

 , Ruofei Feng 2,*
, Ruofei Feng 2,*1 College of Life Science and Engineering, Northwest Minzu University, 730030 Lanzhou, Gansu, China
2 Key Laboratory of Biotechnology and Bioengineering of State Ethnic Affairs Commission, Biomedical Research Center, Northwest Minzu University, 730030 Lanzhou, Gansu, China
†These authors contributed equally.
Abstract
Background: Understanding the mechanisms through which interferon (IFN)
signaling is negatively regulated is crucial for preserving the equilibrium of
innate immune reactions, as the innate immune system functions, such as the
original barrier, combat threats to the host. Although the function of the
encephalomyocarditis virus (EMCV) viral proteins in antagonizing innate immunity
has been related to earlier studies, the precise mechanism underlying the role of
viral protein 3 (VP3) in type I IFN has yet to be fully illuminated.
Methods: VP3 expression and many other adaptor molecules belonging to
type I IFN pathway expression levels were evaluated using Western blotting. The
IFN and other antiviral genes, such as interferon-stimulated genes (ISGs) 15 and
56, were assessed by real-time quantitative polymerase chain reaction (RT-qPCR).
A 50% tissue culture infectious dose (TCID
Keywords
- EMCV viral protein 3
- interferon
- mitochondrial antiviral signaling protein
- autophagy
Encephalomyocarditis virus (EMCV) is a RNA virus belonging to the Picornaviridae family [1]. The EMCV genetic material comprises approximately 7861 nucleotides and comprehends a single open reading frame responsible for encoding a polyprotein [2]. Virus-encoded proteases post-translationally process this polyprotein into four structural proteins, viral protein 1 (VP1) to VP4, along with eight nonstructural proteins: L, 2A, 2B, 2C, 3A, 3B, 3C, and 3D [3]. EMCV 2C, 3C, and VP2 are crucial in inhibiting interferon (IFN) production and subsequent antiviral signaling [4, 5, 6, 7]. However, the mechanism through which EMCV viral protein VP3 restricts the natural immune signaling pathway remains poorly understood.
The innate immunity system contributes to the host’s initial defense against
invading microorganisms. Recognition receptors such as retinoic acid-inducible
gene-I (RIG)-like receptors (RLRs), (RIG-I, melanoma differentiation-associated
protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2)), cyclic
GMP-AMP synthase (cGAS), and nucleotide-binding oligomerization domain (NOD)-like
receptors (NOD2) are essential in innate immunity by interacting with
pathogen-associated molecular patterns (PAMPs) to produce IFN that is effective
against viruses [8]. Viruses must overwhelm the host’s innate immunologic
reaction to build a constructive infection and counteract the host’s antiviral
defenses for sustained viral replication. Following the identification of
virus-related RNA, RIG-I brings in the adapter protein mitochondrial antiviral
signaling protein (MAVS) to trigger TANK-binding kinase 1 (TBK1) and the
inhibitor of nuclear factor-
MAVS, also called VISA, is primarily found within the cell’s mitochondria. In
addition to being distributed at mitochondrion, MAVS is found locally at
peroxisomes and mitochondria-associated membranes (MAM) [12, 13]. Furthermore, the
aggregation of MAVS is facilitated by its localization to the membrane.
Specifically, proper activation of the protein necessitates mitochondrial
localization of MAVS; upon activation, MAVS forms a functional prion-like
structure on mitochondria through aggregation [14]. The platform is essential for
assembling the MAVS signalosome, then initiates the rousing of TBK1 and
IKK
EMCV has developed various immune escape strategies to prevent IFN-I from
spreading quickly and effectively at the initial infection site. Numerous EMCV
proteins are involved in evading the host’s immune system. EMCV proteases
3C
This research aimed to investigate the impact of VP3 on the natural immune system reaction. VP3 negatively regulated EMCV-elicited innate immunity response and strengthened viral replication in vitro. Detailed research discovered that VP3 repressed IFN production by interacting with MAVS and promoting its autophagic degradation. The discovery revealed a novel role of VP3 in suppressing natural immunity reactions.
Baby Hamster Kidney (BHK-21) and Human Embryonic Kidney 293 (HEK-293) cells were acquired from ATCC (Manassas, VA, USA). All cell lines
were validated by STR profiling and tested negative for mycoplasma. Cells were
cultured in high glucose Dulbecco’s modified Eagle medium (DMEM, CGM101.05,
Bailing, Lanzhou, China) with 10% newborn bovine serum (NBS, SA301.02.V,
Cellmax, Beijing, China) at 37 ℃ in a 5% CO
All antibodies were bought from specified suppliers. Sangon Biotech (Shanghai,
China) provided horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG
(D110058) and HRP-conjugated goat anti-mouse IgG (D110087). Antibodies for MAVS
(14341-1-AP), HA-tag (51064-2-AP), and IRF3 (11312-1-AP) were bought from
Proteintech (Wuhan, China). Cell Signaling Technology (Beverly, MA, USA) provided
the antibodies for STING (13647S), TBK1 (3013S), and Myc-tag (2276S). Beyotime
Biotechnology (Shanghai, China) provided the monoclonal antibody for
In-house construction of plasmids containing VP3 (Myc-tag) was completed. The SYBR Green Pro Taq HS qPCR kit (ROX plus) (AG11718) and Pro Taq HS Premix Probe qPCR kit (AG11704) were bought from Accurate Biology (Changsha, China). Invitrogen sold Lipofectamine® 3000 (L3000008, Invitrogen, Carlsbad, CA, USA).
The Myc-VP3 plasmid was introduced into HEK293 cells through
Lipofectamine® 3000 reagent following the guidelines provided by
the manufacturer. Radio-Immunoprecipitation Assay Lysis Buffer (RIPA) from
Solarbio (Beijing, China) was added to collected cells to prepare whole-cell
extracts. Cell lysates underwent sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), and polyvinylidene fluoride (PVDF) membranes from
Millipore were used for protein trans-blotting. PBS/Tween®
20-diluted skim milk (P1033, Solarbio, Beijing, China) was utilized to prevent
non-specific antibody binding, and then unambiguous primary and secondary
antibodies labeled with HRP were applied. Proteins were detected using an
electrochemiluminescence (ECL) reagent from BioRad in the Hercules, CA, USA, with
Transfection of HEK293 cells was carried out using
Lipofectamine® 3000 following the producer’s guidelines. A total
of 100 50% tissue culture infectious dose (TCID
Real-time quantitative polymerase chain reaction (RT-qPCR) was performed for
IFN-
Viral RNA was isolated using the Viral Genomic RNA Extraction kit (TIANGEN,
Beijing, China) and performed as previously described [7]. The standard curve
method was used to quantify EMCV genomic numbers. The 3D gene was used to design
primers and probes targeted for RT-qPCR assay. The primer sequences of EMCV 3D
gene and viral probe are listed as follows: 3D: (forward:
5
BHK-21 cells were used in these experiments. Five replicates of BHK-21 cells were infected with 10-fold serial dilutions of EMCV, and fresh DMEM was added after 1 h at 37 °C. The virus titers were determined after 72 h at 37 °C using the Reed–Muench method.
Cells were lysed using NP40 (Beyotime, Shanghai, China) and phenylmethylsulfonyl fluoride (PMSF; Beyotime, Shanghai, China) and then exposed to specified antibodies at 4 °C for 10 hours. Afterward, the lysates were exposed to 10 µL of protein G agarose slurry (P2053) from Beyotime in Shanghai China. Following a 4-hour incubation at 4 °C, the samples were spun at 2500 revolutions per minute (rpm) for 5 minutes. The beads were collected after being rinsed five times in ice-cold PBS. The solid was coupled with SDS loading buffer (P1040, Solarbio, Beijing, China) and heated at 95 °C for 5 minutes. After centrifugation at 6000 rpm for 1 minute, the liquid above the sediment was examined.
