





 , Ziming Wang 1,†, Yixuan Duan 1, Chang Liu 1, Sihai Zhao 2, Jie Deng 1,*
, Ziming Wang 1,†, Yixuan Duan 1, Chang Liu 1, Sihai Zhao 2, Jie Deng 1,*
1 Department of Cardiology, The Second Affiliated Hospital of Xi’an Jiaotong University, 710004 Xi’an, Shaanxi, China
2 Laboratory Animal Center, Xi’an Jiaotong University School of Medicine, 710061 Xi’an, Shaanxi, China
†These authors contributed equally.
Abstract
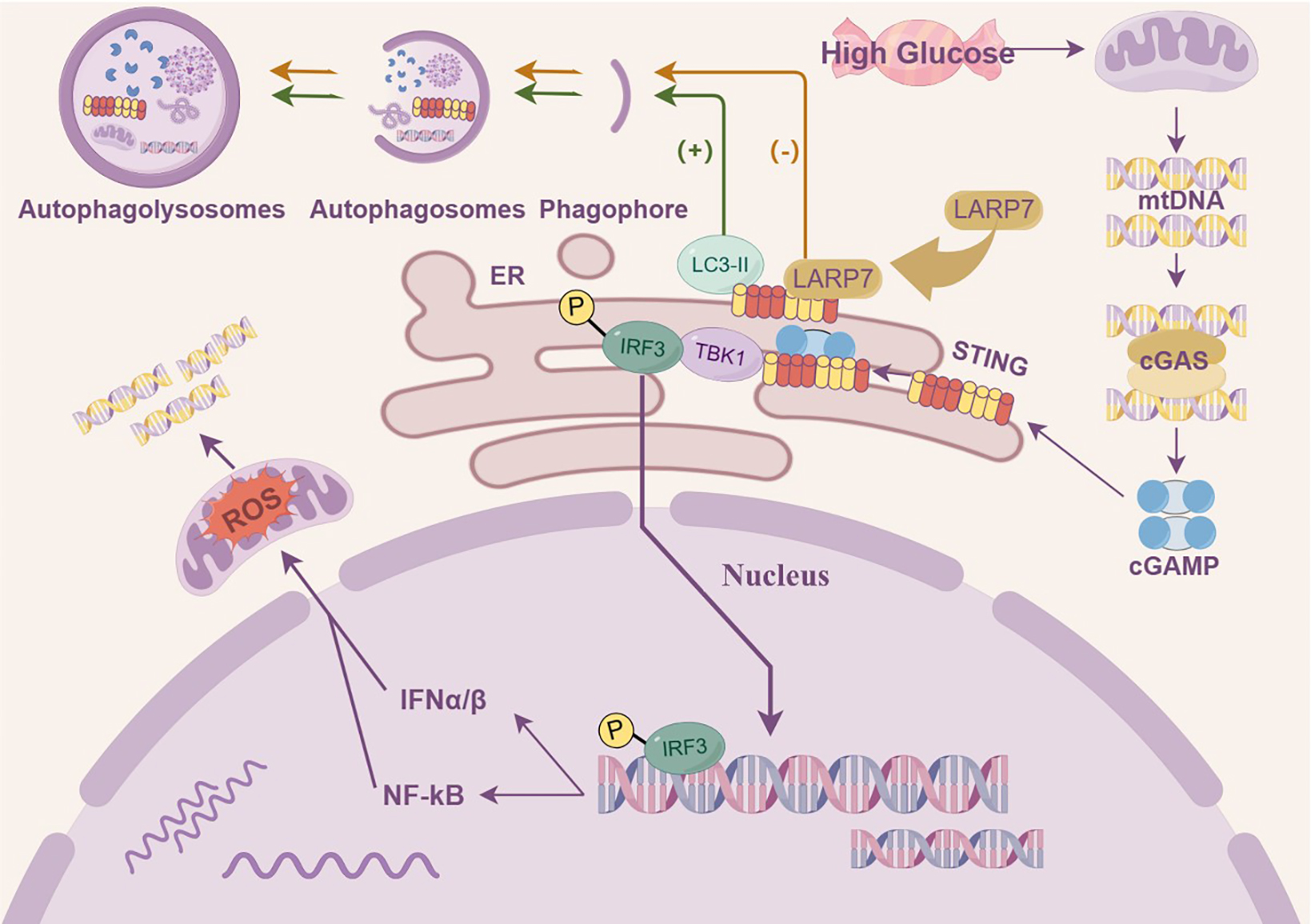
Background: Diabetic cardiomyopathy (DCM) is an important cause of heart failure in diabetic patients. The aim of this study was to investigate the pathogenesis of DCM and to identify potential therapeutic targets. Methods: A mouse model of type 1 DCM was constructed by continuous intraperitoneal injection of streptozotocin (STZ). Systolic and diastolic functions were measured by ultrasound. The expression of La-related protein 7 (LARP7), the stimulator of interferon genes (STING) pathway and light chain 3 (LC3) in myocardial tissue was detected by Western blot and immunofluorescence analyses. Neonatal mouse ventricular cardiomyocytes (NMVCMs) were isolated and cultured. An in vitro type 1 diabetes mellitus (T1DM) model was established by treatment with high glucose. Knockdown/overexpression of LARP7 and STING was achieved by adenovirus transduction, C-176 (a potent covalent inhibitor of STING), and plasmid transfection. The expression, activation, and localization of STING and LARP7 in cardiomyocytes was evaluated, as well as the interaction between the two. The effect of this interaction on the STING-dependent autophagy‒lysosomal pathway was also explored. In addition, the fibrosis and apoptosis of cardiomyocytes were evaluated. Results: High glucose was found to increase the expression and activation of STING and LARP7 in mouse myocardial tissue. This was accompanied by myocardial fibrosis, impaired autophagy degradation function and impaired cardiac function. These findings were further confirmed by in vitro experiments. High glucose caused LARP7 to translocate from the nucleus to the cytoplasm, where it interacted with accumulated STING to inhibit its degradation. Inhibition of STING or LARP7 expression significantly improved myocardial injury induced by high glucose. Conclusions: Targeted inhibition of LARP7 or STING expression may be a potential therapeutic strategy for the treatment of DCM.
