


1 Second Clinical Medical College, Shaanxi University of Chinese Medicine, 712046 Xianyang, Shaanxi, China
2 Department of Gastroenterology, The First Affiliated Hospital of Xi’an Medical University, 710077 Xi'an, Shaanxi, China
3 Department of Pediatrics, Tangdu Hospital, Air Force Medical University, 710032 Xi'an, Shaanxi, China
4 School of Basic Medical Sciences, Xi'an Jiaotong University Health Science Center, 710061 Xi'an, Shaanxi, China
†These authors contributed equally.
Abstract
Colitis-associated cancer (CAC) is the most serious complication of inflammatory bowel disease. In recent years, the incidence of CAC has increased worldwide. Oxidative stress (OS) is involved in the development of CAC through oxidative damage to biomolecules or activation of inflammatory signaling pathways. Exosomes are extracellular vesicles that act as messengers to deliver signals and macromolecules to target cells, making them important mediators of intercellular communication and exchange of biologically active molecules between cells. MicroRNAs (miRNAs) carried by exosomes regulate the pro- and anti-inflammatory pathways of OS and play a key role in communication between OS and cancer cells. This review describes the correlation between OS and exosomal miRNAs with the goal of identifying a novel therapeutic method for CAC.
Keywords
- exosome microRNAs
- colitis-associated cancer
- oxidative stress
Colorectal cancer (CRC) ranks as the third most common cancer among both men and women in the United States, and is the third leading cause of cancer-related mortality. In 2023, about 153,020 people were diagnosed with CRC, and 52,550 people are expected to die from the disease [1]. Inflammatory bowel disease (IBD), comprising ulcerative colitis (UC) and Crohn’s disease, is a chronic inflammatory condition of the gastrointestinal tract [2]. IBD-associated CRC is worse than non-IBD-associated CRC [3]. Unlike sporadic CRC, which develops from normal colonic mucosa via the progressive accumulation of genetic alterations, colitis-associated cancer (CAC) follows an inflammation–dysplasia–carcinoma progression track. The incidence rate of CRC in IBD patients increases with duration of the illness, as confirmed by a meta-analysis, which showed that the cumulative risk of CRC for patients with UC was 2% at 10 years, 8% at 20 years, and 18% at 30 years [4]. The exact cause of IBD progression to CRC remains unknown; however, the inflammatory reaction of the intestinal mucosa upon exposure to oxidative stress (OS) is considered to be a significant factor in the transition from IBD to CAC [5, 6, 7].
OS is caused by an imbalance between free radicals and antioxidants [8]. Under physiological conditions, cells have a certain degree of antioxidant capacity. However, when the increase in reactive oxygen species (ROS) and reactive nitrogen species (RNS) is beyond the tolerance level of the cells, OS occurs. This results in DNA, protein, and lipid damage, inducing gene mutation, protein degradation, and lipid peroxidation, respectively, causing physiological and pathological reactions in cells and tissues. Patients with cancer have higher OS levels than the healthy population. Cancer cells have increased levels of nicotinamide-adenine dinucleotide phosphate (NADP), which protects cells from ROS by generating reduced forms of glutathione (GSH) and thioredoxin (Trx) [9, 10].
Recently, extracellular vesicles originating from cell membranes have received much attention. Exosomes are small extracellular vesicles that are secreted by cells when multivesicular endosomes fuse with the plasma membrane. They are abundant in bioactive molecules such as proteins, lipids, and nucleic acids, serving as vehicles for cell-to-cell communication [11, 12]. During cancer progression, tumor cells secrete exosomes containing cancer-causing proteins and nucleic acids that act on other cells, affecting the function of those cells and reprogramming them. The secretion of microRNA (miRNA) by tumor-associated exosomes not only promotes tumor growth but also changes the tumor microenvironment (TME), providing a favorable environment for further tumor growth [13, 14].
MiRNAs are a group of non-coding RNAs composed of 18–22 nucleotides, which play
a pivotal role in gene expression regulation by targeting mRNAs. They suppress
mRNA translation through specific sequence motifs, typically found in the mRNA
3
This review seeks to clarify the roles of OS and extracellular exosomal miRNAs in CAC, as well as the mechanisms by which OS regulates extracellular exosomal miRNAs, with the aim of identifying novel preventive strategies for CAC.