HEK293 cells were seeded into a 12-well cell plate. Cells were fixed with 4%
paraformaldehyde at room temperature, rinsed with 1
GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA, USA) was used to
analyze the data. Data were expressed as the mean
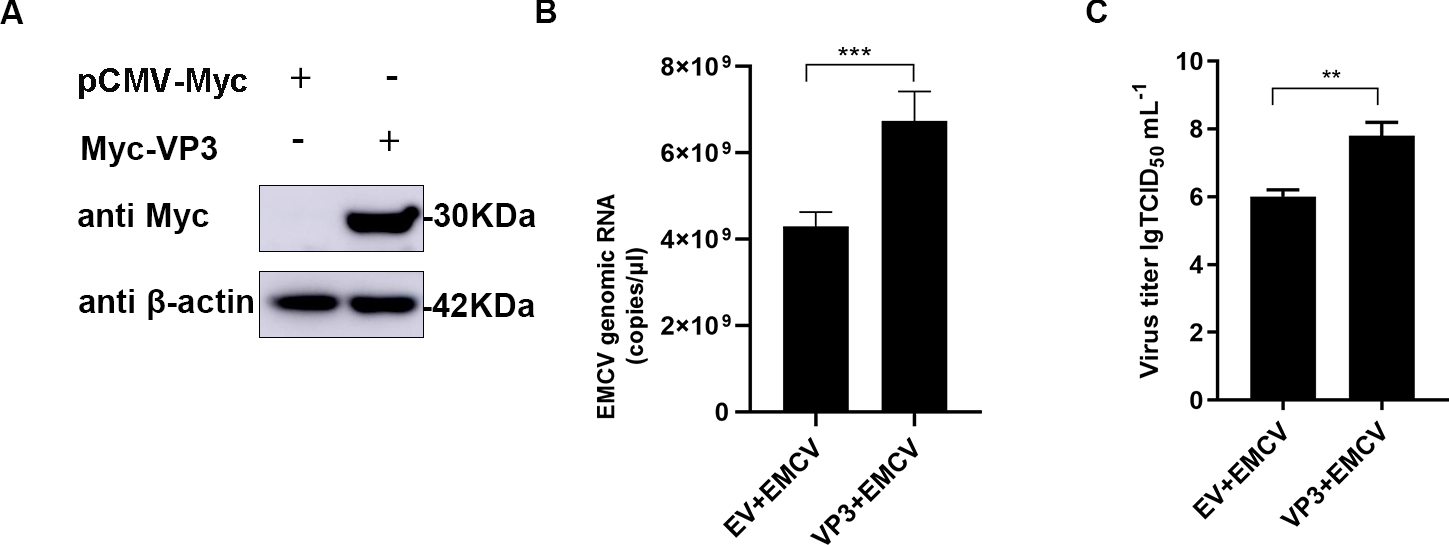
The impact of VP3 on EMCV replication was investigated by transfecting a Myc-tagged VP3 expression plasmid into HEK293 cells (Fig. 1A), followed by EMCV infection for 24 hours. The findings indicated that VP3 overexpression notably boosted EMCV copy numbers (Fig. 1B) and titers (Fig. 1C), indicating an ability of VP3 to enhance EMCV proliferation within host cells.
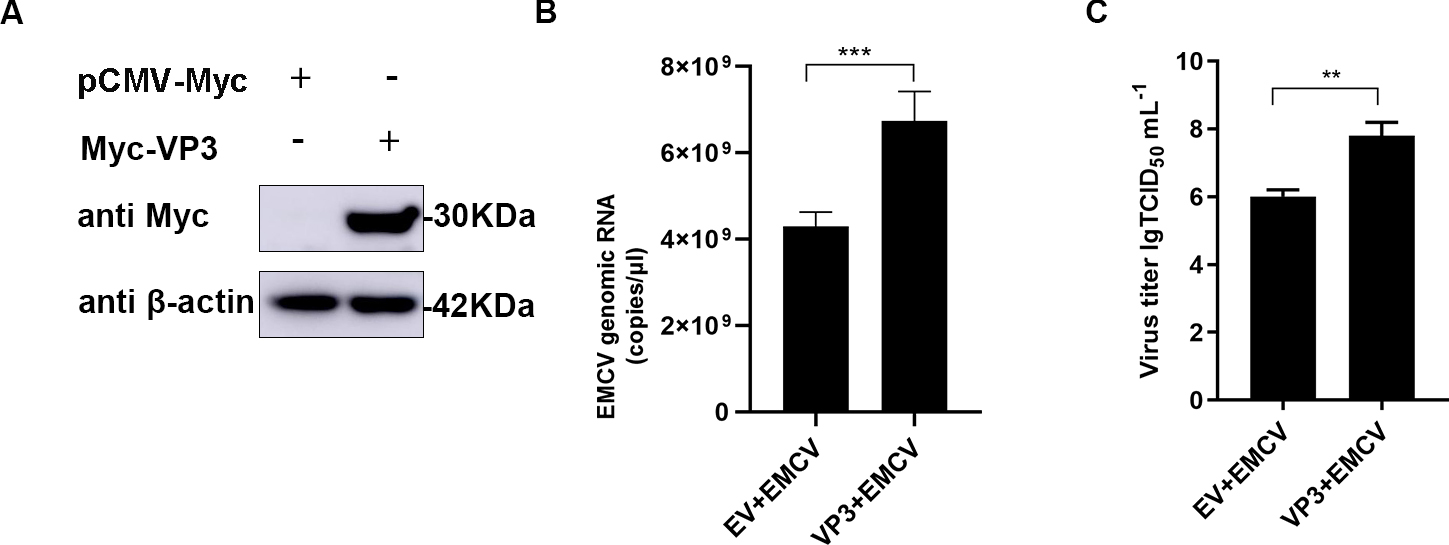 Fig. 1.