Graphical Abstract
Keywords
- diabetic cardiomyopathy
- LARP7
- STING
- autophagy
- apoptosis
- fibrosis
Diabetes mellitus (DM) is a major chronic disease that endangers human health. The cardiovascular damage caused by DM has become the main cause of death in diabetic patients [1]. Diabetic cardiomyopathy (DCM) is one of the cardiac complications of DM and refers to impaired cardiac filling and decreased systolic function, which ultimately induces heart failure [2, 3]. Current treatment methods for DCM focus on controlling blood sugar, delaying ventricular remodeling, and correcting heart failure. However, there is still a lack of effective treatments for DCM. Conventional treatments place a heavy financieal burden on patients and have limitations. Despite the standardized application of drugs such as sodium-dependent glucose transporters 2 (SGLT2) inhibitors, spironolactone, angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs), many diabetic patients still suffer end-stage heart failure due to DCM [4, 5, 6, 7].
Stimulator of interferon gene (STING) is a signal transduction molecule closely
associated with the innate immune response. It is induced by virus invasion and
cell damage and activates the inflammatory response through the induction of type
I interferons (IFN-I) to resist exotic pathogens [8]. Of note, pathogenic
microbial DNA can induce the expression and activation of STING. Moreover,
endogenous DNA release caused by various intracellular toxic stresses can have
the same effect [9]. Increasing evidence suggests that STING plays an important
role in regulating insulin sensitivity and inducing ventricular remodeling in
diabetic patients. On one hand, activation of the STING pathway can affect the
homeostasis of glucose metabolism by mediating pancreatic
LARP7 is a member of the La-related protein (LARP) family. It acts as an
important negative transcriptional regulator of RNA polymerase II and is also
involved in the induction of ventricular hypertrophy [18, 19]. More than 50% of
LARP7 in human cells forms part of the 7SK complex, which includes 7SK RNA,
methyl phosphate capping enzyme (MePCE), hexamethylene
bis-acetamide inducible (HEXIM1/2), and positive transcription elongation factor
b (P-TEFb). LARP7 binds to the C-terminal UUU-3
In the present study, a high glucose-induced DCM cell model was used to investigate the role of free LARP7 in the process of glucose-induced cardiac dysfunction, apoptosis and fibrosis. The results indicate that high glucose inhibits the degradation of STING through the autophagy-lysosomal pathway. Additionally, LARP7 expression was upregulated and its intracellular localization was altered. Moreover, the degradation of STING was inhibited by its interaction with LARP7. The accumulation of STING in vivo not only caused inflammation and immune responses, but also induced myocardial remodeling and cardiac dysfunction by inducing cardiomyocyte fibrosis and apoptosis. Downregulation of STING or LARP7 expression significantly inhibited the occurrence of fibrosis and apoptosis. This study identified a regulatory effect of free LARP7 on the STING pathway, thus providing new insights into the pathogenesis of DCM. Furthermore, it suggested that targeting of STING or LARP7, either alone or in combination, could serve as a novel therapeutic strategy for the treatment of DCM.
Male C57BL/6J mice (6–8 weeks old) were purchased from the Experimental Animal
Center, Fourth Military Medical University (Xi’an, Shaanxi, China). Mice were
randomly assigned to a control group and to a Type 1 diabetes mellitus (T1DM)
group. An Streptozotocin (STZ) solution (Sigma-Aldrich, St. Louis, MO, USA, 60 mg/kg) was injected
intraperitoneally for 5 consecutive days to construct a type 1 DCM mouse model.
The control group was injected with an equal volume of citrate buffer. Blood
samples were collected via the tail vein on days 3, 5 and 7 after the last
injection. Mice with a random blood glucose level
Mice were anesthetized with 2% isoflurane. Echocardiography was used to calculate cardiac function with the UBM system (Vevo 770, Toronto, Canada). Left ventricular fractional shortening (FS) and the ejection fraction (EF) were assessed using Vevo Analysis software (version 3.0.0, VisualSonics, Toronto, Canada). To assess diastolic function, an apical four-chamber view of the left ventricle was obtained. Maximal early (E) and late (A) transmitral velocities in diastole were analyzed to measure the mitral inflow velocity spectrum. Echocardiography was conducted by investigators who were blinded to the study.
After the T1DM model was successfully constructed, mice were sacrificed by cervical dislocation. Left ventricular specimens were obtained, fixed with 4% paraformaldehyde and embedded in paraffin. Tissue samples from the myocardium were then cut into serial sections (5 µm thickness) from the mid-transverse area. The sections were heated to retrieve antigen and then incubated with anti-LC3 antibody overnight at 4 °C. After incubating with a universal two-step kit (anti-rabbit/anti-mouse PV-9000 secondary antibodies) at room temperature for 1 h, diaminobenzidine was used for color rendering and hematoxylin to stain the nuclei. Sections were then cleared in xylene and sealed with neutral gum. An isotype control antibody was used as the negative control. A digital scanner (Pannoramic MIDI, 3DHISTECH, Budapest, Hungary) was used to analyze the immunohistochemistry results. The sections were stained with picrosirus red to display the collagen components. Statistical analysis was carried out using the grading system described in the immunohistochemical technique method of the Chinese Pathological Society.
CY3-labeled STING, FITC-labeled LARP7, and 4′,6-diamidino-2-phenylindole (DAPI) were used in the immunofluorescence analysis. An anti-fluorescence quencher was used for mounting, and a laser confocal workstation was used for immunofluorescence photography. Semi-quantitative analysis of fluorescence images was performed using ImageJ software (version 1.6.0, National Institutes of Health (NIH), Bethesda, MD, USA).
