






 , Dheepika Venkatesh 1, Thirukumaran Kandasamy 1, Siddhartha Sankar Ghosh 1,2,*
, Dheepika Venkatesh 1, Thirukumaran Kandasamy 1, Siddhartha Sankar Ghosh 1,2,*
1 Department of Biosciences and Bioengineering, Indian Institute of Technology Guwahati, 781039 Guwahati, Assam, India
2 Centre for Nanotechnology, Indian Institute of Technology Guwahati, 781039 Guwahati, Assam, India
Abstract
Breast cancer, a heterogeneous and intricate disease, ranks among the leading causes of mortality in women. Restricted therapeutic choices, drug resistance, recurrence, and metastasis are the predominant conditions that lead to mortality. Accumulating evidence has shown breast cancer initiation and progression happen through a multifaceted and intricate process that involves numerous genetic and epigenetic alterations. The modulation of gene expression through epigenetic modifications, encompassing DNA methylation, histone alterations, and non-coding RNA regulation, has emerged as a fascinating field that represents a new avenue for breast cancer therapy. This review emphasizes various aberrant epigenetic regulations implicated in the onset and advancement of breast cancer. The critical epigenetic modifications closely associated with estrogen signaling, epithelial-to-mesenchymal transition (EMT), cancer stemness, and drug resistance have been discussed extensively. Moreover, it highlights current epi-drugs, including DNA modifying agents, histone acetyltransferase inhibitors, histone deacetylase inhibitors, histone methyltransferase inhibitors, and histone demethyltransferase inhibitors used for breast cancer treatment. Nonetheless, we described current investigations pertaining to combination therapy employing epi-drugs and future challenges.
Keywords
- epigenetics
- breast cancer
- epidrugs
- epithelial-to-mesenchymal transition
- drug resistance
Breast cancer is a complex disease and the most prevalent cancer among women worldwide [1]. Owing to its heterogeneous nature, it represents a major therapeutic challenge. Breast cancer is classified into multiple categories based on molecular subtypes, which include hormone receptor-positive (HR+), human epidermal growth factor receptor 2 (HER2+) positive, and triple-negative breast cancer (TNBC). The HR+ subtype comprises cancer cells expressing estrogen receptor (ER) and progesterone receptor (PR), promoting the growth of cancer cells. Targeting these receptors via hormonal therapy is often an effective treatment strategy for this subtype [2]. In the case of the HER2-positive subtype, overexpression of the HER2 receptor is prominent and endows more aggressiveness [3]. Another major subtype, TNBC, is characterized by the lack of estrogen, progesterone, and HER2/neu receptors. The absence of these receptors makes TNBC resistant to conventional hormonal therapy and drugs targeting these receptors [4].
A comprehensive approach that combines radiotherapy, surgery, hormonal, and targeted therapy is generally used to combat breast cancer. Despite significant improvements in the efficacy of various treatment strategies over the decades, numerous challenges have emerged. These challenges include severe side effects, the development of multidrug resistance, intratumoral heterogeneity, and metastatic illness [5]. In addition to the extensively studied genetic alterations, epigenetic changes play a significant role in driving the development of breast cancer by inducing abnormal gene expression. The emerging understanding of epigenetic regulation in cancer offers many new therapeutic prospects for breast cancer treatment [6]. The successful use of epigenetic inhibitors or epidrugs in managing hematological malignancies has generated interest in their potential application for treating solid tumors. Based on preclinical data, these drugs potentially sensitize resistant cancer cells to conventional treatments. Additionally, they can eradicate cancer stem cells (CSCs), which are pivotal in metastasis and the emergence of multidrug resistance to therapy [7, 8]. This review aims to discuss the significant epigenetic alterations linked to the development and advancement of breast cancer. Additionally, we emphasized current accomplishments and potential future applications of epidrug, including various inhibitors and drugs for breast cancer treatment.
Hereditary breast cancer accounts for approximately 10% of all cases. Mutations in tumor suppressor genes such as TP53, BRCA1, and BRCA2 are linked to the development of breast cancer [9]. Alongside genomic alterations, a series of epigenetic modifications contribute to the development and progression of breast cancer. Epigenetic changes are heritable DNA modifications that impact gene expression without altering the DNA sequence. Epigenetic modifications primarily include DNA methylation, histone tail modifications, nucleosomal remodeling, and non-coding RNAs mediated regulatory activity. These mechanisms are crucial for regulating gene transcription and genomic stability, as well as maintaining cellular growth, development, and differentiation (Fig. 1). Furthermore, epigenetic modifications have a significant impact on the development and progression of multiple malignancies, including breast cancer [10]. These alterations are believed to be involved in the early stages of breast cancer carcinogenesis and often serve as biomarkers for early identification, prognostic assessment, and therapy response. Moreover, a thorough grasp of the epigenetic alteration in breast cancer provides an alternative avenue for drug development [11].
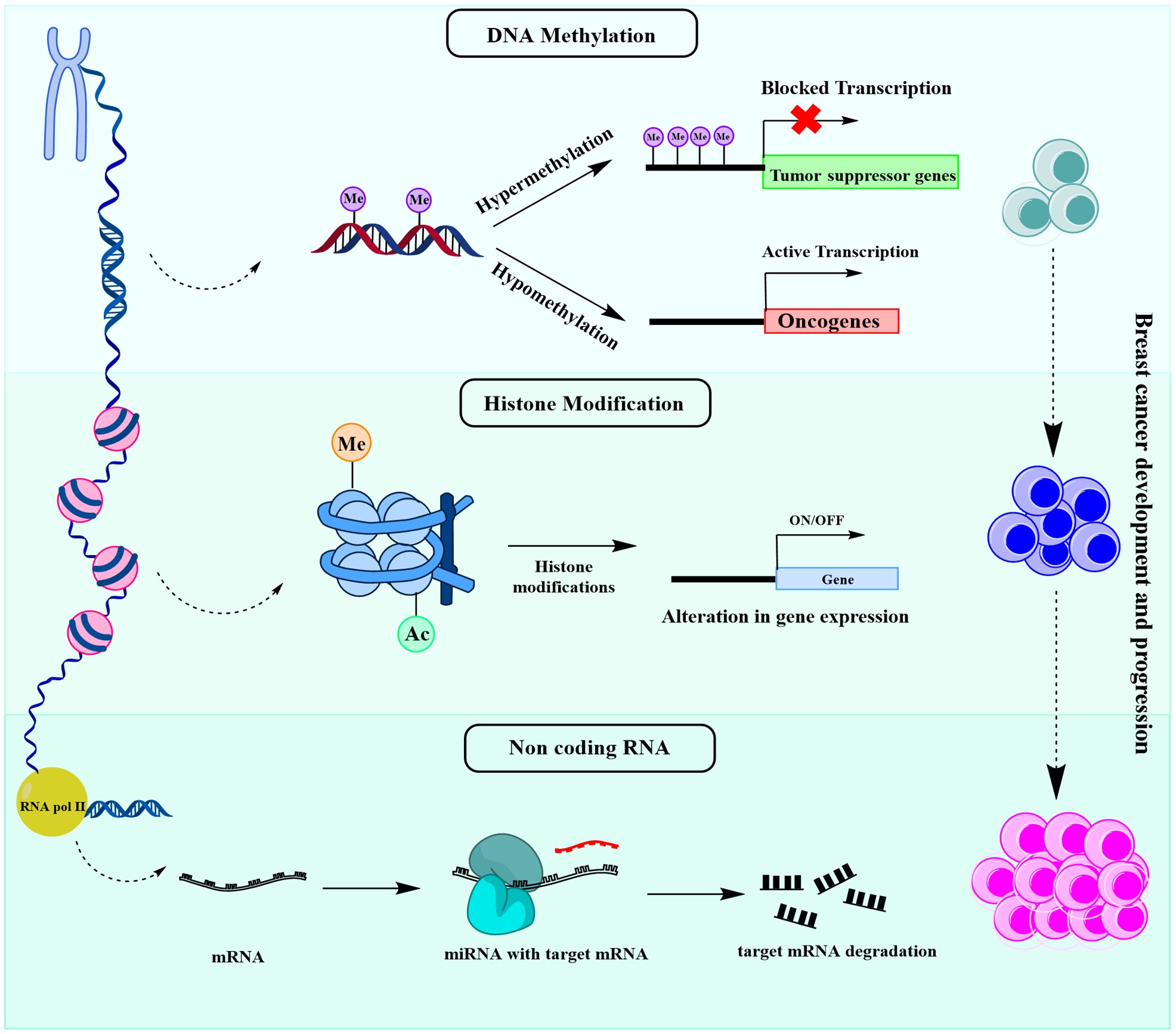 Fig. 1.