Fig. 1.VP3 overexpression promotes EMCV replication. (A) HEK293 cells
were seeded in 6-well plates and transfected with Myc-VP3 (1 µg). After 24
h, samples were collected for Western blot analysis. (B,C) HEK293 cells were
cultured in 6-well plates, followed by Myc-VP3 (1 µg ) or empty vector (1
µg) transfection for 24 h. Cells were infected with EMCV (100 TCID
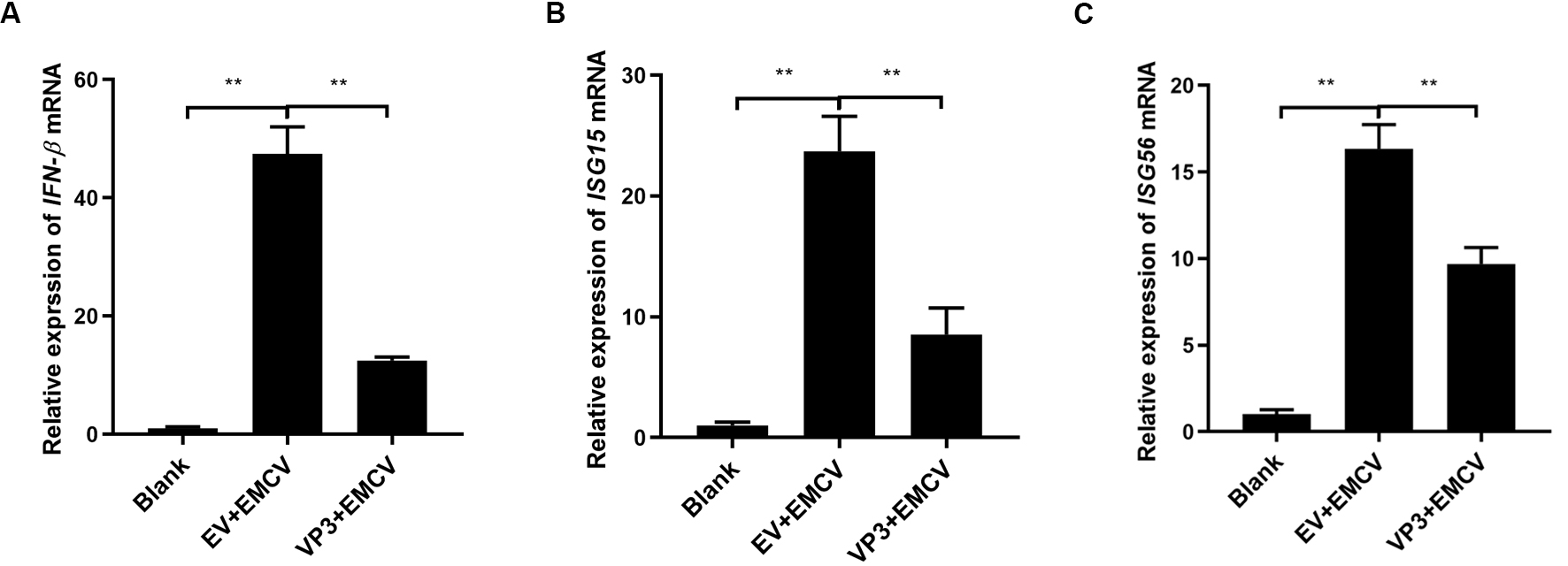
To further discuss the effect of EMCV VP3 on the type I IFN signaling cascade,
we monitored the IFN-
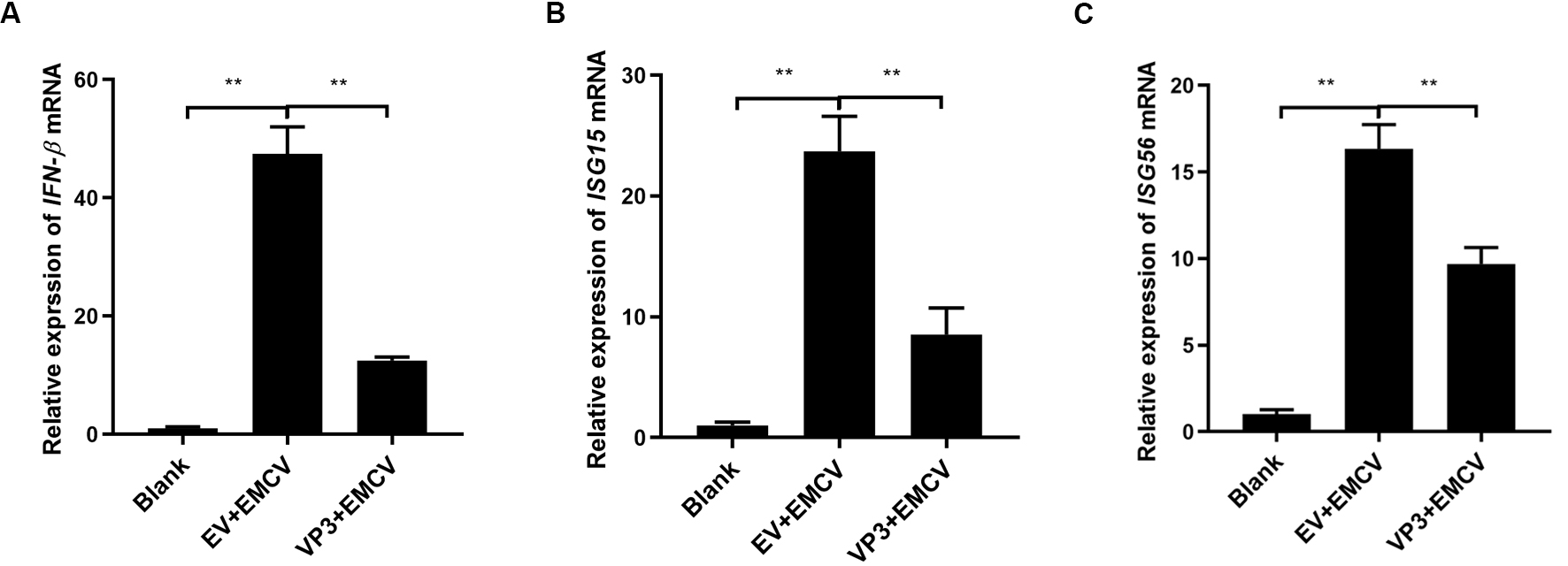 Fig. 2.
Fig. 2.VP3 inhibits EMCV-induced type I IFN signaling activation. (A)
HEK293 cells were transfected with Myc-tagged VP3 plasmid (1 µg) or
pCMV-Myc empty plasmid (EV, 1 µg) for 24 h and then stimulated with EMCV
(100 TCID
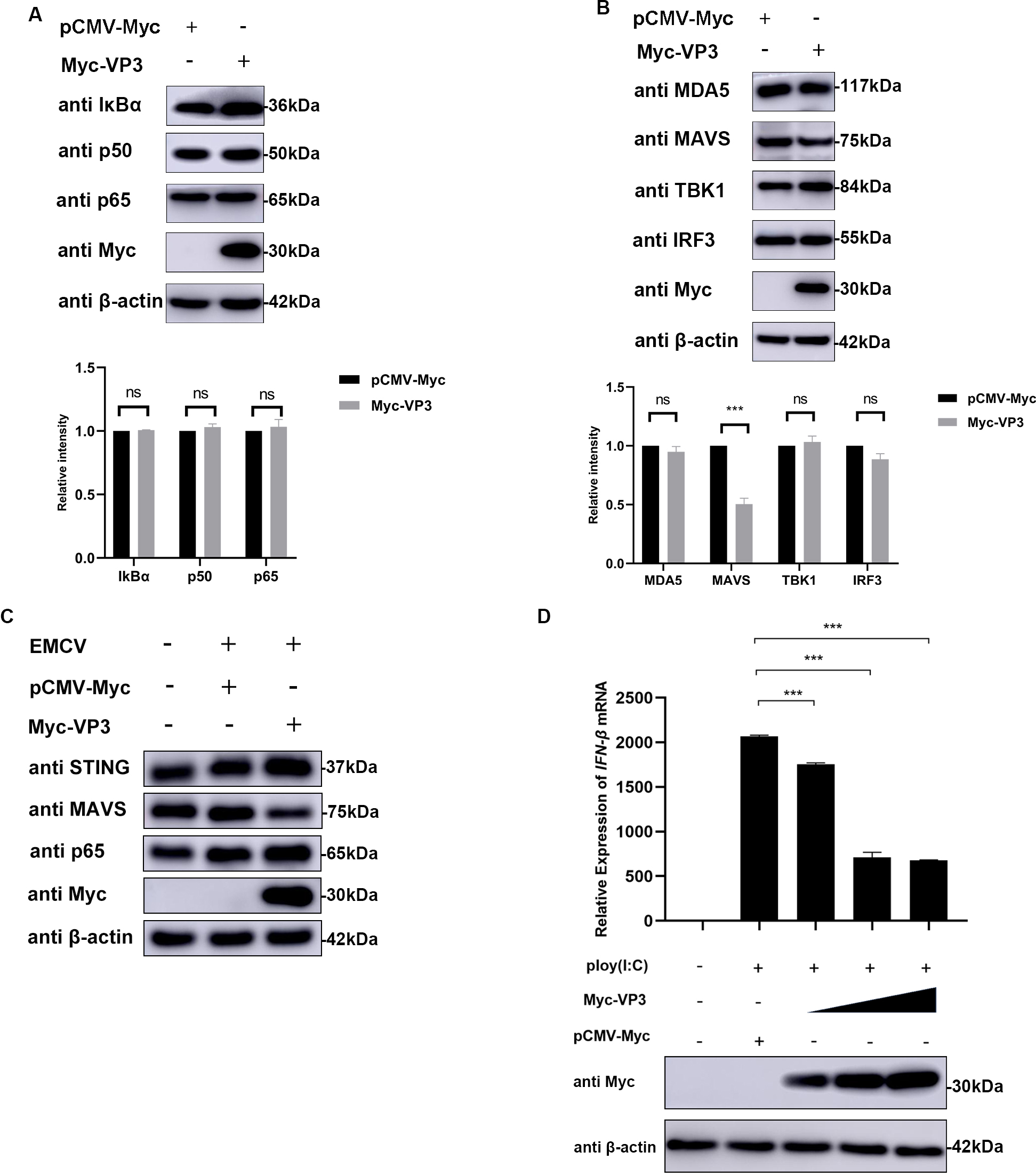
The structural protein VP3 of EMCV has a crucial function in inhibiting antiviral reactions, yet the specific molecular mechanism remains unknown. Host sensor molecules are known to recognize a viral infection and trigger the IFN pathway, leading to the initiation of antiviral defenses. We assessed the impact of EMCV VP3 on innate immune reactions by examining the expression levels of components associated with the IFN signaling pathway. In the case of VP3 overexpression, only the expression level of MAVS was affected, regardless of EMCV infection (Fig. 3A–C). Additionally, we investigated how VP3 impacts the transcription of IFN triggered by Polyinosinic-polycytidylic acid (poly(I:C)). Following transfection of HEK293 cells with varying amounts of the VP3 plasmid, the suppressive impact of VP3 on IFN production exhibited a correlation with dosage (Fig. 3D). These findings indicate that EMCV VP3 inhibits antiviral reactions, leading us to believe that VP3 may target MAVS signaling to suppress IFN responses.