Cardiomyocytes were obtained from the ventricles of 1–3-day old neonatal
C57BL/6 mice. The mice were sterilized with 75% ethanol and the hearts removed
and rinsed in ice-cold phosphate-buffered saline (PBS, Servicebio, Wuhan, China). The myocardial samples
were then cut into small pieces and digested with collagenase type 2
(Sigma‒Aldrich, Saint Louis, MO, USA) until the tissue pieces had
dissolved. The suspension was then mixed with dulbecco’s modified eagle medium
(DMEM) (ScienCell, San Diego, CA, USA) to stop digestion. The mixture was
centrifuged (800
For the knockdown or overexpression of LARP7, NMVCMs were infected with
adenovirus carrying negative control (NC-small interfering RNA (siRNA)), knockdown of rat LARP7
gene (LARP7-siRNA), or overexpression of the LARP7 gene (m-LARP7) at an
MOI of 100 from Hanbio Tech (Shanghai, China). STING was exogenously
overexpressed in NMVCMs via plasmid transfection (m-3*Flag-STING). The
corresponding sequences were: LARP7-siRNA (5
The cell medium from NMVCMs was discarded and the cells washed with PBS. The
samples were then homogenized in pre-cooled protein lysis buffer containing
phenylmethylsulfonyl fluoride (PMSF) (1 mM, pH = 7.4) (HY-B0496, MedChemExpress,
Shanghai, China), lysed by ultrasound, and centrifuged for 30 min to extract
total protein. Protein quantification was performed using the
bicinchoninic acid assay (BCA) method. Total protein (20
µg) was electrophoresed on a 10% denaturing polyacrylamide gel and
the separated proteines then transferred onto a polyvinylidene fluoride (PVDF)
membrane. After blocking with 5% non-fat milk, the membrane was incubated
overnight at 4 °C with the following primary antibodies: cGAS, p-STING
(Ser365), STING, LARP7, INF-
To evaluate the extent of cardiomyocyte remodeling,
Cardiomyocyte apoptosis was assessed using the DNA ladder assay, terminal deoxynucleotidyl transferase mediated nick end labeling (TUNEL) staining, and Western blot analysis of Cl-caspase3. TUNEL staining was used to stain myocardial tissue sections, as recommended by the instructions provided with the apoptosis detection kit (Roche, Basel, Switzerland). After mounting with an anti-fluorescence quenching agent, the sections were observed and photographed under a laser confocal microscope (Leica TCS SP8, Leica, Hessen, Germany), thus allowing the cell apoptosis rate to be determined [24].
In the DNA ladder assay, myocardial DNA was extracted using a nucleic acid extraction kit (IsoQuicks, Microprobe, Carlsbad, CA, USA). The extracted DNA (10 mg) was added to a 2% agarose gel containing ethidium bromide and electrophoresed in Tris-borate-EDTA (TBE) buffer at 100 V for 2 h. The DNA ladders were then photographed.
The colocalization in cardiomyocytes of STING and LC3, and of STING and LARP7, were evaluated separately by immunofluorescence. NMVCMs were seeded into confocal dishes and then treated. The culture medium was discarded and the cells washed three times with sterile PBS, incubated in permeabilization buffer (0.1% Triton X-100 in PBS) for 15 min to rupture cell membranes, and incubated in a BSA solution for 60 min to block nonspecific binding. The cells were then incubated overnight at 4 °C with