activation of T cells and endowed with the repertoire of functional skills to recognize and eliminate neoplastic cells [277]. Thus, ICD- and DC-based immunogenic tumoricidal response is critical for translational advancement in cancer research and patient care.
Autophagy is a homeostatic and catabolic process responsible for the degradation and recycling of cellular components. Autophagy serves as cellular housekeeping and metabolic functions and constitutes a regulatory mechanism for several cellular functions. Dysregulation of autophagy is associated with tumorigenesis, tumor-stroma interactions, and resistance to cancer therapy. Autophagy affects the TME and constitutes a key factor in the function of APCs, macrophages, and T cells. Autophagy is found to be closely associated with the various functions of DCs under physiological and pathological conditions. Presentation of neo-antigens derived from tumor cells by both MHC-I and II in DCs results in functional activity of immune cells by creating T cell memory, as well as in cross-presentation of neo-antigens for MHC-I presentation and the internalization process. Autophagy plays a crucial role in immunotherapy via the presentation of neo-antigens constituting a potential target in order to strengthen or attenuate the effects of immunotherapy against different types of cancer. This is promising for long-term responses for patients who lack the ability to respond to immune checkpoint inhibitors for malignant tumors [278].
In Pancreatic ductal adenocarcinoma (PDAC) patients, the tumor cells are resistant to immune checkpoint inhibitors and have a significant reduction in MHC-I expression. Yamamoto et al. [279] showed that NBR1-mediated selective macroautophagy/autophagy of MHC-I represents a novel mechanism that facilitates immune evasion by PDAC cells. Autophagy or lysosome inhibition reinstates MHC-I expression, resulting in increased anti-tumor T cell response besides improvement in response to immune checkpoint inhibition in a syngeneic mouse model of transplanted tumors [279]. In addition, autophagy also influences the recruitment of APCs in TME and their maturation, leading to tumor evasion from immune surveillance via autophagy-driven activation of the STAT3 signaling pathway [280]. Autophagy also acts as a major regulator of MHC-I/II protein molecules, facilitating antigen presentation and targeting T-cell activation besides autophagy degradation of the molecules. MHC-II is degraded by membrane-associated RING-CH1 (March1) E3 ubiquitin ligase in myeloid-derived suppressor cells (MDSCs), while autophagic degradation of MHCI is induced by NBRI, resulting in tumor immune evasion. MHC-I degradation in DCs is mediated by adaptor-associated protein kinase 1 (AAK1), involving receptor-mediated endocytosis (RME), leading to impaired antigen presentation and T-cell stimulation [281, 282]. In addition, opsonization in DCs autophagy induced by nanomaterials could be conducive and reasonable for a deeper understanding of the plasticity of DCs function, which may have a positive role in promoting tumor adaptive immune responses and constitute a potential strategy for novel DC vaccines [283].
It is essential to reduce the knowledge gap in the translational potential of DC-targeted therapies, and we need to have significant findings on the importance of the cDC1 subset for effector killer cell-mediated immunity against tumors. Major shifts should focus on the success of checkpoint inhibitor therapy to realize the binary therapy for durable, complete responses. Understanding of molecules that drive the antigen presentation and stimulation of DCs as important ‘adjuvants’ to augment the therapeutic efficiencies in poorly immunogenic tumors. Dissecting out of tumor driven immunosuppressive TME which modify the phenotype and functions of tumor associated DC (TADC) and thus prevention of dysfunctional or tolerogenic state and impaired T cell activation and priming in the TME. Understanding the roles TADC subset to enhance tumoricidal potential and sharpen the efficacy of existing immunotherapies including wide range applicability of checkpoint blockade in addition to adoptive cell therapies (ACT). Investigations are required for inadequacy in T cell priming for cold tumors (TME lacks T lymphocyte infiltration) and unresponsiveness to immune checkpoint blockade (ICB). Exploring new strategies for manipulating cDC1 aided CD8+ T cell cross-priming, including increase in the number of cDC1s and heightening their cross-priming capacity in tumors and tumor draining LNs in addition to efficacy of ICB and ACT. Understanding and bridging the knowledge gap in the activation signals and the immunosuppressive conditions within tumors to improve immune recognition for translational promise of DC-targeted therapies. Importance of cDC1 subset in antigen delivery to the lymph nodes for anti-tumor T-cell response needs extensive analysis. Exploration of cytokine blockers as potentially promising therapeutic