Fig. 1.
Schematic illustration of fundamental epigenetic mechanisms involved in breast cancer development and progression. Me, Methylated; Ac, Acetylated. Created with ChemDraw Professional (version: 16.0, PerkinElmer Inc., Waltham, MA, USA).
DNA methylation is the primary epigenetic mechanism extensively studied for its significant role in regulating gene expression. The process of DNA methylation entails the covalent addition of methyl group to the C-5 position of the cytosine in cytosine-guanine (CpG) dinucleotides by DNA methyltransferases (DNMTs). DNMT1 maintains the pre-existing methylation patterns during replication, while DNMT3A and DNMT3B govern de novo methylation [12]. Methylation of CpGs at the promoters of genes leads to suppression of gene expression, whereas methylation levels in the gene body positively correlate with expression. DNA methylation is critical in maintaining chromosome stability, X chromosome inactivation, transposable element suppression, aging, and genomic imprinting. Moreover, it influences several disease conditions, including cancer and autoimmune disorders [12].
Breast cancer cells often exhibit disruptions of normal methylation patterns.
The hypermethylation of promoter region leads to inactivation of tumor suppressor
genes, such as BRCA1, APC, CDH1, CCND2, and
CTNNB1. On the other hand, breast cancer exhibits global DNA
hypomethylation, with up to 50% of instances exhibiting lower 5-methylcytosine
content than their counterparts in normal tissue [13]. Hypomethylation in
repetitive sequences and pericentromeric satellite DNA is prominent in cancer
cells, and it generally remains highly methylated in normal breast cells. For
example, retrotransposons, including long interspersed nuclear elements (LINEs),
are hypomethylated in breast cancer [14]. In addition, Sat2 and Sat
Another key epigenetic mechanism is the function of non-coding RNAs (ncRNAs), which constitute approximately 62–75% of the genome [19, 20]. To date, numerous ncRNAs have been found, including circular RNA (circRNA), short hairpin RNA (shRNA), small nucleolar RNA (snoRNA), piwi-interacting RNA (piRNA), small-interfering RNA (siRNA), and long non-coding RNA (lncRNA). These ncRNAs have emerged as an essential source of biomarkers and targeted therapies due to a growing understanding of their role in various diseases [21].
The dysregulation of miRNAs, one of the crucial ncRNAs, has come to light in the emergence of multifactorial disorders, including breast cancer. miRNAs are short (18–22 nucleotides) endogenous single-stranded RNA molecules that attach to the target mRNA to limit translation. Aberrant miRNA activities are associated with breast cancer onset and progression [22]. For instance, metastatic breast cancer has been associated with overexpression of miR-21 and miR-155 [23]. Similarly, patients with higher expression levels of miR-1307-3p, miR-940, and miR-340-3p were observed to experience decreased overall survival rates [24]. Furthermore, miR-497 regulates cell growth and invasion of breast cancer cells through inhibiting the expression of cyclin E1 [25].
LncRNAs, another important class of non-coding RNAs ranging from 200 nucleotides
to 100 kilobases in length, can regulate gene expression at various levels by
interacting with DNA, RNA, and proteins [26]. Dysregulation in lncRNA regulatory
activity facilitates breast cancer development and dissemination. For example,
growth arrest-specific 5 (GAS5) remains highly downregulated in breast cancer and
regulates the expression of several tumor-associated genes. Employing various
pathways, such as mitochondrial signaling pathways and cell death receptors, GAS5
can induce apoptosis in breast cancer. Moreover, it plays a critical role in
regulating PI3K/AKT/mTOR, NF-
Histones serve as essential DNA packaging proteins, thereby maintaining chromatin structure. Histone proteins (H2A, H2B, H3, and H4) assist in generating nucleosomes by wrapping octamers around 147 bp of DNA. Post-translational modifications (PTMs) of histone cause changes in the state of chromatin, subsequently leading to gene expression regulation [29]. Specific residues of the amino and carboxy ends of the histone tails undergo several modifications, including acetylation, phosphorylation, methylation, sumoylation, ubiquitination, ADP-ribosylation, glycosylation, and carbonylation. Different enzymes, such as histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone demethylases (HDMs), catalyze these modifications. Alterations in the expression of these enzymes also implicate the onset and advancement of cancer [29]. Numerous alterations have been identified in breast cancer, including the upregulation of p300, HBO1, HDAC1, HDAC2, HDAC3, and HDAC6 [30].
Histone methyltransferases, including lysine methyltransferase 2 (KMT2), were found to be linked with the proliferation and metastasis of breast cancer cells. KMT2 activates oncogenes and pro-metastatic genes by methylating H3K4 at both enhancer and promoter regions [31]. Amplification and overexpression of a crucial histone methyltransferase enhancer of zeste homolog 2 (EZH2) are prominent in breast cancer. By catalyzing H3K27 methylation, EZH2 facilitates transcriptional silencing of several genes and persuades epithelial to mesenchymal transition (EMT) and metastasis in breast cancer [32, 33]. Another prominent HMT, disruptor silencing 1 like (DOT1L), is also known to potentiate metastatic behavior of breast cancer cells [34].