rabbit-derived STING, LC3 and LARP7 antibodies. Subsequently, the cells were incubated with an appropriate secondary antibody for 90 min at room temperature. Different molecules are labeled with different fluorescence which was evaluated using a laser confocal microscope. The extent of merging of different groups of fluorescent particles was analyzed and calculated using Image-Pro PLUS software (Media Cybernetics, Silver Spring, MD, USA) to evaluate the colocalization of STING and LC3, as well as that of STING and LARP7.
COIP was conducted using a Proteintech immunoprecipitation (IP) kit (KIP-2).
NMVCMs were pre-processed and grouped. To fully lyse cardiomyocytes, a total of
100 µL of precooled IP lysis buffer (containing 1
The immunoprecipitation (IP) procedure used for the preparation of samples for Mass Spectrometry (MS) was the same as that described above. The IP samples from each group were electrophoresed to obtain gel strips, and the peptides in these strips were then enzymatically hydrolyzed. The hydrolyzed peptides were separated by column chromatography and subsequently injected into a tandem MS for primary and secondary MS analyses.
Ventricular cardiomyocytes pretreated with high glucose were fixed with 3.0% glutaraldehyde and 1.5% paraldehyde, washed 3 times with PBS, and finally fixed with 1% osmium tetroxide for 1 h to produce osmium black. Samples were then dehydrated with ethanol and embedded in epoxy resin. A transmission electron microscope (H-7650, Tokyo, Japan) was then used to observe the number of myocardial autophagosomes and autophagolysosomes in myocardial tissue, as well as the damage to mitochondria, endoplasmic reticulum and other organelles.
Continuous variables were presented as the mean
The expression of STING and LARP7 was evaluated in myocardial cells from a mouse
model of type 1 DCM, while cardiac systolic and diastolic function were evaluated
by echocardiography. The systolic function index in the T1DM group was
significantly lower than in the control group. High glucose levels induced LV
enlargement. Meanwhile, the E/A ratio was markedly decreased in the T1DM group,
indicated the mice had cardiac diastolic dysfunction (Fig. 1A). In addition, the
expression of STING and LARP7 in the myocardial tissue of mice from the T1DM
group was significantly higher than in the control group (Fig. 1B–E). This
finding was further confirmed by Western blot analysis, which revealed
significant upregulation of the upstream signaling molecule cGAS and downstream
effector molecule INF-
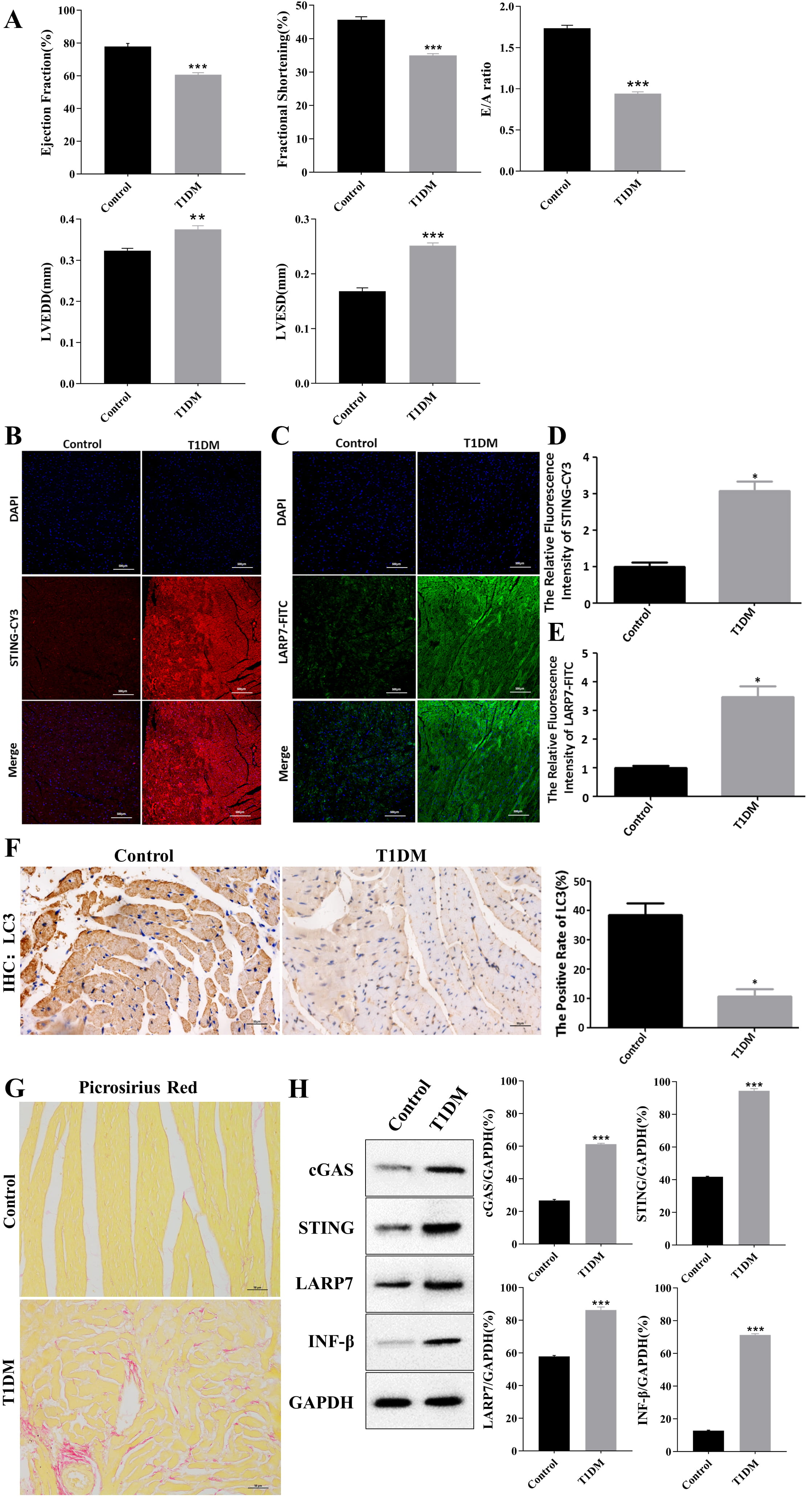 Fig. 1.
Fig. 1.Increased expression of STING and LARP7 in the myocardial tissue
of mice with type 1 DCM, and inhibition of autophagy. (A) High glucose levels
induced systolic and diastolic dysfunction in T1DM mice. Bar charts show
decreased EF, FS, left ventricular end-diastolic internal diameter (LVEDD), left
ventricular end-systolic internal diameter (LVESD) and E/A ratio after STZ
injection. **p
Western blot analysis showed that the expression and activation levels of STING
were significantly upregulated in the HG group. This effect occurred in parallel
with increases in
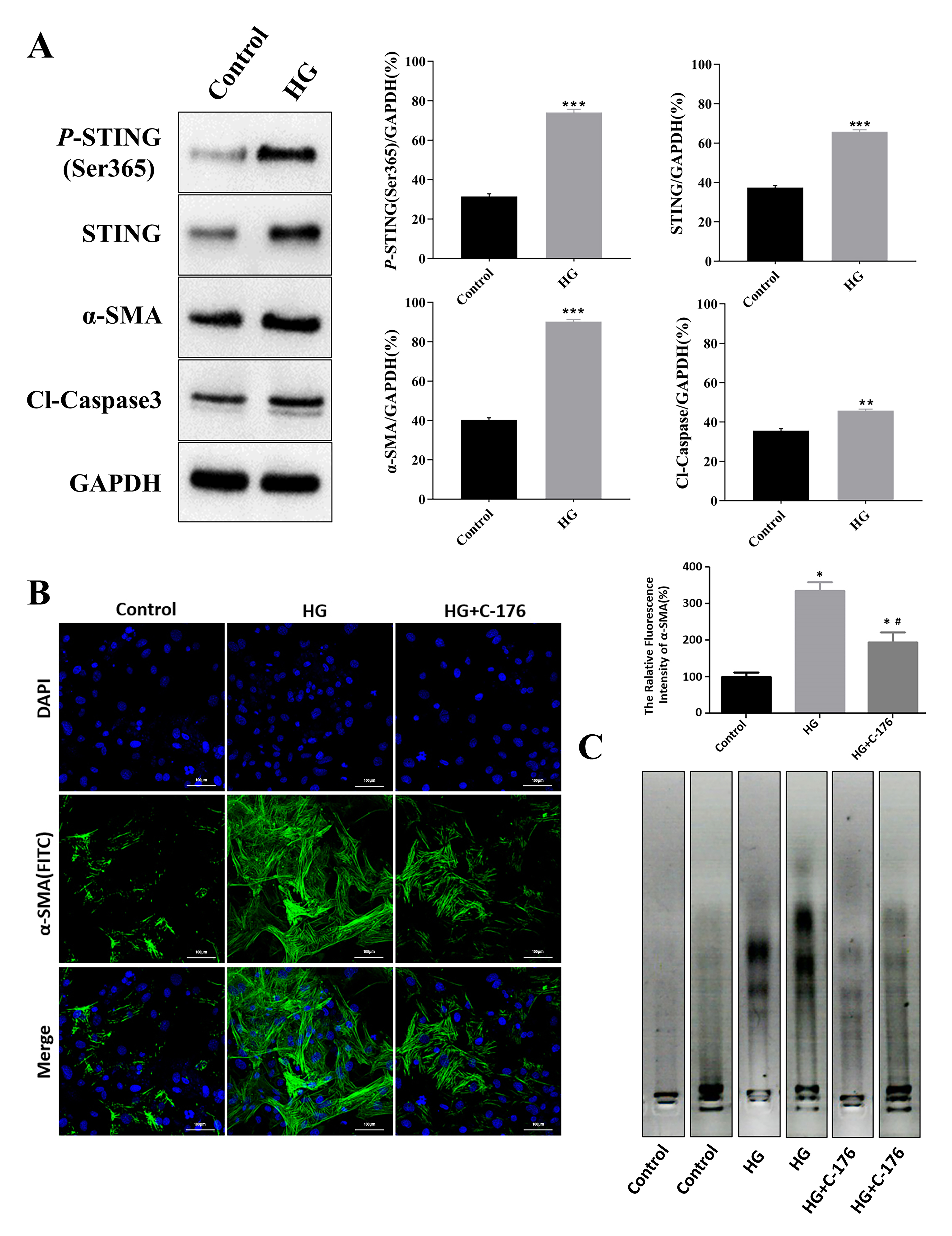 Fig. 2.