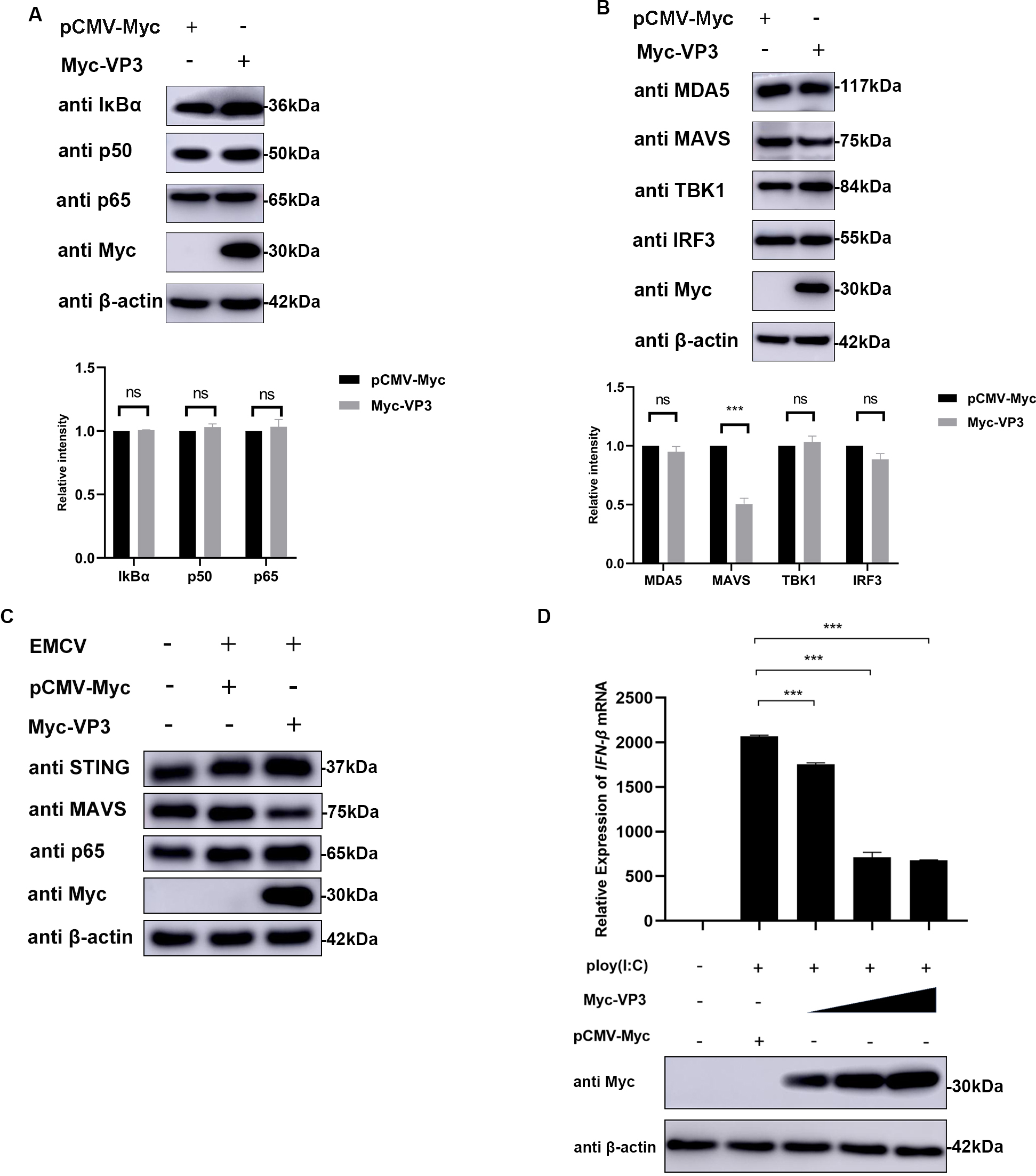 Fig. 3.