Several histone demethylases, including Lysine-specific demethylase 4A (KDM4A),
Lysine-specific demethylase 4B (KDM4B), and Lysine-specific demethylase 4C
(KDM4C) are also associated with breast cancer. ER
Estrogens, the predominant sex hormones in women, play a crucial role in
reproductive maturity, bone growth, and energy homeostasis. The five primary
subtypes of estrogen are estrone (E1), 17-
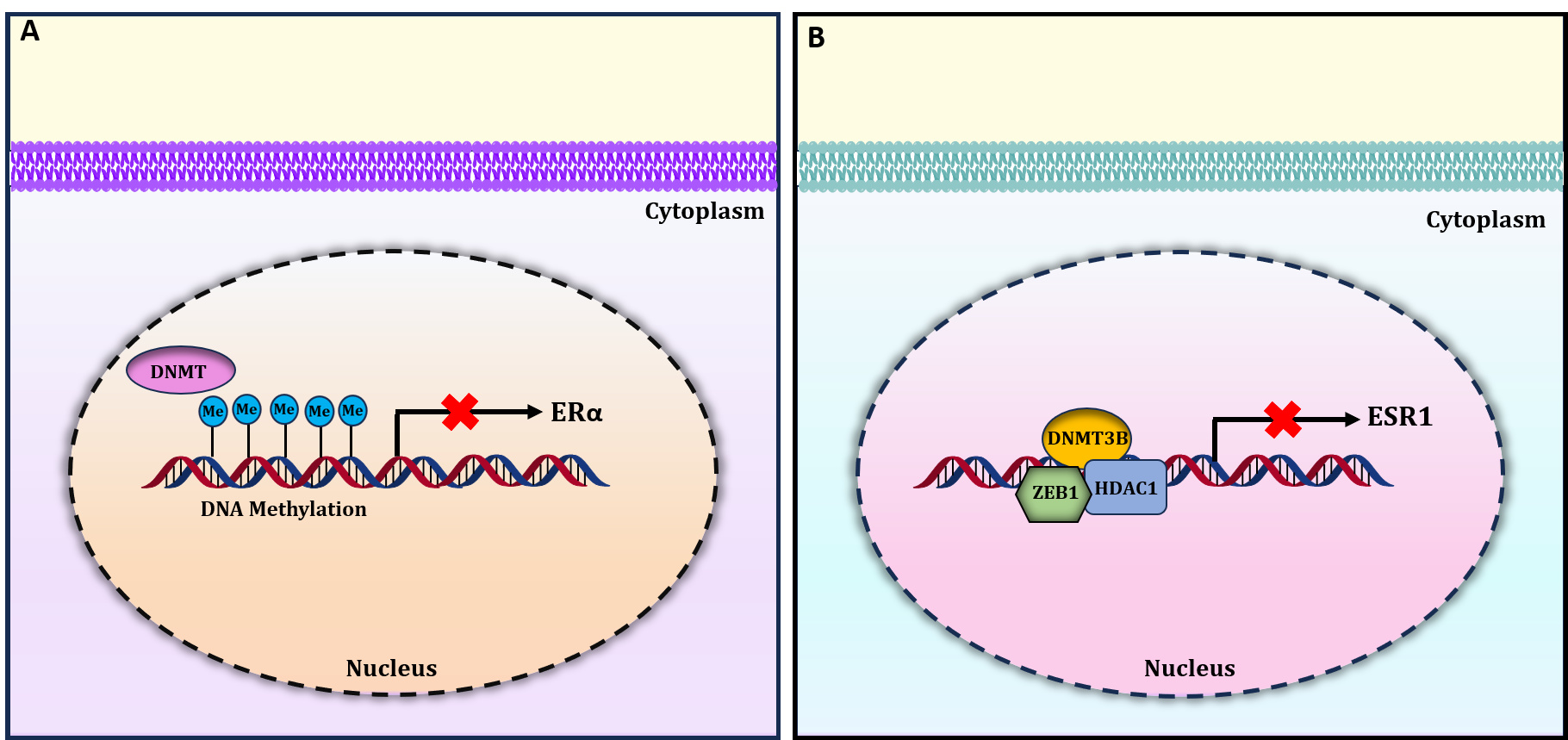 Fig. 2.
Fig. 2.
Epigenetic mechanisms regulating the expression of ER
Upon binding of E2, dimerization of ER
Several studies demonstrated a significant role of miRNAs in suppressing ER
expression [45, 46]. For instance, overexpression of miR-221 and miR-222 results
in post-transcriptional suppression of ER
EMT is a cellular process wherein cells transform, losing their epithelial
traits and adopting mesenchymal characteristics. EMT plays a pivotal role in
wound healing, development, and progression of cancer. It makes cancer cells more
migratory and endows drug resistance, stemness, and recurrence. Different growth
factors (Transforming Growth Factor
For example, in breast cancer, DOT1L collaborates with c-Myc and p300 to promote the methylation and acetylation of H3K79 in the promoter areas of EMT transcription factors, which leads to increased expression of these genes (Fig. 3). Consequently, this process promotes EMT-associated cancer stemness characteristics [47]. Furthermore, invasive breast cancer cells exhibit aberrantly high levels of methylation in CDH1, which is associated with downregulation in the expression of E-cadherin [48]. E-cadherin is a tumor suppressor that facilitates cell-to-cell adhesion between neighboring cells [49]. G9a silences transcription at the E-cadherin promoter in breast cancer by interacting with Snail (Fig. 3) [50].