OS is triggered by an imbalance between ROS production and antioxidant defense
activity. ROS include hydrogen peroxide (H
CAC correlates with weakened antioxidant defenses and increased oxidative damage to proteins and DNA [20]. During the onset of IBD, inflammatory cell infiltration and crypt abscesses in the small intestine occur. Infiltrating neutrophils produce a large amount of ROS, which in turn cause OS. Because OS damages the intestinal mucosal barrier, harmful bacteria can infiltrate and inflammation will worsen [21]. Under normal circumstances, intestinal ROS play a key role in defenses against invading pathogens. However, when the host produces too much ROS, it can damage proteins, lipids, and nucleic acids, eventually leading to cancer. Prolonged OS can result in the oxidation of biomolecules or the activation of inflammatory signaling pathways [22].
ROS have the potential to cause DNA and protein damage, lipid peroxidation, genetic mutations, and cell death. ROS/RNS can react with protein amino acid residues to undergo protein oxidative modification, which is divided into two categories: reversible and irreversible oxidation. The protein adducts modified by oxidation include carbonylation, 3-nitrohydrocarbons, s-sulfidation, s-nitrosylation, s-GSH, and disulfide formation [23]. Cysteine is the residue in protein that most frequently undergoes reversible redox reactions with ROS/reactive halogen species [24].
ROS can disrupt double-stranded DNA; distort purines, pyrimidines, and deoxyribose; and induce transcriptional inhibition or activation, replication, and errors, all of which are linked to carcinogenesis [25]. The DNA damage caused by inflammatory OS is the main reason why patients with IBD are prone to cancer. One of the most researched oxidative metabolites, 8-hydroxy-2-deoxyguanosine, is thought to be a biomarker of DNA oxidative damage [26].
ROS is easily bound to unsaturated fatty acids, triggering free radical chain reactions of fatty acid radicals, peroxyfatty acid radicals, and electrophilic carbonyl groups [27]. Phospholipids are rich in unsaturated fatty acids and mainly participate in the formation of cell and organelle membranes. When OS occurs, excessive ROS leads to mitochondrial damage, oxidative respiratory chain disorders produce more ROS, and membrane damage causes the release of more ROS into the cytoplasm, resulting in a vicious cycle that leads to cell death and tissue damage [28]. Endoplasmic reticulum membrane peroxidation, in which calcium enters the cytoplasm, can promote the generation of nitric oxide, further exacerbating OS [29]. OS leads to lipid peroxidation, and the final product of the free radical chain reaction, malondialdehyde (MDA), reacts with DNA to form an MDA–DNA complex. MDA–DNA complexes can induce mutations in oncogenes and tumor suppressor genes in tumor cells; thus, MDA can serve as a biomarker for OS-mediated lipid peroxidation [30, 31].
OS is essential for the development of CAC. Tumor growth in the gut is
initiated by DNA alterations in intestinal epithelial cells caused by
H
OS can affect many signaling molecules, such as nuclear facto kappa B
(NF-
The pathophysiology of colitis involves activation of the NF-
A key component in the prevention of CAC is Nrf2 [42, 43]. Nrf2 is a transcription factor that regulates the redox balance and the expression of antioxidant enzymes, and mediates the induction of phase II detoxification reactions in mammalian cells [44]. Nrf2 is composed of seven functional Neh domains (Neh1–7), of which the Neh2 domain contains two ETGE and DLG motifs that specifically bind to Kelch-like ECH-associated protein 1 (Keap1), an inhibitory protein. Keap1 is responsible for the continuous degradation of Nrf2 by the ubiquitin-proteasome pathway under physiological conditions, maintaining Nrf2 at a low level [45]. The Nrf2/antioxidant response element (ARE) signaling pathway plays a crucial role in protecting cells from carcinogenesis [46]. AREs include NAD(P)H, NAD(P)H dehydrogenase [quinone] 1, superoxide dismutase, glutathione S-transferase, GSH peroxidase (GSH-Px), heme oxygenase-1, glutamate-cysteine ligase, catalase and Trx. When OS occurs, Nrf2 dissociates from Keap1. Then Nrf2 translocates into the nucleus and forms a heterodimer with small Maf proteins, facilitating the binding of Nrf2 to ARE/electrophilic reactive elements, thereby alleviating inflammation-related OS [47, 48, 49]. The activation of low-level Nrf2 is believed to inhibit OS and inflammation, which can prevent the development of CAC [50]. However, under the condition of a large amount of ROS, continuous activation of Nrf2 causes it to accumulate in the nucleus and bind to the kinesin family member 9 promoter, leading to ROS elevation and cell death [51].