Fig. 2.High glucose enhanced the expression and activation of STING in
cardiomyocytes. (A) Western blot results for STING, p-STING,
STING contains 7 regions (LIR1-7) that bind directly to the autophagy molecule LC3. This region induces LC3 lipidation and activates autophagy (Fig. 3A). To explore the effect of high glucose on autophagy, immunofluorescence colocalization was used to study the intracellular expression of LC3 and STING. LC3 expression and autophagy were lower in the HG group compared to the control group (Fig. 3B). COIP experiments further confirmed that high glucose reduced the binding ability of LC3 to STING (Fig. 3C). Transmission electron microscopy revealed the presence of significantly fewer autophagosomes, autophagolysosomes, and lysosomes in cardiomyocytes from the HG group compared to the control group. In addition, many of the mitochondria were swollen, damaged, and ruptured (Fig. 3D). Together, these results indicate that high glucose inhibits autophagy. This was further confirmed by Western blot analysis, which showed a significant decrease in the expression of autophagy signature proteins such as LC3II/I, Beclin-1 and P62 in NMVCMs treated with high glucose (Fig. 3E). Treatment of cardiomyocytes with the autophagy inhibitor 3-MA significantly increased the expression of STING in the cytoplasm. This was accompanied by cardiomyocyte shrinkage and poor cytoskeletal continuity. The number of apoptotic cardiomyocytes detected by the TUNEL assay was also significantly increased (Fig. 3F–I). In summary, high glucose interfered with STING degradation through the autophagy-lysosomal pathway by inhibiting the binding of STING to LC3. This resulted in accumulation of STING, leading to structural damage and increased cardiomyocyte apoptosis.
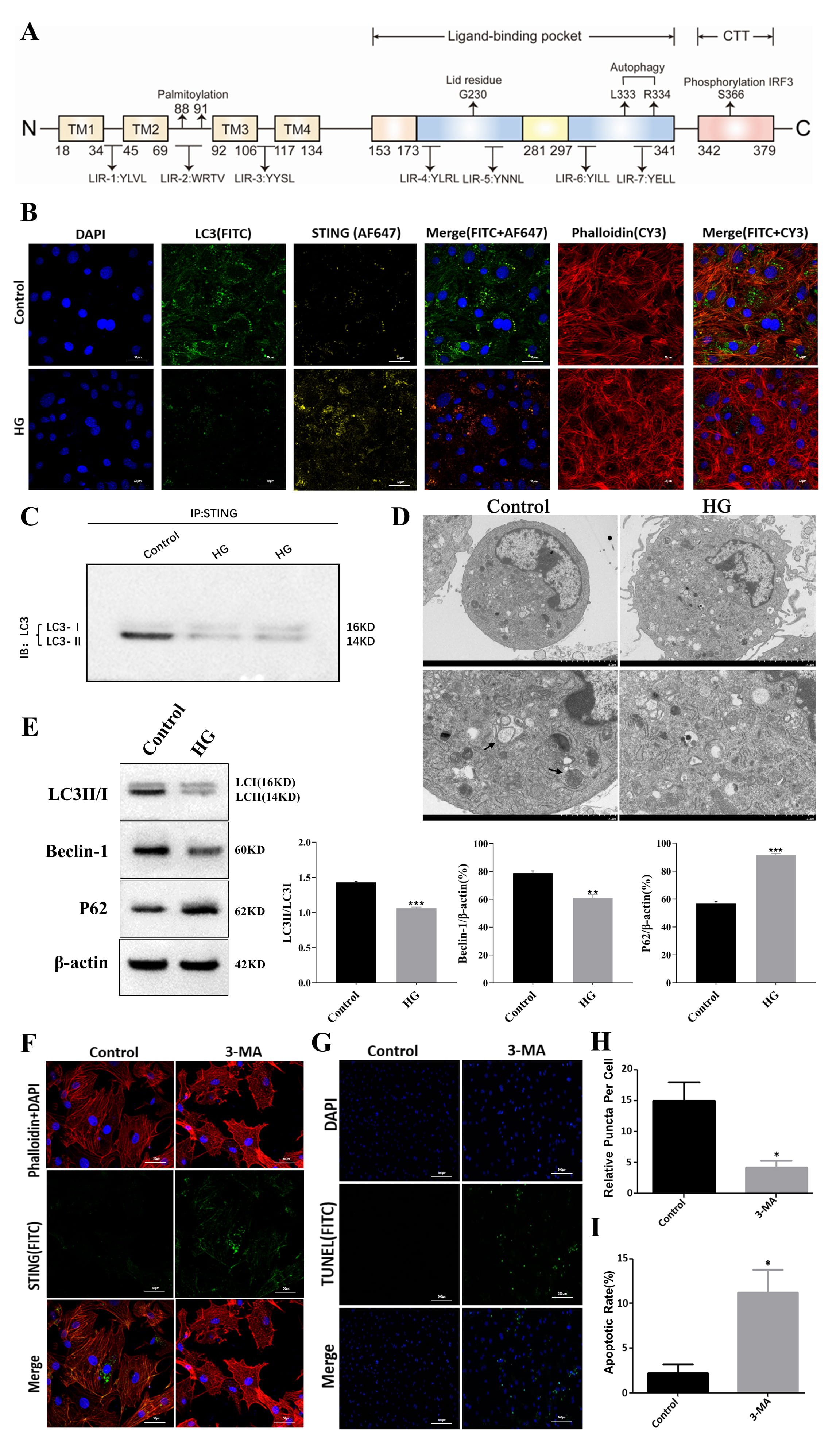 Fig. 3.