Fig. 3.VP3 inhibits type I IFN signaling by targeting MAVS. (A,B)
HEK293 cells were transfected with pCMV-Myc (1 µg) or Myc-VP3 (1 µg)
for 24 h; then cells were collected for p50, p65, I
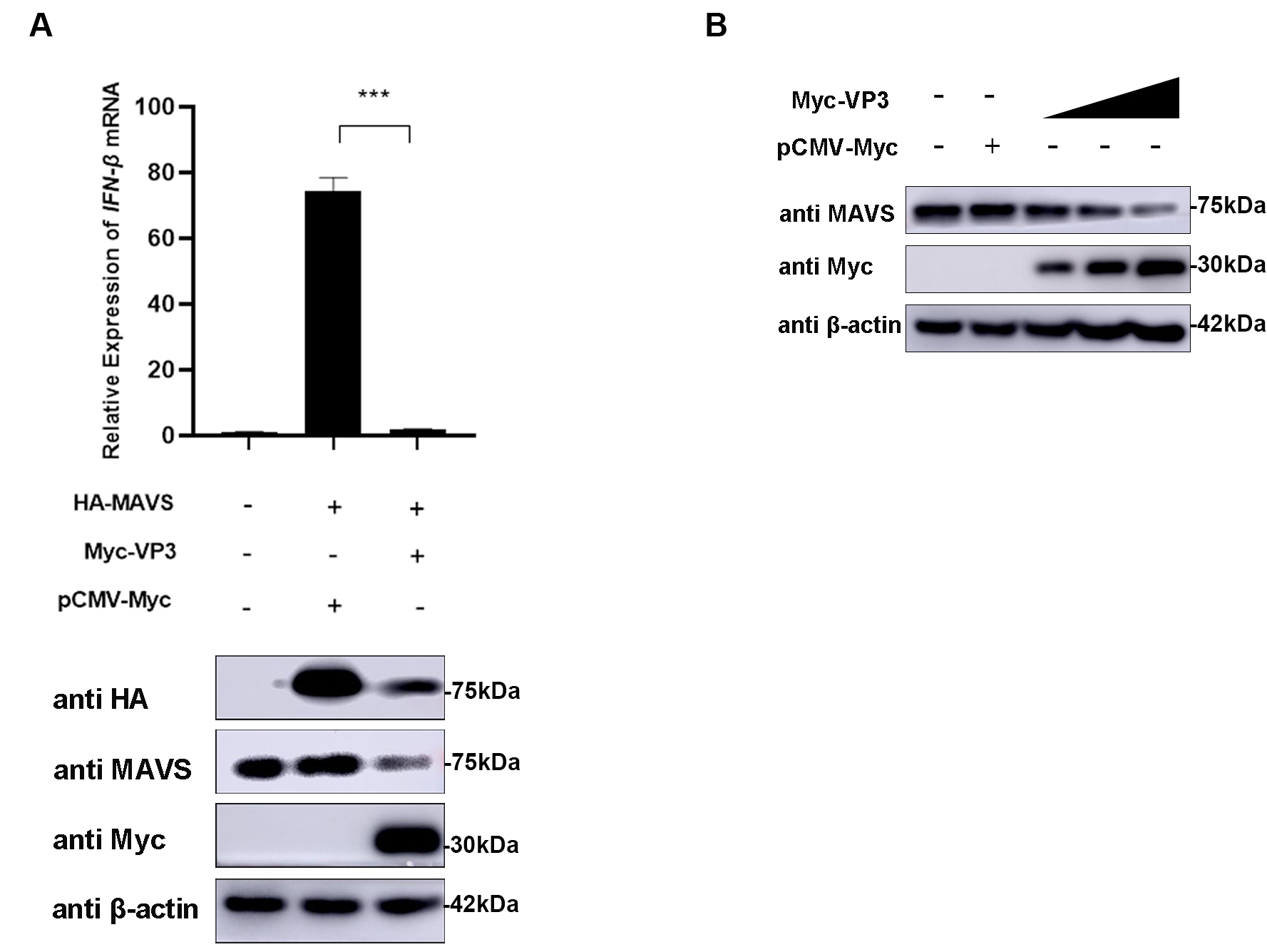
MAVS was confirmed as the target of EMCV VP3 by co-transfecting VP3 and MAVS
expression plasmids into HEK293 cells and detecting the IFN-
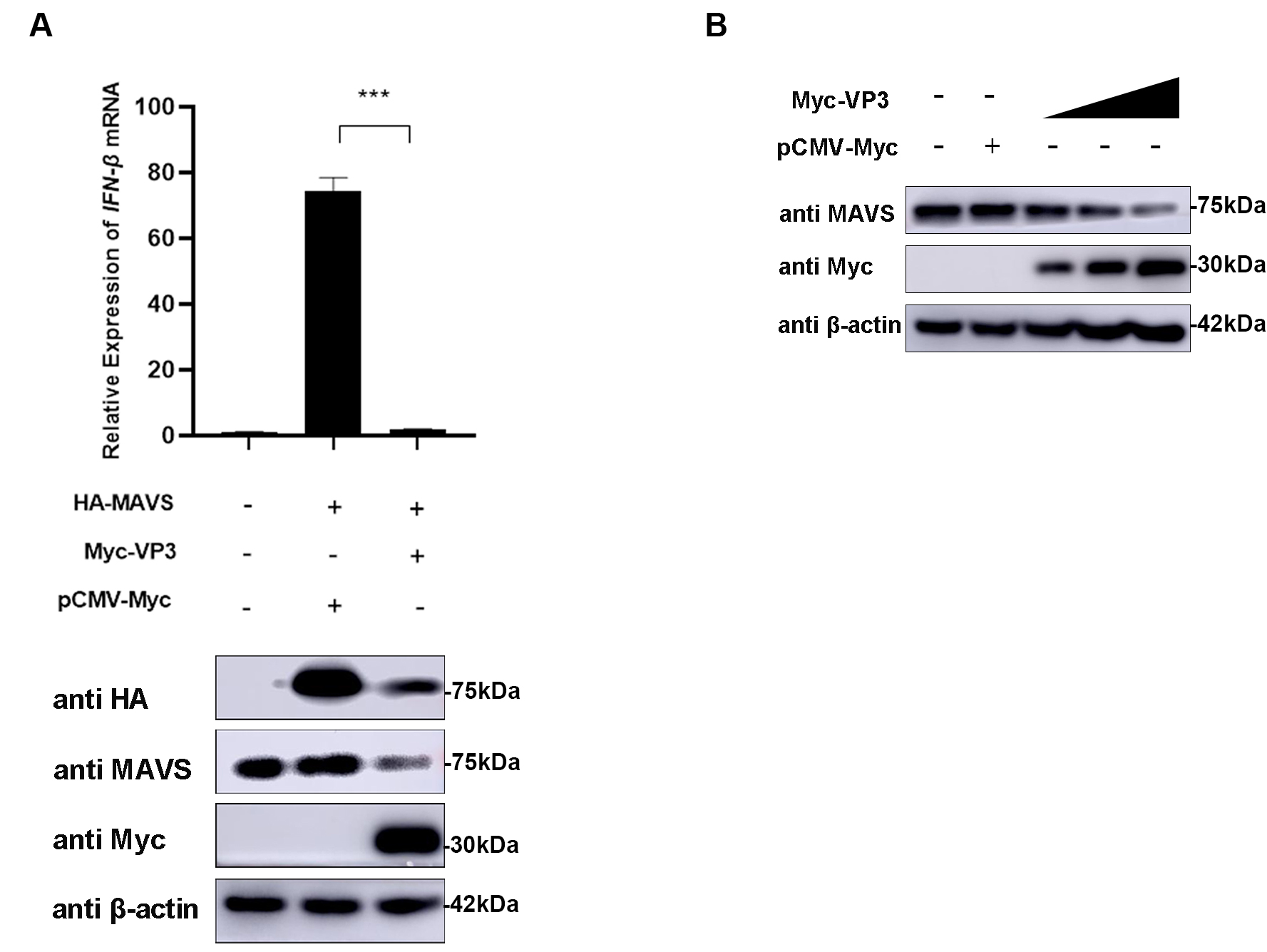 Fig. 4.
Fig. 4.MAVS is the target molecule of EMCV VP3. (A) HEK293 cells were
co-transfected with HA-MAVS (500 ng) and Myc-VP3 (1 µg) for 24 h; HA-MAVS
(500 ng) and pCMV-Myc (1 µg) were transfected as the positive control.
Then, cells were collected, and Myc-tagged VP3 and HA-tagged MAVS protein
expression was assessed.