Both OS and exosome-derived miRNAs are closely related to the development of CAC. Previous studies have shown that OS can alter the expression level of many miRNAs. Conversely, miRNAs can also regulate signaling pathways involved in OS responses, thereby promoting cancer development [52]. Therefore, OS and miRNAs both play critical roles in the development of CAC. In this section, we discuss the crosstalk between exosomal miRNAs and OS in CAC (Fig. 1; Table 1, Ref. [52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63]).
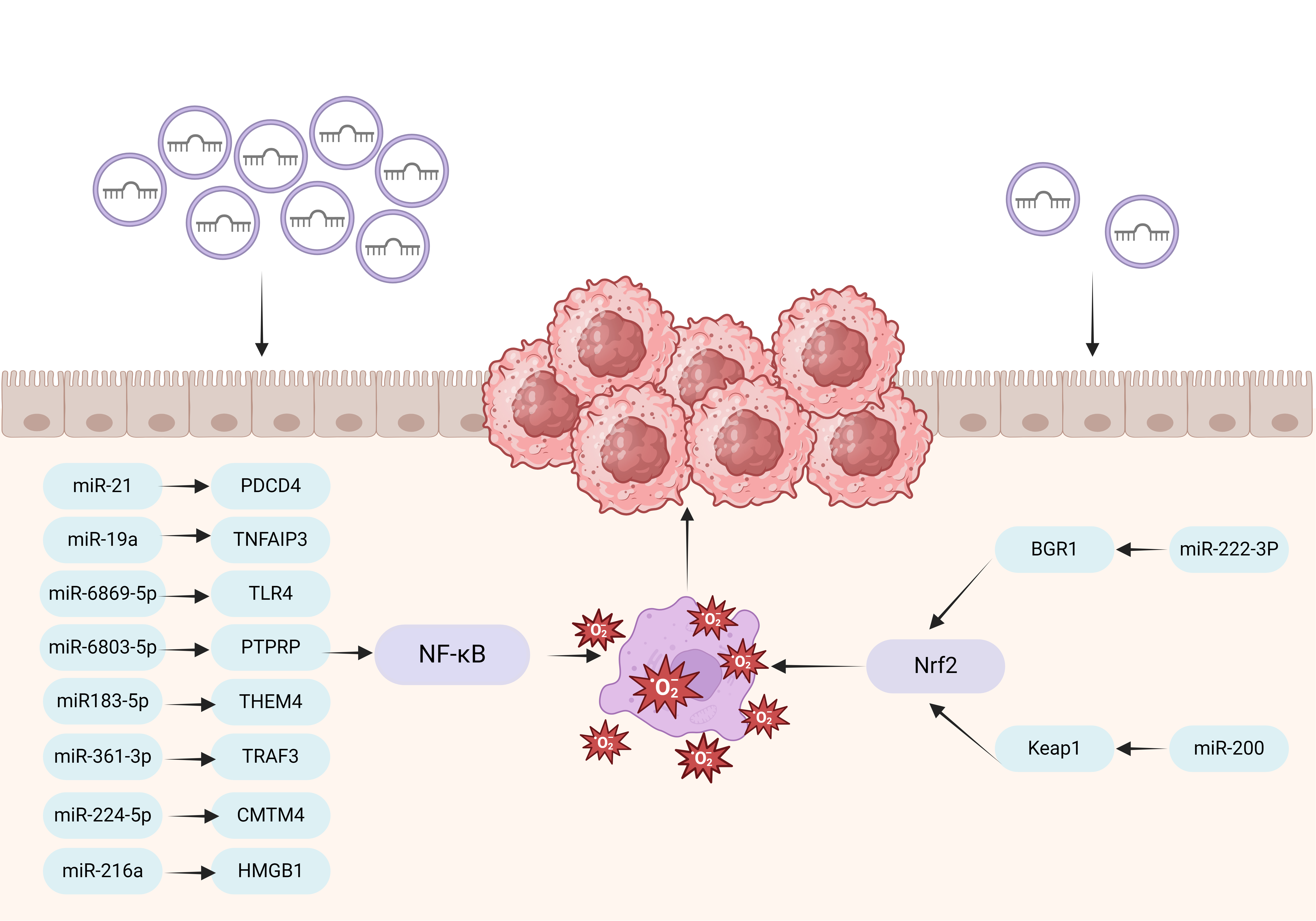 Fig. 1.