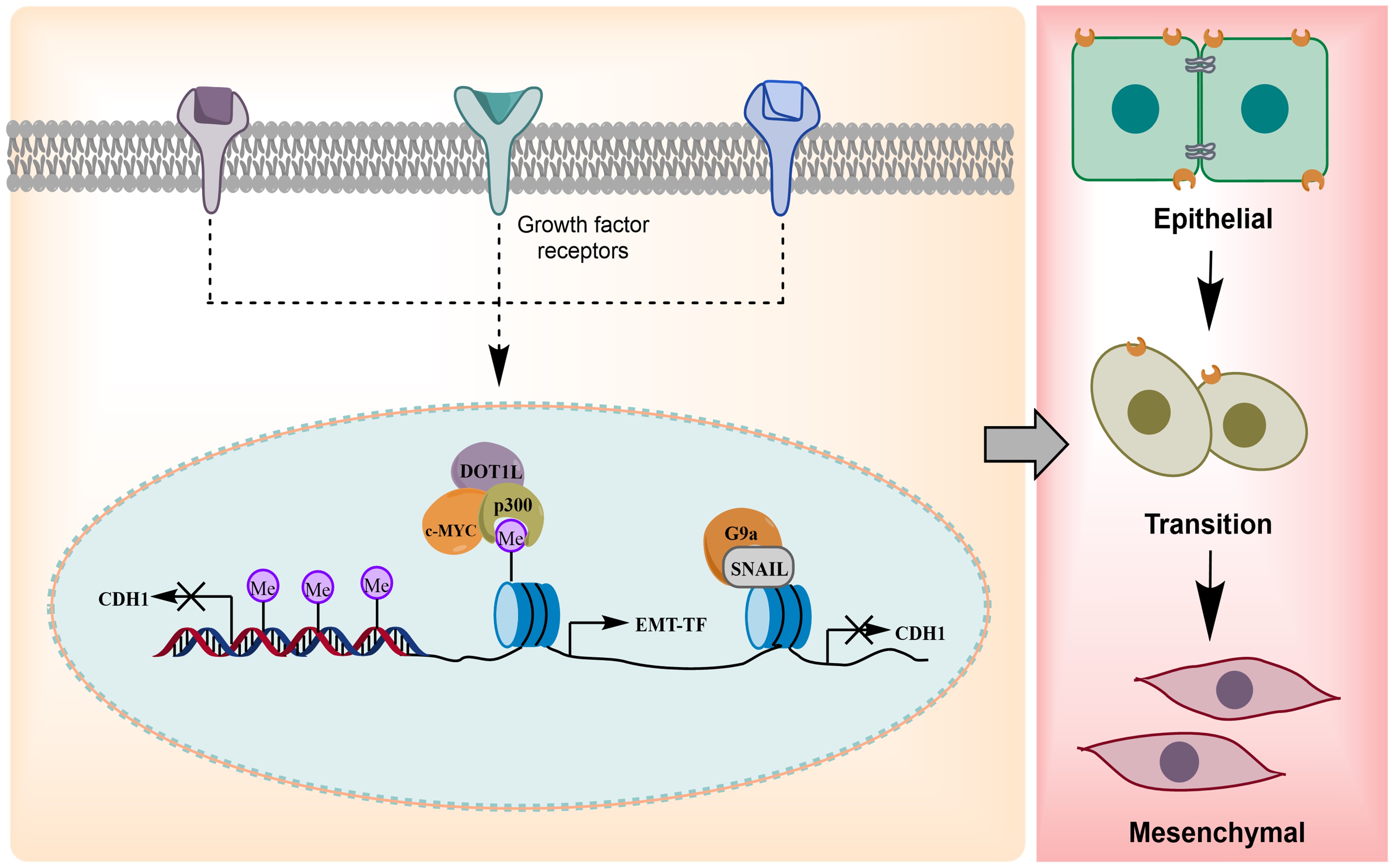 Fig. 3.
Fig. 3.
Role of epigenetic mechanisms in epithelial to mesenchymal transition (EMT) dynamics of breast cancer. CDH, Cadherin; DOT1L, Disrupter of telomere silencing protein 1; c-MYC, Cellular Myelocytomatosis Oncogene; EMT-TF, Epithelial to Mesenchymal Transition Transcription Factor; G9a/EHMT2, Euchromatic Histone Lysine Methyltransferase 2; SNAIL: Snail Family Transcriptional Repressor. Created with ChemDraw Professional (version: 16.0, PerkinElmer Inc., Waltham, MA, USA).
Histone deacetylase inhibitor Trichostatin A (TSA) reverses EMT in breast cancer by inhibiting Slug expression [51]. Additionally, inhibiting Bromodomain protein 4 (BRD4) leads to suppression of Gli1, which is necessary for the transcriptional activation of Snail. This suggests that BRD4 regulates the aggressiveness of breast cancer cells through both transcriptional regulation of Snail and post-translational mechanisms [52]. Thus, understanding the interplay between EMT and epigenetics offers new avenues for cancer treatment.
Emerging evidence suggests that a tiny population of cancer cells promotes the recurrence and dissemination of the disease known as cancer stem cells (CSC). Like other CSCs, breast cancer stem cells (BCSCs) comprise a subset of diverse breast cancer cells with substantial potential for proliferation and self-renewal. Several studies demonstrated that BCSCs are the main contributory factor of drug resistance, recurrence, metastasis, and the onset of the disease [53]. During development, epigenetic machinery is crucial in reprogramming stem cells to facilitate differentiation into specific cellular and tissue lineages. In several malignancies, including breast cancer, aberrant epigenetic alterations often potentiate the generation of CSCs, which lack differentiation capacity [54].
Epigenetic mechanisms are pivotal in regulating the expression of numerous genes
involved in signaling pathways associated with cancer stemness, including Wnt,
Notch, Hedgehog, and Hippo/yes-associated protein 1 (YAP) [55]. In breast cancer cells, the promoters of
genes involved in Wnt/
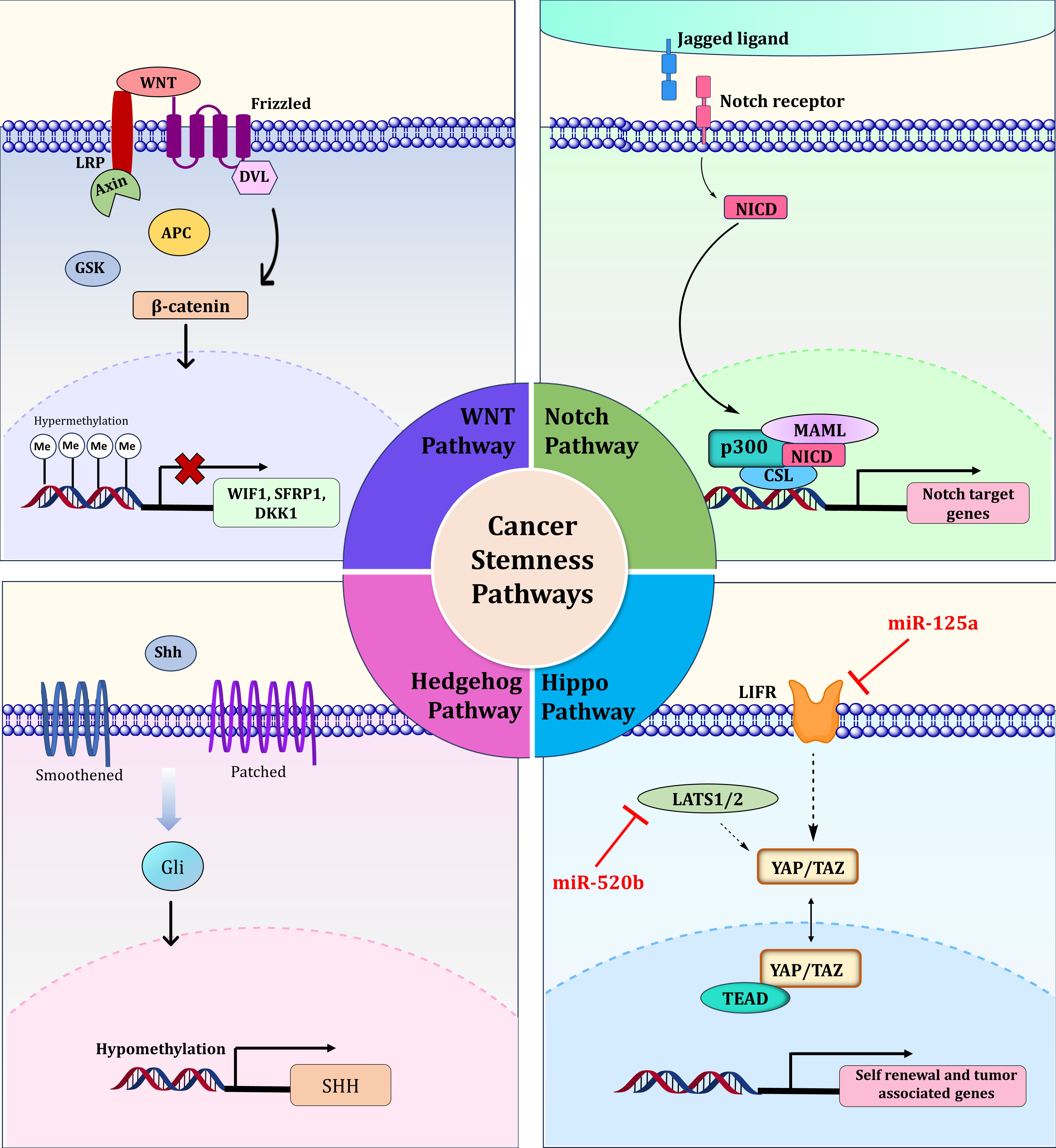 Fig. 4.