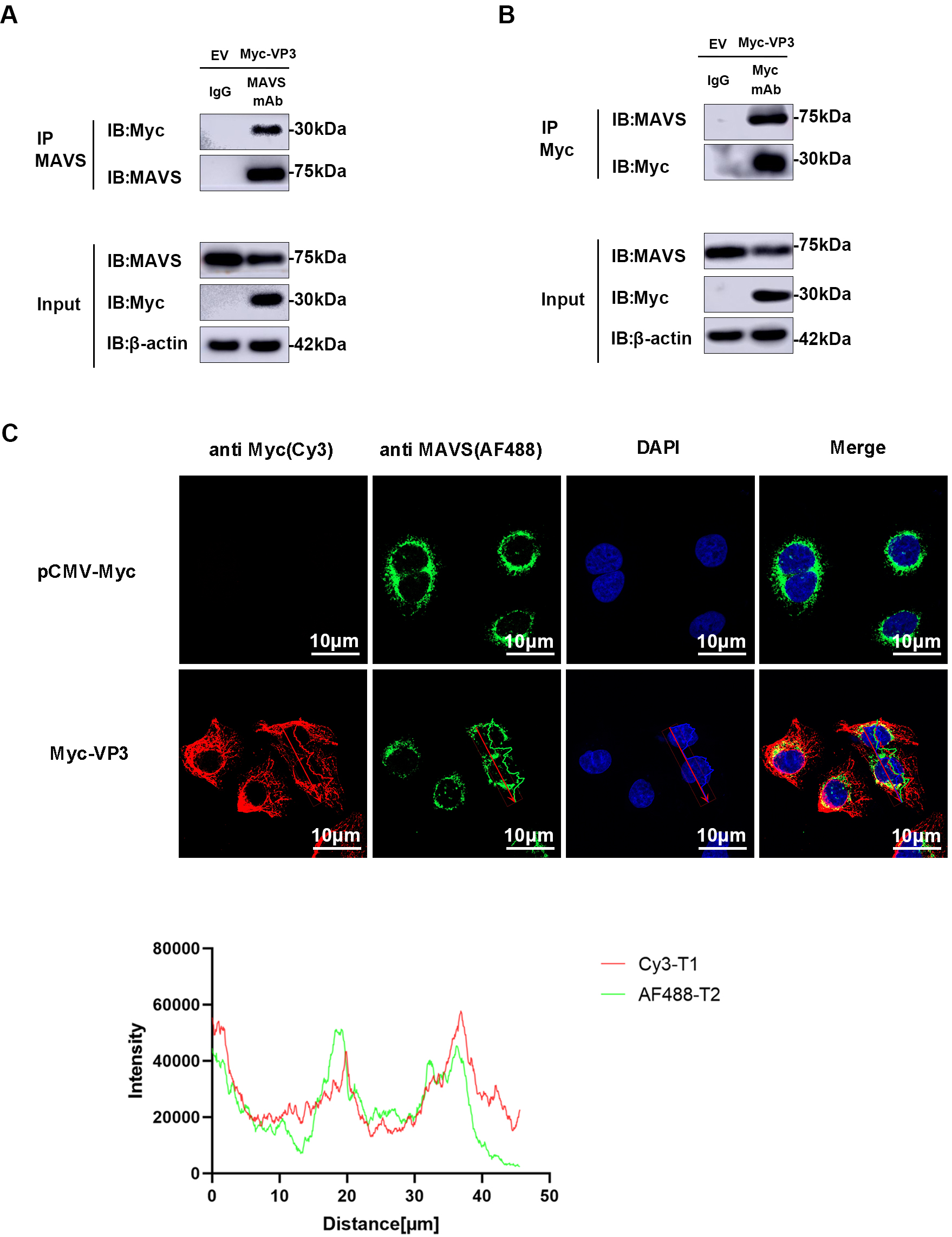
Based on previous findings indicating the role of VP3 in MAVS degradation, we hypothesized a potential interaction between VP3 and MAVS. As anticipated, an interaction was detected in HEK293 cells between EMCV VP3 and endogenous MAVS. Following immunoprecipitation using an anti-MAVS antibody, VP3 can be detected by an anti-Myc antibody (Fig. 5A). Subsequently, after immunoprecipitation with an anti-Myc antibody, MAVS was identified via Western blotting (Fig. 5B). Furthermore, we verified the co-localization of VP3 and MAVS using confocal microscopy (Fig. 5C).
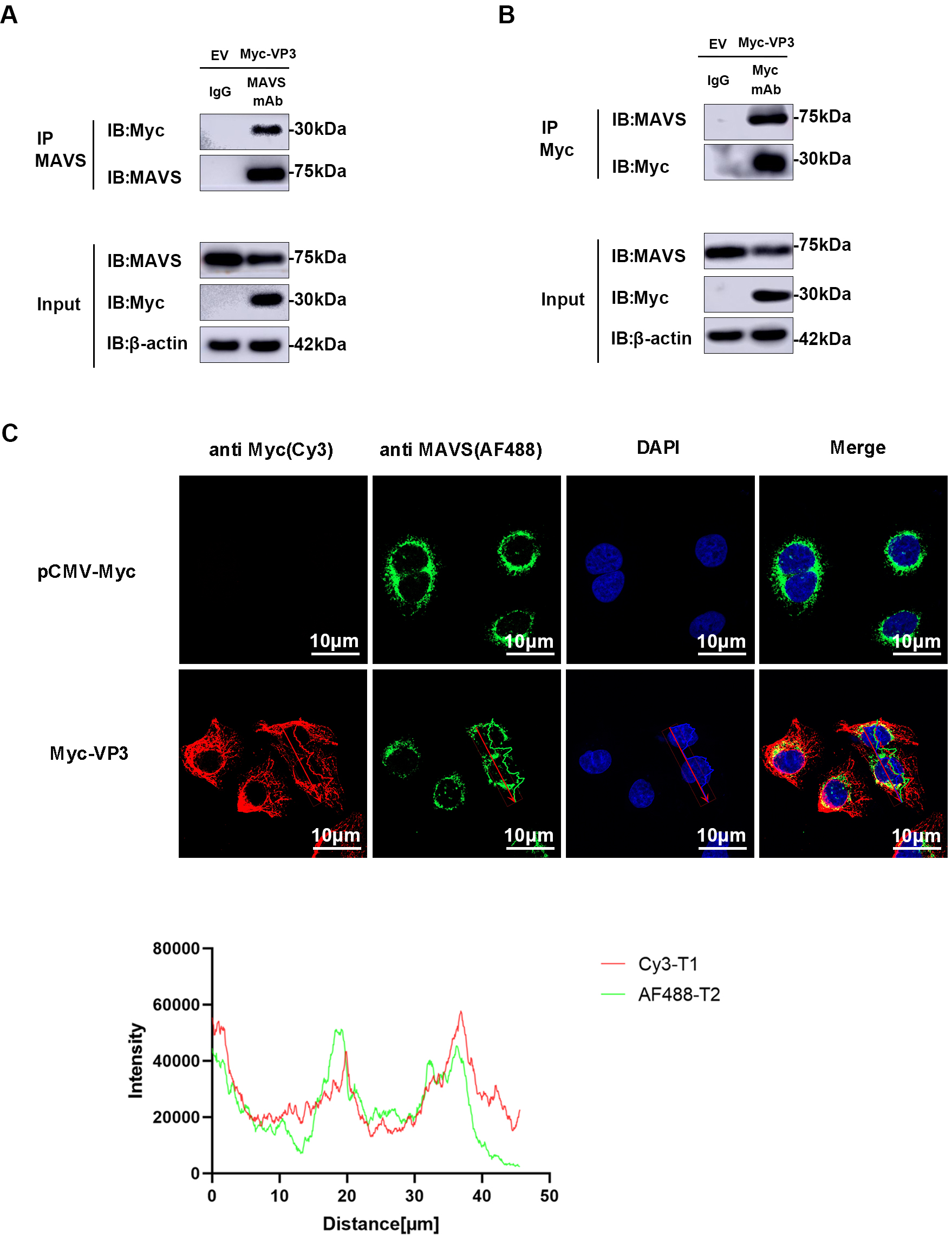 Fig. 5.