agents against tumor progression irrespective of concern regarding toxicity. Up gradation in control rate of Immune checkpoint blockade (ICB) by targeting of cytokine blockers, reduction in toxicity, and optimizing their combination via application of steroid hormones. Multi center and interdisciplinary research with large samples are imperative for achieving this goal. Designing new dynamics for the clinical use of DCs in combination with neoantigen, targeting immune checkpoint inhibitors and conditioning the tumor immunosuppressive mechanisms. Rational and logical selection of adjuvants or maturating agents (e.g., TLR stimulation) for manipulation of the blood DC population may be a key approach for enhancing mechanisms. Establishment of biomaterial-based scaffolds for in situ recruitment and accentuation of functions of tissue-resident cDC1 subsets appears to be novel strategies with true potential for clinical translation. Exploration of immune suppression associated with TME appears to be relevant for success in therapy. Combination/binary application of DC vaccination plus appropriate anti-cancer treatments may modulate the TME for pro-inflammatory/stimulatory status for improving efficacy. Integration of ICD-based cancer treatment with in situ DC-vaccination or in vivo DC targeting needs further investigation. Inoculation of ex vivo generated autologous MODCs may resurge the prospect of ICD inducers and improve tumor antigen presentation to abnegate immune suppression in favor of immune stimulation. Standardization of DC-based vaccines for inducing a durable response and enhancing long-term survival. Optimization of next-generation DC-based vaccines for use in individual patients selectively based on their disease state. This includes harvesting enough tumor material; DC precursors; and sound understanding of highly heterogeneous tumors with TAAs and tumor-specific antigens (TSAs). High-throughput sequencing and bioinformatics analysis of big data for identification of novel and immunogenic tumor antigenic determinants, such as neoantigens. This includes TSAs specifically expressed in tumor for targeting by DCs in vivo of TSA derived peptide. Designing of flexible platforms for the targeting vector with self-adjuvant properties to enable realization of personalized vaccines. Development of new adjuvants or stimulants like TLR ligands for selective stimulation of CD8+ CTL responses is critical for cancer immunotherapy in future despite the possibility of various adverse effects including fever, tissue damage and inflammation at the injection site etc.
Despite significant advancement and breakthrough, the overall interpretation on DC-mediated cytotoxicity and cross-priming needs more study to decipher and link the precise molecular mechanisms governing ICD with particular relevance to T-and NK-cell cytotoxicity. As a professional APC, DCs are viewed as sentinels of the immune system which collect and phagocytose apoptotic cells resulting in elimination of tumor cells by cytotoxic effectors or spontaneous death. Tumors on the other hand opt for unsettling of the mechanisms of immunogenic cytotoxicity and cross-priming, responsible for destabilization and termination of malignancies. DCs are proficient in processing TAAs and are superiorly adept for cross-presenting them to CTLs. In addition, a growing and significant body of literature reports the direct tumoricidal activity of DCs, indicating that DC subsets are capable of detecting signs of cellular stress via expression of NKG2D and TRAIL. Beside identification of these specialized DC subsets for recognition and killing of the therapeutically targeted tumor cells, deciphering counteracting immunosuppressive mechanisms that may downregulate the efficiency of cDC1 cross-priming in tumor tissue may attribute for the development of precise biomarkers for immunotherapies. By mimicking or enforcing cytotoxicity in part of the tumor lesions, endogenous vaccines can be designed against complicated malignancies. Identification of strong and durable cDC1 maturation stimuli, including TLR agonist or co-stimulatory CD40 assault, could render super-effective results. With all the things considered, the interplay of cytotoxic lymphocytes and cDC1 mediated antigen cross-priming will be a fertile field of research to harvest novel biomarkers and immunotherapy in addition to available options. Cytotoxicity by DC is a form of ICD having significant implications for organ-specific neoplasias besides other dysfunctions like autoimmunity and viral infections.
PPM, PC and PS contributed in conceptualization, and writing the review. PC and PS written the preliminary draft and contributed for analysis and making the figures. PPM contributed to overall supervision including editing of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by IOE grant No. R/Dev/D/IOE/ Incentive/2021–22/32275 to Partha Pratim Manna by Banaras Hindu University (BHU).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.