Fig. 3.High glucose levels disrupted the STING-triggered autophagy
“negative feedback loop”. (A) Molecular structure of STING. (B)
Immunofluorescence colocalization of STING and LC3 in NMVCMs treated with high
glucose: green fluorescent particles represent LC3, yellow fluorescent particles
represent STING, and yellow fluorescent particles that overlap with green
fluorescent particles suggest that STING directly interacts with LC3 (scale bar = 50
µm). (C) Coimmunoprecipitation (COIP) results for LC3 and STING. (D) TEM
image of cardiomyocytes treated with high glucose. The black arrow represents
autophagolysosomes (scale bar = 5 µm (overview); scale bar = 2 µm (inset)). (E) Western blot results for LC3II/I,
Beclin-1 and P62 in NMVCMs treated with high glucose. Two-tailed Student
t test; **p
To directly explore the role of free LARP7 in DCM, the expression of LARP7 and STING were inhibited with siRNA and C-176, respectively. Western blot results showed that STING expression was significantly upregulated in the HG group. In the groups in which LARP7 or STING were inhibited, activation of the STING pathway was attenuated compared with the HG group, and cardiomyocyte apoptosis was reduced. The inhibition of LARP7 by siRNA following the inhibition of STING by C-176 had a more obvious inhibitory effect on the STING pathway (Fig. 4A). In addition, cardiomyocyte apoptosis was further reduced. These findings suggest that a regulatory mechanism between free LARP7 and STING mediates the expression and activation of STING, thereby aggravating cardiomyocyte damage.
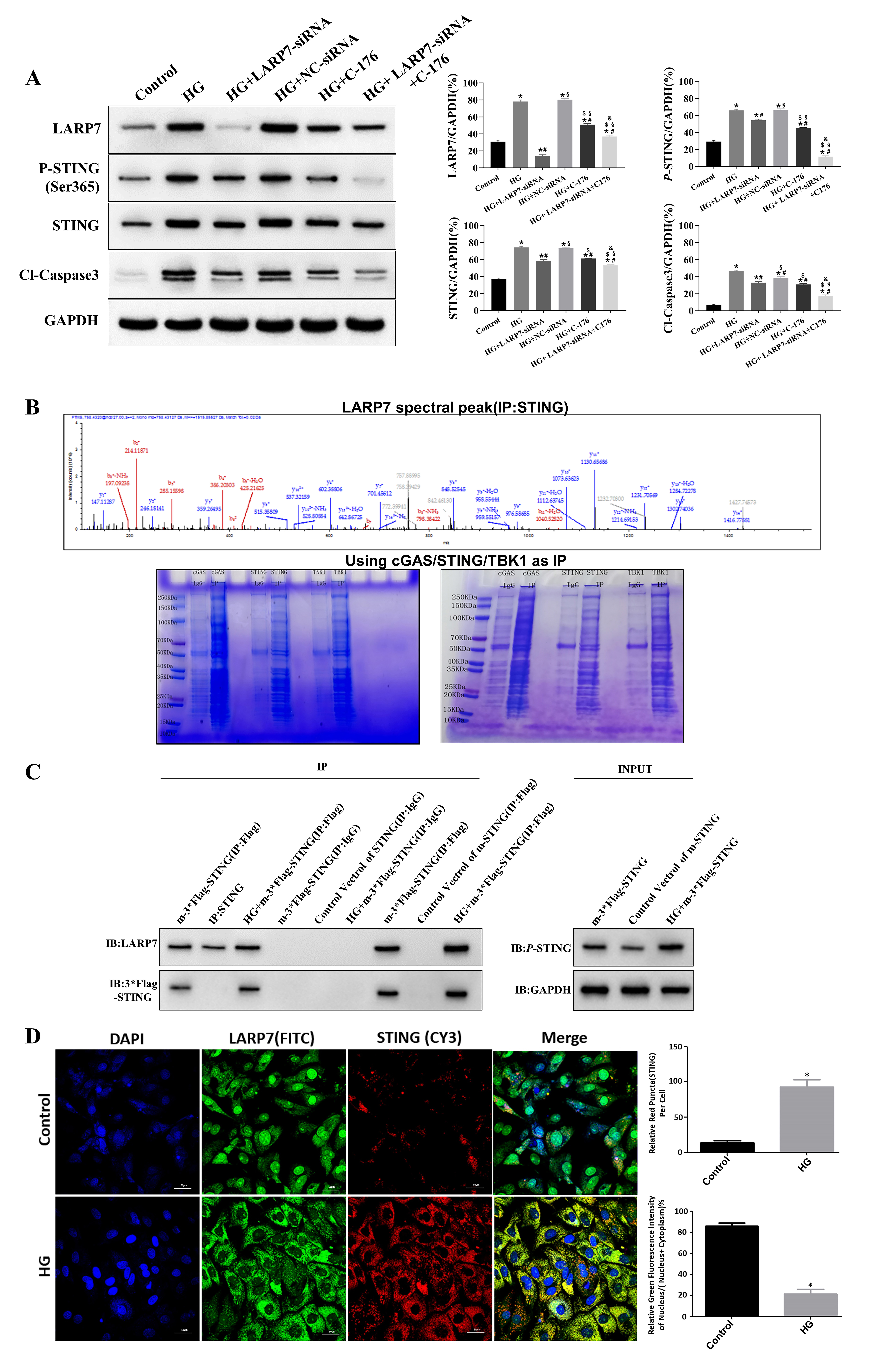 Fig. 4.
Fig. 4.LARP7 translocates from the nucleus to the cytoplasm, where its
interaction with STING results in the aggravation of myocardial injury. (A)
Western blot results for LARP7, STING, p-STING, and Cl-caspase3 in NMVCMs in
which the expression of LARP7 and STING was inhibited by siRNA and C-176,
respectively. One-way ANOVA with the Bonferroni-corrected t test;
*p
MS analysis of three important molecules in the STING signaling pathway, cGAS-STING-TBK1, revealed a very high abundance of LARP7 in the final STING IP solution compared with the negative control (Fig. 4B). COIP experiments confirmed that both exogenously overexpressed STING and endogenous STING interacted with LARP7, and that high glucose enhanced this interaction (Fig. 4C). Next, LARP7 and STING were fluorescently labeled to visualize their intracellular localization. After high glucose treatment, a large amount of LARP7 was observed to translocate from the nucleus to the cytoplasm, where it colocalized with STING (Fig. 4D). Taken together, these results indicate that high glucose induced the intracellular translocation of LARP7, and that its interaction with STING led to accumulation in the cytoplasm where it mediated cardiomyocyte apoptosis.