Fig. 5.VP3 interacts with MAVS. (A,B) MAVS and VP3 immunoprecipitation (IP) assay. All these experiments were performed in HEK293 cells transfected with Myc-VP3 (1 µg) or pCMV-Myc empty vector (EV, 1 µg). An anti-MAVS or anti-Myc antibody was incubated with protein G agarose, and IgG was used as the control. Input and IP complexes were analyzed using Western blotting. (C) Confocal laser scanning microscopy images of VP3 (anti-Myc (red)) and MAVS (anti-MAVS (green)). Nuclei were stained with DAPI (blue). Scale bars = 10 µm. MAVS, mitochondrial antiviral signaling protein; DAPI, 4′,6-diamidino-2-phenylindole.
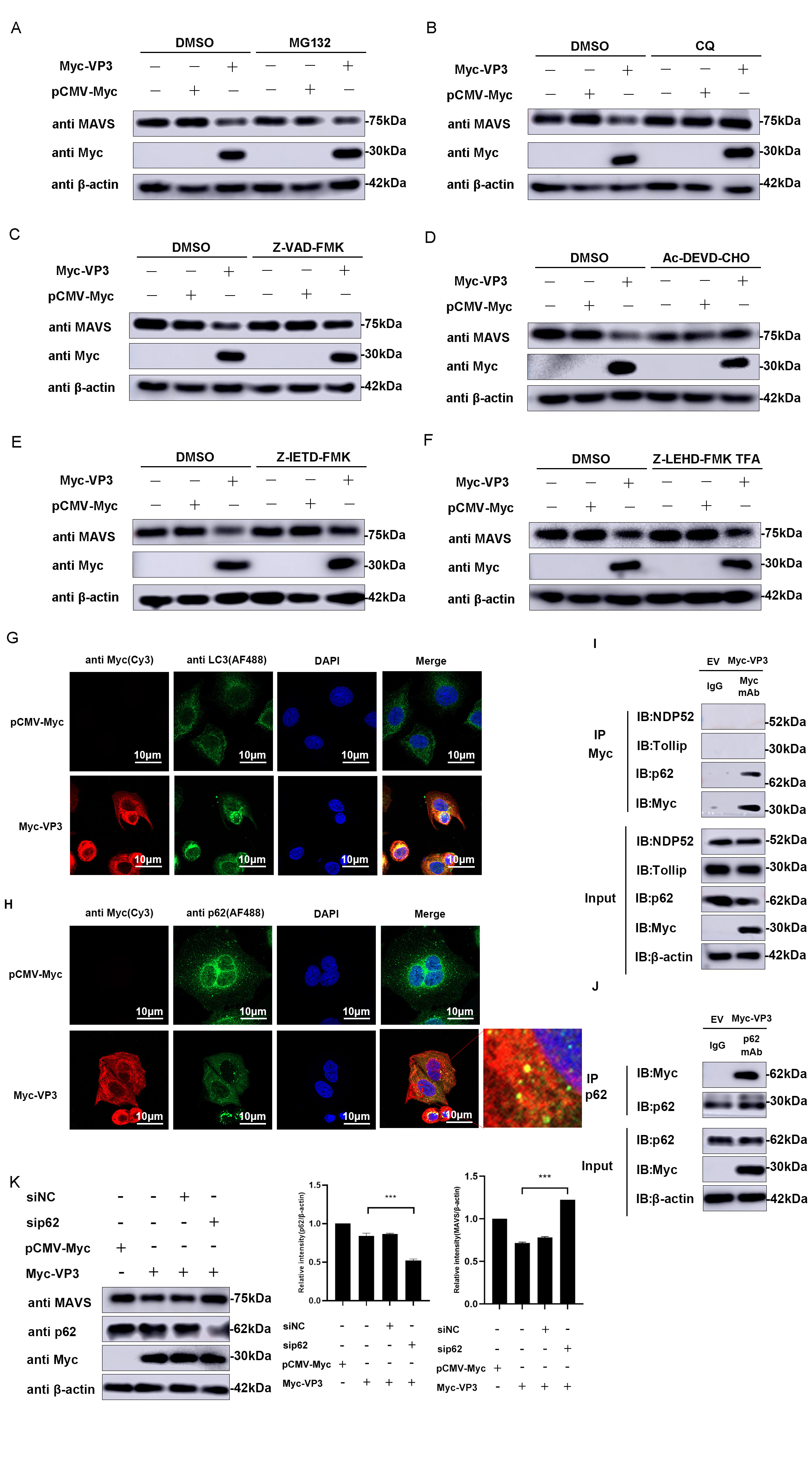
Various degradation pathway inhibitors were introduced to the cultured cells transfected with VP3 to determine the specific pathway involved in EMCV infection. Fig. 6A–C demonstrates that the presence of chloroquine (CQ), an autophagy–lysosomal pathway inhibitor, resulted in a notable increase in MAVS expression. In contrast, the presence of MG132, an inhibitor of the ubiquitin–proteasome pathway, did not lead to a noticeable improvement in MAVS expression. However, Z-VAD-FMK-treated cells also rescued the MAVS expression. Then, we used caspase 3 (Ac-DEVD-CHO), caspase 8 (Z-IETD-FMK), and caspase 9 inhibitors (Z-LEHD-FMK TFA) to treat cells with VP3 overexpression. Results showed that VP3-mediated MAVS degradation was rescued by caspases 3 and 8 (Fig. 6D–F). These data showed that the EMCV VP3 protein promotes the breakdown of MAVS through an autophagic process and caspase-dependent manner.
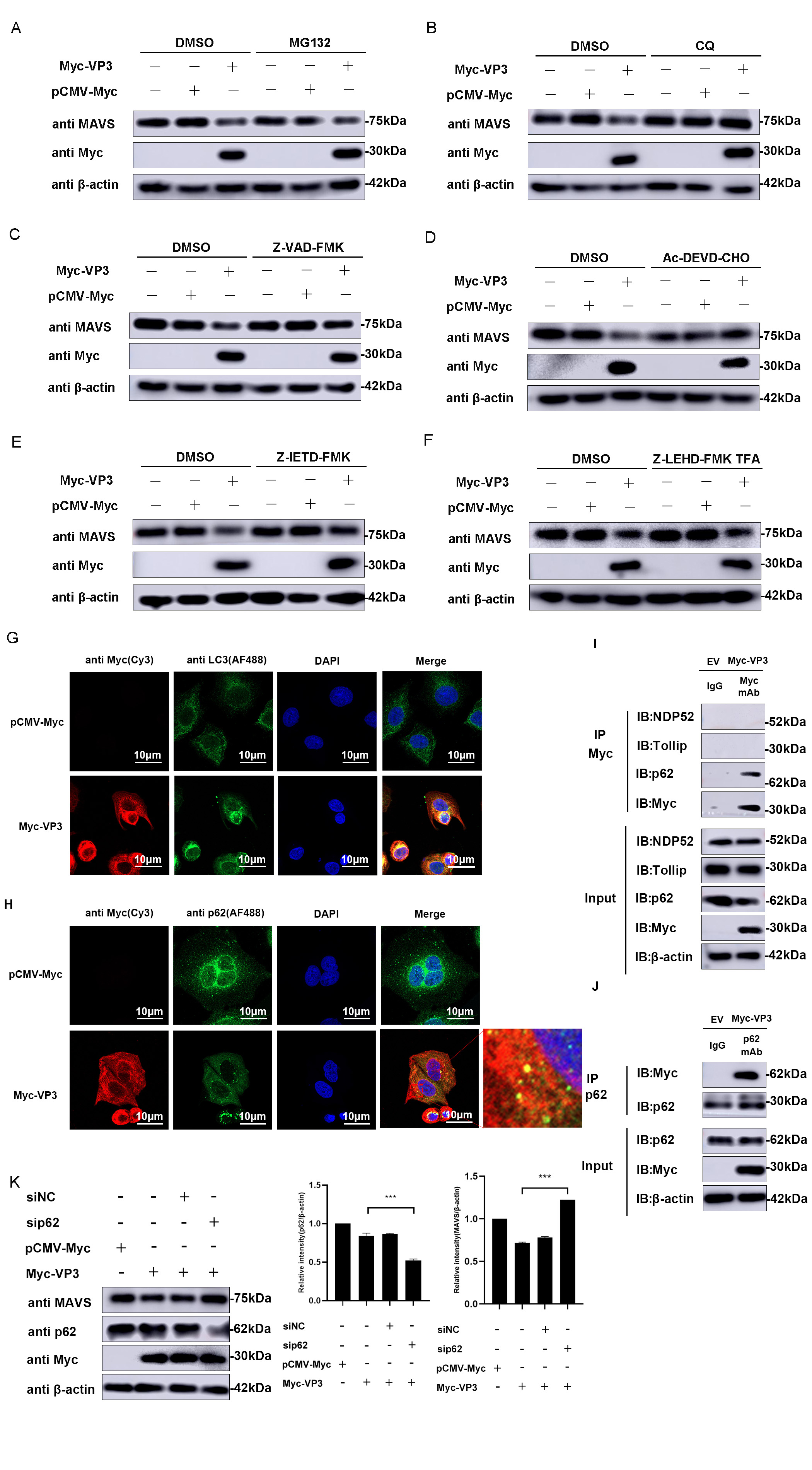 Fig. 6.