Fig. 1.
Interaction of exosomal miRNA and OS in CAC. Exosomal miR-21,
miR-19a, miR-6869-5p, miR-6803-5p, miR-183-5p, miR-361-3p, miR-224-5p, and
miR-216a on the left side of the figure act on NF-
| miRNA | Target | Pathway | Ref. |
| miR-21 | PDCD4, TPM1, PTEN | NF- |
[52] |
| miR-19a | TNFAIP3 | NF- |
[53, 54] |
| miR-6869-5p | TLR4 | NF- |
[56] |
| miR-6803-5p | PTPRP | NF- |
[57] |
| miR-183-5p | THEM4 | NF- |
[58] |
| miR-361-3p | TRAF3 | NF- |
[55] |
| miR-224-5p | CMTM4 | NF- |
[60] |
| miR-216a | HMGB1 | NF- |
[62] |
| miR-222-3p | BRG1 | Nrf2 | [59, 63] |
| miR-200 | Keap1 | Nrf2 | [61] |
PDCD4, programmed cell death 4; TPM1, The shortlisted genes, tropomyosin 1; PTEN, phosphatase and tensin homolog; TNFAIP3, Tumor necrosis factor alpha induced protein 3; TLR4, Toll-like receptor 4; PTPRP, protein tyrosine phosphatase receptor type R; THEM4, thioesterase superfamily member 4; TRAF3, tumor necrosis factor receptor-associated factor 3; CMTM4, CKLF like MARVEL transmembrane domain containing 4; HMGB1, high mobility group box 1 protein; BRG1, brahma-related gene 1.
CAC mice deficient in miR-21 have upregulated expression of its target gene
programmed cell death 4 (PDCD4), which affects NF-
miR-19a is markedly upregulated in the serum of CRC patients [56]. miR-19a
directly increases NF-
Decreased levels of miR-6869-5p have been found in serum exosomes from CRC
patients. MiR-6869-5p directly targets Toll-like receptor 4 (TLR4) and inhibits
cell proliferation and inflammatory cytokine production in CRC cells through the
TLR4 NF-
The exosome miR-6803-5p is increased in CRC and is negatively correlated with
protein tyrosine phosphatase receptor type O (PTPRO). PTPRO can exacerbate UC
inflammation by activating the TLR4/NF-
The proliferation and invasion of CAC cells are enhanced by overexpression of
the exosome miR-183-5p, which is generated from M2 macrophages. By focusing on
thioesterase superfamily member 4 (THEM4), miR-183-5p activates the
AKT/NF-
A crucial regulator of the advancement of cancer is the hypoxic TME.
Hypoxia-inducible factor 1 alpha causes CRC metastasis and the substantial
upregulation of miR-361-3p in hypoxic exosomes. The NF-
It has been discovered that the overexpression of miR-224-5p strongly suppresses
OS and increases the migration, invasion, and proliferation of normal human colon
epithelial cells. CKLF like MARVEL transmembrane domain containing 4
(CMTM4) is a tumor suppressor gene and in cancer, CMTM4 is a
key regulator of NF-
By encouraging macrophage polarization towards the M2 phenotype, exosomes
produced from adipose-derived stem cells may be able to reduce colitis
inflammation. Exosomes from hypoxia contain more miR-216a-5p than those from
normoxia, and the treatment is better. MiR-216a-5p can regulate macrophage
balance by regulating the high mobility group box 1 protein
(HMGB1)/TLR4/NF-
The expression levels of all members of the miR-200 family have been observed in mesenteric vein exosomes at higher levels than in plasma exosomes in Spanish patients with CRC [73]. In dextran sulfate sodium-induced colon injury, miR-200a targets Keap1 and activates Nrf2-regulated antioxidant pathways, alleviating excessive inflammatory activation, intestinal cell apoptosis, and colon dysfunction. OS and inflammatory responses are key factors in the pathogenesis of UC [63].
The integrity of the intestinal barrier and innate immunity depend on IECs.