Fig. 4.
Regulation of signaling pathways associated with cancer stemness, including Wnt, Notch Hedgehog, and Hippo pathways by epigenetic mechanisms. LRP, Low-Density Lipoprotein Receptor-Related Protein; WNT, Wingless-Type Integration Site; DVL, Dishevelled Segment Polarity Protein; GSK, Glycogen Synthase Kinase; APC, Adenomatous Polyposis Coli; WIF1, WNT Inhibitory Factor 1; SFRP1, Secreted Frizzled-Related Protein 1; DKK1, Dickkopf-Related Protein 1; NICD, Notch Intracellular Domain; MAML, Mastermind-Like Protein; CSL, Suppressor of Hairless; LIFR, Leukemia Inhibitory Factor Receptor; LATS1/2, Large Tumor Suppressor Kinase 1/2; YAP, Yes-Associated Protein; TAZ, Transcriptional Coactivator with PDZ-Binding Motif; TEAD, TEA domain transcription factor; SHH, Sonic Hedgehog Signaling Molecule. Created with ChemDraw Professional (version: 16.0, PerkinElmer Inc., Waltham, MA, USA).
Dysregulation in Hippo signaling has been linked to increased stem cell proliferative and self-renewal capacities of cancer cells. It has been reported that by altering Leukemia inhibitory factor receptor (LIFR) expression, miRNA-125a indirectly regulates the activity of Transcriptional coactivator with PDZ-binding motif (TAZ), an effector molecule in the Hippo pathway [63]. Furthermore, another crucial miRNA, miR-520b, activates Hippo/YAP signaling by targeting Large tumor suppressor kinase 2 (LATS2), thereby promoting stemness in breast cancer patients (Fig. 4) [64].
Innate and acquired resistance towards conventional chemotherapeutic drugs
facilitates cancer recurrence and poor prognosis. The occurrence of cancer stem
cells (CSCs) and the EMT process are thought to be the leading causes of cancer
therapeutic resistance. According to accumulating evidence, drug resistance in
several cancers, including breast cancer, is also influenced by aberrant
epigenetic regulation [65]. Tamoxifen resistance in breast tumors is caused by
EZH2-mediated suppression of Growth Regulating Estrogen Receptor Binding 1 (GREB1) expression, an ER
Epidrugs or epigenetic drugs represent a promising field of medicine that focuses on the chemical compounds utilized to rectify epigenetic modifications by targeting epigenetic modulators. These drugs open up new avenues for personalized medicine and have the potential for precise targeting [6]. They can regulate gene activity by correcting aberrant gene expression associated with cancer, neurodegenerative diseases, and autoimmune conditions [10]. Epidrugs can be broadly classified as DNA methyl transferase (DNMT) inhibitors, Histone Acetyltransferase (HAT) inhibitors, Histone deacetylase (HDAC) inhibitors, Histone methyltransferase (HMT) inhibitors, and Histone Demethylase (HDM) inhibitors (Fig. 5) [6]. The need for effective epigenetic inhibitors led to the development of the first generation of epi-drugs, which includes DNMT inhibitors such as pyridine analogs and HDAC inhibitors that target specific histone classes. The first-generation epidrugs fall short in terms of poor pharmacokinetic properties, selectivity, and bioavailability, which paves the way for the advancement of second-generation epidrugs with a long half-life, lower cytotoxicity, side effects, and potent inhibitory action. The third-generation epidrugs target epigenetic factors categorized as writers, erasers, and readers. Writers add methyl or acetyl groups to DNA or histones, erasers remove these modifications, and readers recognize and regulate the binding interactions [75]. The list of epidrugs used for breast cancer therapy is provided in Table 1 (Ref. [75, 76, 77]).