To further explore the mechanism by which LARP7 and STING interact to induce
myocardial injury, the expression of LARP7 was first inhibited with siRNA. STING
was then overexpressed via 3
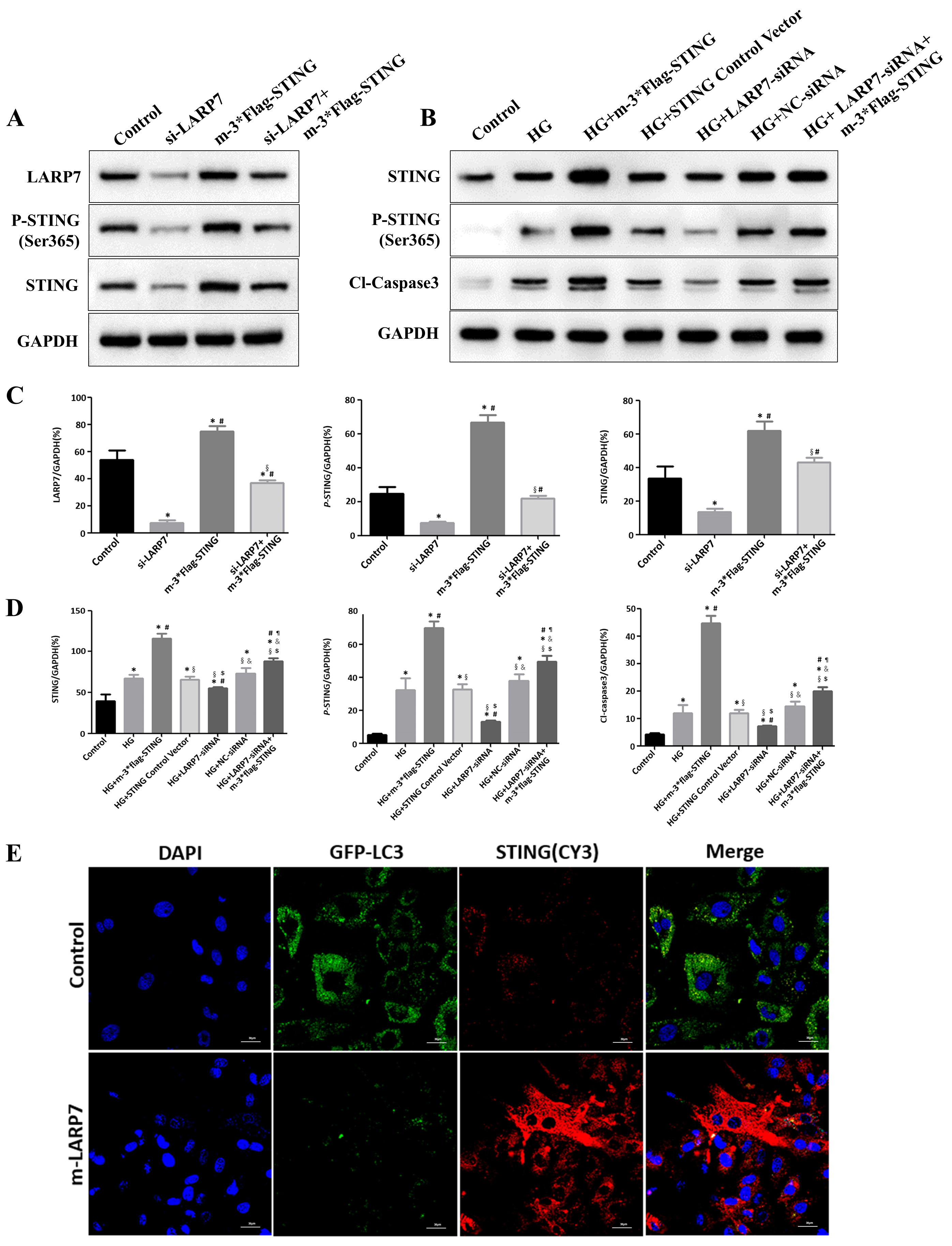 Fig. 5.
Fig. 5.LARP7 mediates myocardial injury by inhibiting the
autophagy-lysosomal degradation pathway. (A,C) Mouse cardiomyocytes were treated
with siLARP7, m-3*Flag-STING, or siLARP7+m-3*Flag-STING. Western blot analysis
was then performed on LARP7, STING, and p-STING (Ser365). One-way ANOVA with the
Bonferroni-corrected t test; *p
To our knowledge, this is the first study to investigate the role and mechanism of LARP7 in glucose-induced cardiac dysfunction, apoptosis and fibrosis. The main finding of this research is that high glucose induces the expression and activation of STING in ventricular myocytes, and inhibits the degradation of STING through the autophagy-lysosomal pathway. In addition, high glucose levels cause LARP7 to dissociate from the 7SK RNP complex and translocate to the cytoplasm where it interacts with STING. This inhibits the degradation of STING and causes it to accumulate and to activate downstream signaling pathways, resulting in cardiac dysfunction, fibrosis and apoptosis. Inhibition of STING or LARP7 expression significantly ameliorated high glucose-induced myocardial injury.
STING is an important signal transduction molecule and has been implicated in various diseases [25, 26, 27]. Under the action of pathogenic factors, STING becomes hyperactivated and initiates a downstream signaling cascade that induces the overexpression of IFN-I and proinflammatory cytokines [28]. STING can also trigger a fibrotic cascade, resulting in increased fibrosis in multiple tissues and organs throughout the body, including the heart [29, 30, 31]. Moreover, it can activate downstream pathways and is also associated with the induction of apoptosis [32]. The results of the present study showed that high glucose induced increased expression and activation of STING in mouse cardiomyocytes, accompanied by increased cardiomyocyte fibrosis and apoptosis. Myocardial fibrosis can impair diastolic and systolic function and eventually lead to refractory heart failure. Most cardiomyocytes are terminally differentiated cells with a limited proliferative capacity. An apoptosis rate of just 0.1% can reduce the number of cardiomyocytes by 37% within one year [33, 34]. Based on this, activation of the STING pathway may cause myocardial injury in the process of DCM by mediating cardiomyocyte apoptosis and fibrosis. The reduction in myocardial fibrosis and apoptosis after treating cells with C-176 supports this inference.