Recent studies have revealed elevated levels of exosomal miR-222-3p in the
bloodstream of CRC patients. An essential function of miR-222-3p is to regulate
OS. Both in vitro and in vivo studies have assessed the effects
of miR-222-3p-mediated OS on UC and CAC in a mouse model of colitis. Targeting
Brahma-related gene 1, suppression of miR-222-3p has been found to activate the
Nrf2/HO-1 signaling pathway in IECs from CAC, which increases GSH-Px levels while
lowering ROS and MDA levels, leading to decreased TNF-
There are numerous medications available for treating CAC [76, 77]. 5-fluorouracil (5-FU) is commonly used in CRC patients. However, the multidrug resistance caused by long-term use of 5-FU severely weakens the treatment effect, and resistance to 5-FU is one of the main reasons for CRC recurrence. Compared with artificially synthesized drug carriers, exosomes, as natural carriers, are rapidly cleared by the monocyte/macrophage system. Therefore, exosomes have advantages such as stable properties, easy immune escape, long circulation time, no obvious toxic side effects, ability to load multiple drugs to the target, specific delivery, and the ability to utilize different intracellular transport pathways to exert their effects [14, 78, 79]. There are two ways to prepare exosomes as drug carriers: modify proteins or nucleic acids in exosomes through genetic modification, and place cells that release exosomes into therapeutic agents, so that the therapeutic agent is included in the exosomes [80]. In summary, exosomes have natural advantages over other synthetic carriers and treatment methods, and have great potential for the treatment of tumor diseases.
Exosome miRNAs can regulate the occurrence and persistence of inflammatory
responses. Inflammation-related miRNA expression levels can be controlled to
prevent the release of inflammatory mediators; lessen inflammation and damage to
intestinal mucosa; promote the growth, differentiation, and repair of IECs;
preserve the integrity of the intestinal epithelial barrier; and ultimately lower
the risk of CRC [81]. The miR-129-5p produced from human umbilical cord
mesenchymal stem cell-derived exosomes (HucMSC-Ex) target acyl-CoA synthetase
long chain family member 4 (ACSL4) and decreases its expression to lessen
intestinal inflammation and repair damage, which helps to relieve IBD. Lipid
peroxidation is positively regulated by ACSL4 [82]. According to previous
research, husMSC-Ex miRNA is crucial in inhibiting the growth of CAC. In a mouse
model of colitis caused by dextran sodium sulfate and azioxymethane, husMSC-Ex
miR-146a prevented the production of small ubiquitin-related modifier 1 and its
attachment to
In recent years, there has been increasing research on exosomes. Extracellular miRNAs from tumors can recode other untransformed cells in the TME. Extracellular miRNAs are more stable than free miRNAs. Extracellular miRNAs protect miRNAs and transport them to target mRNA, playing a key role in cancer progression by acting as tumor inhibitors or oncogenes.
Detecting the changes in exosomal miRNA before and after treatment can improve the efficiency of treatment. However, exosome extraction presents some challenges because of its small size and low density. Extracellular vesicle separation has been achieved through the development of six techniques: precipitation, size exclusion chromatography, ultrafiltration, ultracentrifugation, immunoaffinity-based capture, and microfluidics [85]. Future studies need to further establish more accurate and reliable detection techniques of exosomal miRNAs.
Several studies have shown that certain exosomes miRNAs can prevent the
development of CAC by suppressing inflammatory responses, regulating the immune
system, and alleviating damage to IECs, among other mechanisms. For example, by
binding to the coding region of large tumor suppressor kinase 1 (LATS1), exosomal
miR-590-3p generated from M2 macrophages downregulate LATS1 expression. The
transcription mechanism mediated by Yesassociated protein/
These review summaries the association between exosomal miRNAs and OS in CAC.
These exosomal miRNAs act on the NF-
IBD, inflammatory bowel disease; ROS, reactive oxygen species; RNS,
reactive nitrogen species; PPP, pentose phosphate pathway;
AMPK, adenosine 5
YFL and HYL reviewed the literature and drafted the manuscript. MLC and YZ: Data curation, Writing-original draft. MXZ and MZZ: Conceptualization, Writing-Review & Edition. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The author sincerely thanks all the people for their help, whose advice and encouragement have made me have a deeper understanding of this aspect. The figure is created with BioRender.com.
Shaanxi Province Key Research and Development Program (2024JC-YBMS-664); The Innovation Team of Xi’an Medical University (2021TD15); Xi’an Science and Technology Plan Project (24YXYJ0143).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.