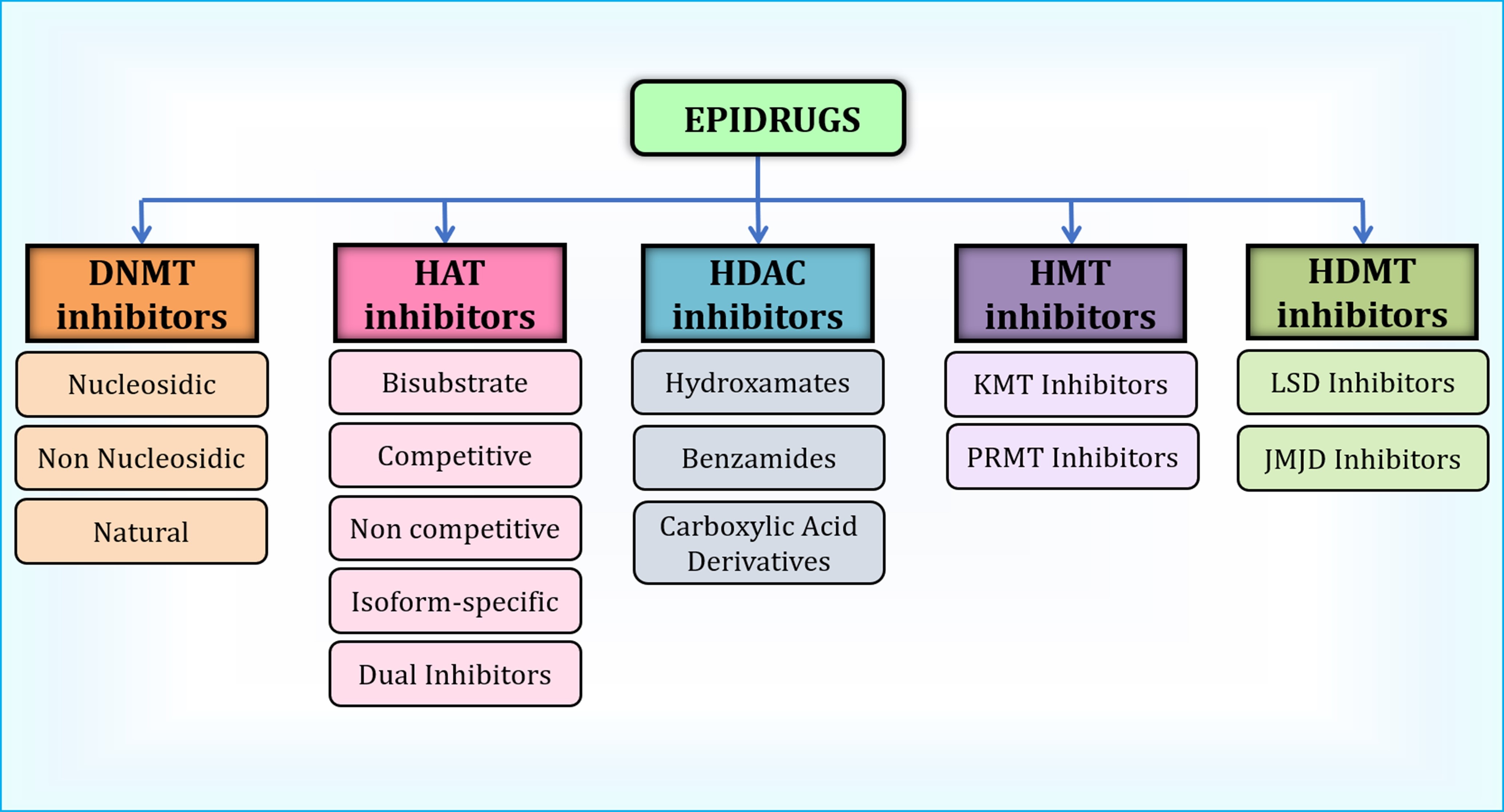 Fig. 5.
Fig. 5.
Classification of Epidrugs used in breast cancer therapy. HAT, Histone Acetyltransferase; HDAC, Histone Deacetylase; HMT, Histone Methyltransferase; HDMT, Histone Demethylase; KMT, Lysine Methyltransferase; PRMT, Protein Arginine Methyltransferase; LSD, Lysine-Specific Demethylase; JMJD, Jumonji Domain-Containing proteins. Created with ChemDraw Professional (version: 16.0, PerkinElmer Inc., Waltham, MA, USA).
| Generation | Type of epidrug | Name | Reference |
| First generation epidrugs | DNMT inhibitor | Decitabine, Azacitidine | [75, 76, 77] |
| HDAC inhibitor | Vorinostat, Trichostatin A, Trapoxin A, Romidepsin | [75, 76, 77] | |
| Second generation epidrugs | DNMT inhibitors | Zebularine, CP-4200, Guadecitabine, Hydralazine | [76, 77] |
| HDAC inhibitors | Belinostat, Dacinostat, Panobinostat, CUDC-101, Quisinostat, Tefinostat, Tacedinaline, Entinostat, Mocetinostat, Chidamide, Butyric acid, Pivanex, Phenylbutyric acid, Valproic acid | [76, 77] | |
| Third generation epidrugs | KDM inhibitors | Tranylcypromine, ORY-101, GSK2879552, 4SC-202, Clorgyline, Pargyline, Bizine, KDM5-C70, JIB-04 | [76, 77] |
| KMT inhibitors | Sinefungine, Pinometostat, GSK2816126, Tazemetostat, GSK3326595, JNJ64619178, GSK3368715, EPZ-6438, CPI360, DZNep, GSK343, EI1, BIX-01294, UNC0638, EPZ004777, UNC0224 | [76, 77] | |
| Bromodomain ligands | I-BET762, CPI-0610, OTX015, RVX-280 | [76, 77] |
DNMT, DNA Methyltransferase; HDAC, Histone Deacetylase inhibitor; KDM, Lysine Demethylase; KMT, Lysine Methyltransferase; DZNep, 3-Deazaneplanocin A; I-BET, inhibitor of BET (bromodomain and extra-terminal) proteins; RVX-280, Resverlogix-280; CP-4200; CUDC-101; ORY-101; GSK2879552; 4SC-202; JIB-04; EPZ-6438; EPZ004777; CPI360; EI1; BIX-01294; UNC0638; UNC0224; OTX015 are names of various small molecule inhibitors or experimental compounds, often used as epidrugs.
DNA methylation entails the transfer of a methyl group to the fifth carbon of cytosine, orchestrated by a set of enzymes known as DNA methyltransferases (DNMTs), comprising DNMT1, DNMT3A, DNMT3B, and DNMT3L. DNA methylation leads to the silencing of gene expression by impeding the binding of transcription factors to the DNA [12]. The DNMT inhibitors work by incorporating themselves between the DNA base pairs and preventing the methylation of the promoter sequence (CpG islands). Decitabine and Azacitidine are well-known first-generation DNMT inhibitors that follow this mechanism [78].
DNMT inhibitors are of three types, namely non-nucleosidic, nucleosidic, and
natural compounds. Natural compounds such as Epigallocatechin-3-gallate (EGCG),
catechin, and quercetin demonstrated the ability to inhibit the activity of
DNMT1. This inhibition leads to the demethylation of DNA, reactivation of tumor
suppressor genes, and reduction of breast cancer cell proliferation [79]. A
non-nucleosidic compound called hydralazine is conventionally used as an
antihypertensive drug but serves to reduce the expression of DNMTs in mammals
[80]. Hydralazine, combined with valproic acid, a mild demethylating drug,
decreases cancer cell survival dose-dependently [81]. Zebularine, a potent
nucleoside DNMT inhibitor, upregulates p21 expression, downregulates cyclin-D
expression, induces apoptosis, and arrests MCF-7 and MDA-MB-231 cells in the
S-phase [82]. Procainamide, an antiarrhythmic drug, is a non-nucleoside DNMT
analog that sensitizes the ER
Histone acetyltransferases (HATs) and Histone Deacetylases (HDACs) are essential enzymes involved in the regulation of gene expression and chromatin remodeling [87]. HATs mediate the addition of an acetyl group from acetyl-CoA to the lysine residues, resulting in the neutralization of the charge of lysine. This process results in the transcriptional activation of the underlying DNA sequence due to the slackened chromatin structure [87]. On the contrary, HDACs catalyze the acetyl group removal from the histones, leading to a more stable and condensed chromatin structure contributing to transcriptional repression [88]. Any dysregulation in the dynamic interplay between these enzymes may result in life-threatening diseases like cancer [75].