Under physiological conditions, STING can be degraded through various pathways after fulfilling its signal transduction function, thereby avoiding tissue damage caused by excessive activation [35, 36]. Several previous studies in diabetic mice have demonstrated the role of impaired proteasome activity in pathological cardiac remodeling. However, the aim of the current study was to further explore the pathogenic mechanism of the autophagy-lysosomal “negative feedback loop” in DCM. First, we confirmed that STING binds to LC3 in cardiomyocytes, thereby triggering the autophagy-lysosomal “negative feedback loop”. However, after 3-MA was administered to inhibit autophagy, significant accumulation of STING was observed in cardiomyocytes. This was accompanied by aggravation of cardiomyocyte injury and a significant increase in the number of apoptotic cells. The above results confirm the important role of autophagy in the degradation of STING. The expression of LC3 in cardiomyocytes from the HG group was significantly lower than in cardiomyocytes from the control group. Transmission electron microscopy further confirmed that high glucose inhibited autophagy, and this was accompanied by significant cardiomyocyte damage. Collectively, these results indicate that high glucose inhibits the STING-dependent autophagy‒lysosomal “negative feedback loop”.
This study found a significant increase in LARP7 expression in high glucose-induced mouse ventricular myocytes. Immunofluorescence colocalization revealed that a large amount of LARP7 translocated from the nucleus to the cytoplasm and was completely colocalized with STING, thus revealing a strong interaction between LARP7 and STING. Previous studies have reported that LARP7 plays an important role in stabilizing the 7SK complex and regulating transcription elongation. In the present study, we hypothesised that LARP7 plays an important role in high glucose-induced cardiomyocyte injury once it is released into the cytoplasm. The results of MS and COIP experiments confirmed that LARP7 can indeed interact with STING, and that high glucose strengthens this interaction. Additional experiments found that inhibiting LARP7 not only decreased apoptosis induced by high glucose, but also attenuated the activation effect caused by upregulating STING. These findings indicate that LARP7 plays a key regulatory role upstream of STING. The decreased expression of LC3 molecules observed after the upregulation of LARP7 expression suggests the existence of a specific regulatory mechanism. This was supported by the results of in vivo animal experiments. LARP7 inhibits the STING-dependent autophagy-lysosomal “negative feedback loop”, thereby causing the accumulation and overactivation of STING. This promotes cardiomyocyte apoptosis and fibrosis, which may contribute to the development of DCM.
The immunomodulatory effect of STING is currently an active area of research in the field of tumor immunotherapy, with some drugs now undergoing clinical trials. Our study also suggests that regulating the expression of LARP7 and STING may help in the treatment of DCM. However, the expression of STING was found to vary in different disease stages of DCM. In the early stage, STING expression is down-regulated due to the overactivation of autophagy. In contrast, inhibition of autophagy during the middle and late stages causes STING to accumulate. These specific pathogenic characteristics may determine stage differences in treatment. For example, Irisin can alleviate cardiomyocyte apoptosis and myocardial damage by regulating STING expression [37], whereas Metrnl negatively regulates the cGAS and STING pathways and exerts an anti-diabetic effect by promoting the degradation of STING after ubiquitination modification [38]. These contrasting treatment strategies suggest that tailoring the treatment to different disease stages is likely to present a major challenge. More experiments are needed to fully explore the characteristics of DCM, thereby allowing better targeted treatment.
In addition to the effects of different disease stages, it is generally accepted that the autophagic responses are distinctly different in type 1 and type 2 DM, whereas cardiac autophagic activity is enhanced in T1DM, it is suppressed in T2DM [39]. In T1DM, ATP deficiency caused by metabolic disorders or AMP accumulation activates AMPK to initiate autophagy. The main characteristic of T1DM is insulin deficiency, and insulin is known to inhibit autophagy by activating the PI3K-Akt/PKB-mTORC1 pathway [40]. T2DM hearts show the opposing conditions regarding autophagy [41, 42]. High intracellular nutrient energy status lead to suppression of autophagy, which shows as the inhibition of autophagosome maturation and reduced lysosomal activity. However, diametrically opposite reports do exist. For example, decreased AMPK activity and subsequent reduction in cardiac autophagy are observed in diabetic OVE26 mice [43]. Such results are same as our study. Furthermore, Zang et al. [44] demonstrated that cardiac autophagic flux is intact at 3 months but is dramatically suppressed at 6 months after onset of diabetes. The results suggest that T1DM can induce glucose-dependent cardiomyocyte death by inhibiting myocardial autophagy over time. Therefore, we suggest that the different results might be related to the autophagy flux assay criteria and the different stages of mouse model construction between the studies. Further investigation is warranted regarding this issue.
Taken together, our evidence indicates that LARP7 is a key molecule in high glucose-induced cardiomyocyte injury. It exerts a pathogenic role by interfering with the STING-dependent autophagy-lysosomal “negative feedback loop”. Inhibition of STING and LARP7 confers a significant cardioprotective effect, and hence the targeting of these proteins may be an effective therapeutic strategy to improve DCM.
Our study demonstrated that under high glucose conditions, LARP7 damages mouse cardiomyocytes by inhibiting STING-dependent autophagy-lysosomal degradation pathways. Targeted inhibition of LARP7 or STING expression may be a potential therapeutic strategy for DCM.
Data presented in this study are contained within this article and in the Supplementary Material, or are available upon request to the corresponding author.
JS and ZW contributed to the preliminary data analysis, interpretation, and manuscript writing. YD participated in the research design, CL and SZ provided help on the feeding of experimental animals and the construction of animal models. JD participated in the research design, provided experimental funding as well as supervision of the entire research process. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services, Publication No.[NIH] 86–23), and were approved by Animal Experimentation Committee of the Second Affiliated Hospital of Xi’an Jiaotong University (Shaanxi, approved number XJTUAE2022-374).
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.