Extensive in silico studies, including expression and survival studies, have recognized p300 acetyltransferase as the critical tumorigenic driver. Screening from 1293 FDA-approved drugs using Molecular Docking (MD) and molecular dynamic simulation (MDS) have identified Netarsudil and Imatinib as potential repurposed drugs against the p300 HAT domain. Further, these drugs exhibited significant anti-proliferative activity against breast cancer cell lines [89]. In addition, several HAT inhibitors from natural sources such as Carnosol, Garcinol, Anacardic acid, and Curcumin are investigated in breast cancer. Carnosol, a polyphenol abundant in rosemary, oregano, and sage plants, serves as a potential inhibitor of p300. It blocks the acetyltransferase activity of p300 by impeding the acetyl CoA binding site, which impels histone hypoacetylation in MDA-MB-231 cells [90]. Another natural compound, Garcinol, is reported to be an effective inhibitor of CBP/p300 mediated acetylation of p53 in Michigan Cancer Foundation-7 (MCF-7) cells. Furthermore, a significant decrease in H3K18 methylation was observed, thereby hindering the MCF-7 cell growth by arresting the cells at the G1 phase. Curcumin also inhibits the activity of p300/CBP, which blocks the MCF-7 cells at the G2/M phase [91, 92]. Anacardic acid is predominantly found in Anacardiaceae members like Amphipterygium adstringens, cashew, ginko, etc. It is a non-selective HAT inhibitor with HSP90 ATPase inhibitory potential to suppress Heat shock protein 90 (Hsp90) oncoproteins overexpressed in TNBC cells [93]. Pentamidine is a chemical compound used to treat protozoal infection. It has been shown to repress DNA and protein synthesis by targeting Tip60, a HAT from the MYST family (MOZ, Ybf2/ Sas3, Sas2 and Tip60 family), thereby dwindling the H2A acetylation [94]. In addition, bisubstrate inhibitor A is more capable of restraining the HAT activity by resembling the HAT substrate possessing a lysine peptide and an acetyl-CoA group [95]. Some other notable HAT inhibitors include NU9056, MG-149, TH1834, and Lys-CoA [94].
The HDAC inhibitors can be subdivided into three groups, namely, Hydroxyamates
(Vorinostat, Belinostat, Dacinostat, Panobinostat, CUDC-10, Quisinostat, and
Tefinostat), Benzamides (Tacedinaline, Entinostat, Mocetinostat, and Chidamide)
and Carboxylic acid HDAC inhibitors (Butyric acid, Pivanex, Phenylbutyric acid,
and Valproic acid). Electrophilic ketones and cyclic peptides are recently
explored for their HDAC inhibitory activity [96]. Vorinostat, commonly known as
suberoylanilide hydroxamic acid (SAHA), is one of the most prevalent
first-generation HDAC inhibitors to be discovered first. It functions by
inhibiting the class I and II family of HDAC and requires a Zn2+ ion for the
catalytic binding. Vorinostat is known to deplete the ER
Histone methyltransferases are essential for adding methyl groups to specific lysine or arginine residues on histone proteins. This process is catalyzed by lysine methyltransferases (KMTs) or arginine methyltransferases (RMTs), which derive the methyl groups from S-adenosyl-l-methionine (SAM). KMTs append 1, 2, or 3 methyl groups to the lysine residues, while RMTs add 1 or 2 methyl groups to the arginine residues. It is further classified based on the presence or absence of Su(var)3-9, Enhancer-of-zeste, and Trithorax domain (SET domain) as SET domain lysine methyltransferases and Disruptor of telomeric silencing 1 (DOT1) domain lysine N-methyltransferase [75]. A few histone methyltransferases include EZH2, G9a, DOT1L, and Protein arginine methyl transferase (PRMT). Chaetocin, derived from Chaetomium fungi, explicitly targets the H3K9 tri-methylation mediated by SUV39H1. In MDA-MB-231 cells, it inhibited cell proliferation and induced apoptosis by activating caspase 3 and Poly (ADP-ribose) polymerase (PARP) proteins [103]. A PRMT5 inhibitor, GSK3326595, is in a phase II clinical trial for treating early-stage HR+ breast cancer. JNJ-64619178 is one of the potent inhibitors of PRMT5, and it is currently under clinical trial for advanced tumors [104]. Pemrametostat is an oral small molecule inhibitor under investigation, which inhibits PRMT5 and reduces the arginine methylation levels of histones H2A, H3, and H4, thus downregulating the cancer cell proliferation. Sinefungin is a natural inhibitor of EHMT1/2 derived from Streptomyces incamatus and S. Griseolus [105]. EZH2 specific inhibitors that inhibit its activity by competing with S-Adenosyl methionine (SAM) are 3-deazaneplanocin A hydrochloride (DZNep), GSK2816126, EPZ005687, GSK343, GSK926, Tazemetostat, CPI-1205, etc. Some inhibitors broadly curb the activity of both EZH1/2, like UNC1999, ORS1/ORS2, and DS-3201b, while the others target the EZH2 degradation, such as GNA022, ANCR, FBW7, ZRANB1 [106].
Histone demethylases are divided into two classes based on their specificity as Flavin adenine dinucleotide (FAD) dependant lysine-specific demethylases (LSDs) and 2-oxoglutarate- ferrous iron and oxygen-dependent demethylases, which encompasses the Jumonji C domain-containing family. The former can demethylate mono and dimethylated substrates, while the latter can demethylate mono, di, and trimethylated substrates [107]. Phenelzine, a monoamine oxidase inhibitor, inhibits the activity of LSD1 irreversibly by downregulating the mesenchymal markers alongside nab-paclitaxel in metastatic breast cancer patients. It was also found to inhibit the cancer stem cells and reduce the number of circulating tumor cells that give rise to metastasis [108]. Pargyline is a monoamine oxidase inhibitor that blocks the activity of LSD1, exhibits a dose-dependent decrease in cell proliferation, and elevated expression of cleaved PARP proteins that consequences apoptosis [109]. The MAO-A gene is primarily associated with depression but is often found to be overexpressed in breast cancer cells, contributing to poor prognosis. Clorgyline, an MAO-A inhibitor, facilitates the mesenchymal to epithelial transition (MET) in metastatic MDA-MB-231 cells by promoting the expression of epithelial markers such as E-cadherin while concurrently altering the expression of mesenchymal markers like vimentin. In addition, it suppresses cell proliferation, anchorage-independent growth, and invasiveness [110]. ORY-1001 (Iadademstat) is another LSD1 inhibitor that suppresses androgen receptor expression in TNBC cells like MDA-MB-231 and BT549. It also abridges the multiplication of cells and provokes apoptosis [111]. 4SC-202, also referred to as Domatinostat is a potent inhibitor targeting both HDAC class I and LSD1 [112]. It demonstrates cytostatic and cytotoxic effects, making it a promising candidate for cancer therapy. Treatment of MDA-MB-231 and 4T1 TNBC cells with 4SC-202 led to cytotoxic effects, prohibited migration, invasion, and mitigated lung metastasis [112].
Combining epidrugs with chemotherapy or immunotherapy offers potential benefits
such as reduced drug resistance and improved overall treatment outcomes [113, 114]. This integrated strategy holds immense potential to fight against breast
cancer, paving the way for personalized and effective therapeutic interventions.
Several such combinations are under clinical trial to optimize the dosage and
evaluate long-term safety and efficacy (Table 2, Ref. [75, 82, 85, 97, 99, 100, 101, 108, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126]). Combining HDAC inhibitors has proven to be more efficient
than using them individually [113, 127]. The co-administration of Estrogen
Related Receptor
| Serial No. | Drugs | Target/Effect of the drug | Reference |
| 1. | Decitabine + C29 | Inhibits DNMT1 and ERR |
[115] |
| 2. | Vorinostat & Valproic acid + Trastuzumab | Reduction in MCL1, heightens ADCC and ADCP | [116] |
| 3. | Entinostat + Lapatinib | Transcriptional activation of FOXO3 and Bim | [117] |
| 4. | Vorinostat + Olaparib | HDAC, PARP | [97] |
| 5. | Valproic acid + Capecitabine | Upregulation of TP, downregulation of TS enzyme, reduction in thymidine synthesis | [118] |
| 6. | Azacitidine + Entinostat | Blocks DNA synthesis and HDAC class I activity | [119] |
| 7. | Panobinostat + Trastuzumab | Triggers NK-cell mediated immune response | [120] |
| 8. | Entinostat + Atezolizumab | Inhibits HDAC activity and PD | [121] |
| 9. | Letrozole + Panobinostat | Sensitizes BC cells to endocrine therapy, enhances H3, H4 acetylation, inhibits aromatase activity | [122] |
| 10. | Vorinostat + Paclitaxel | Acetylation of histone, |
[123] |
| 11. | Entinostat + Exemestane | HDAC class I inhibitor and Aromatase inhibitor | [75] |
| 12. | Tacedinaline + UNC0638 | Targets class I HDAC and G9a, suppresses BIRC5 and upregulates GADD45A | [99] |
| 13. | Capecitabine + Mocetinostat | Triggers apoptotic pathway by downregulating BCl2, Akt, HDAC1, PI3K, c-myc and upregulating Bax, Pten, C-Parp, Cas-7, Cas-9, p53 and Cas-3 | [100] |
| 14. | Chidamide + Exemestane | Targets HDAC 1,2,3&10 and aromatase activity | [101] |
| 15. | Hydralazine + Magnesium valproate + doxorubicin + cyclophosphamide | Demethylates DNA and inhibits HDAC activity, global decrease in C5me content | [124] |
| 16. | Phenelzine + Nab-Paclitaxel | Suppresses the generation of CSCs by inhibition of mesenchymal markers | [108] |
| 17. | Romidepsin + Gemcitabine + Cisplatin | TNBC cell death via ROS generation | [125] |
| 18. | Decitabine + Doxorubicin | Mitigates tumor proliferation, DNMT1 activity, and DNA methylation | [126] |
| 19. | Zebularine + Decitabine/Vorinostat | Dysregulation of cell proliferation and colony formation potential | [82] |
| 20. | Liraglutide + Paclitaxel/Methotrexate | Induction of global demethylation through the abrogation of DNMTs and transcription of CDH1, ESR1, and ADAM33 genes, resulting in reduced migration and viability | [85] |
DNMT, DNA Methyltransferase; MCL1, Myeloid Cell Leukemia 1; ADCC, Antibody-Dependent Cellular Cytotoxicity; FOXO3, Forkhead Box O3; HDAC, Histone Deacetylase; PARP, Poly ADP-Ribose Polymerase; TP, Thymidine Phosphorylase; TS, Thymidylate Synthase; NK, Natural Killer cell; PD, Programmed Death; BC, Breast Cancer; HDAC, Histone Deacetylase; CSC, Cancer Stem Cells, TNBC, Triple-Negative Breast Cancer; ROS, Reactive Oxygen Species; ADCP, Antibody-Dependent Cellular Phagocytosis; Bim, BCL2 Interacting Mediator of Cell Death; TP, Thymidine Phosphorylase; TS, Thymidylate Synthase; NK, Natural Killer; UNC0638, A specific chemical compound, a G9a and GLP inhibitor; BIRC5, Baculoviral IAP Repeat Containing 5; GADD45A, Growth Arrest and DNA-Damage-Inducible Alpha; BCl2, B-Cell Lymphoma 2; Akt, Protein Kinase B; PI3K, Phosphoinositide 3-Kinase.
Moreover, Letrozole, a widely recognized aromatase inhibitor, when paired with
Panobinostat, successfully reinstated ER
Breast Cancer is a heterogeneous disease considered dreadful, challenging to treat, nonresponsive to conventional treatment options, and likely to recur. The extensive research conducted in the recent decade on the role of epigenetic dysregulation in various cancers reveals the significant potential of epidrugs for clinical application. The advancement of breast cancer is significantly influenced by epigenetic regulators, encompassing DNA methylation, histone modification, and non-coding RNAs. Changes in the epigenome frequently contribute to metastasis, the acquisition of cancer stem cell traits, resistance to drugs, and disease recurrence. Moreover, epigenetic regulators and epithelial to mesenchymal transition transcription factors (EMT-TFs) have an intricate connection; subsequently, reversing the EMT process by targeting epigenetic enzymes is a viable and effective strategy. Whereas, regarding the therapy of solid tumors, including breast cancer, epi-drugs in monotherapy often represent substantial problems due to their poor tolerability, limited efficacy, and potential off-target effects. Therefore, it is necessary to adopt a more systematic strategy for integrating epi-drugs in combination with other cancer drugs. Utilizing appropriate delivery systems, coupled with the advantages offered by the latest generation of epi-drugs, holds the potential to improve the efficacy of breast cancer and other cancer therapies.
Conception and Design: SS, and SSG. Data acquisition and interpretation: SS, DV and TK. Drafting of the manuscript: SS, DV and TK. supervised the study: SSG. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
Not applicable.
We would like to acknowledge the Department of Biosciences and Bioengineering, and Centre for Nanotechnology at IIT Guwahati. We also acknowledge the School of Health Science and Technology of IIT Guwahati.
This work was supported by the Centre for Nanotechnology at IIT Guwahati.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.