Fig. 6.VP3 promotes MAVS degradation via autophagy. MAVS degradation
pathway screening was performed in HEK293 cells transfected with Myc-VP3 (1
µg) or pCMV-Myc (1 µg). Proteasomal inhibitor MG132 (7.5
µM) (A), autophagy–lysosomal inhibitor CQ (50 µM) (B),
caspase inhibitor Z-VAD-FMK (50 µM) (C), caspase 3 inhibitor
Ac-DEVD-CHO (20 nM) (D), caspase 8 inhibitor Z-IETD-FMK (20 nM) (E) or caspase 9
inhibitor Z-LEHD-FMK TFA (20 nM) (F) were used to treat related cells. MAVS and
Myc-tagged VP3 expressions were detected.
The transformation of soluble microtubule-associated protein 1 light chain 3 (LC3) to membrane-bound LC3 plays a crucial role in autophagy. LC3, which is attached to the membrane, regulates various important functions within autophagy, such as the development and enlargement of the phagophore, the gathering of materials, and the merging of autophagosomes with lysosomes [21]. Confocal microscopy analysis was performed to verify the expression of LC3 and investigate if VP3 promotes autophagy. As shown in Fig. 6G, the expression of LC3 in VP3-transfected cells transferred from the cytoplasm to the perikaryon and exhibited punctate distribution compared to the control group, implying the formation of autophagosomes. Further, confocal images also directly revealed the obvious co-localization between VP3 and LC3.
In addition to LC3, autophagy can be monitored by assessing p62 levels. p62 is specifically included in autophagosomes by directly binding to LC3 and is effectively broken down by autophagy, leading to an inverse relationship between the overall cellular levels of p62 and autophagic activity. The p62 protein level notably decreases when VP3 is transfected compared to the control group (Fig. 6H). Subsequently, an interaction between VP3 and p62 was identified through immunoprecipitation tests (Fig. 6I,J). Furthermore, siRNA knockdown of p62 alleviated the inhibitory effect of VP3 on MAVS (Fig. 6K). The above data showed that VP3 triggers MAVS autophagic degradation through the p62-mediated autophagy pathway.
The initial defense against RNA or DNA viruses is provided by the innate immune
response triggered by RLRs, Toll-like receptors (TLRs), NOD-like receptors
(NLRs), or cGAS. Following viral infection, the IFN-
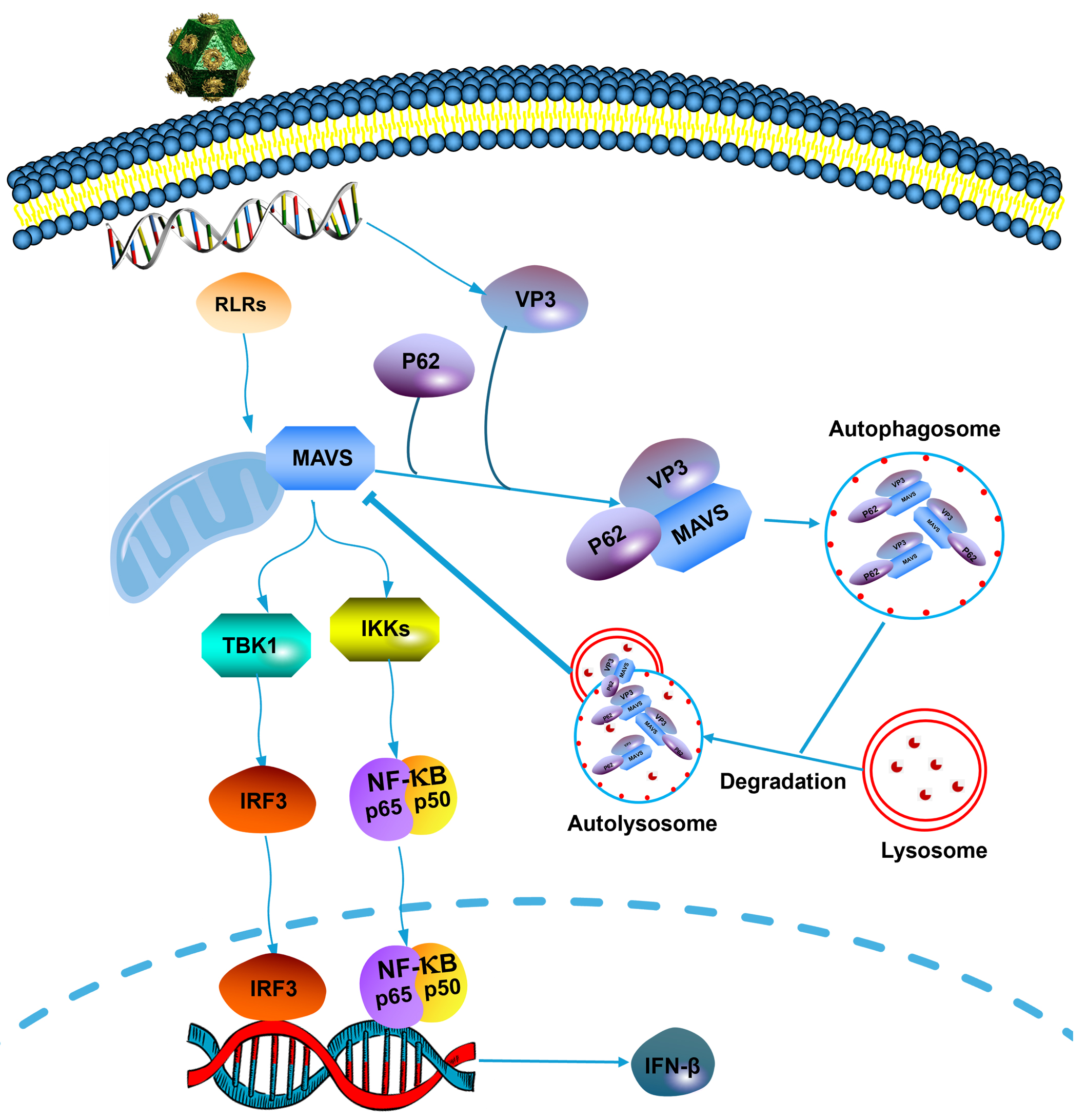
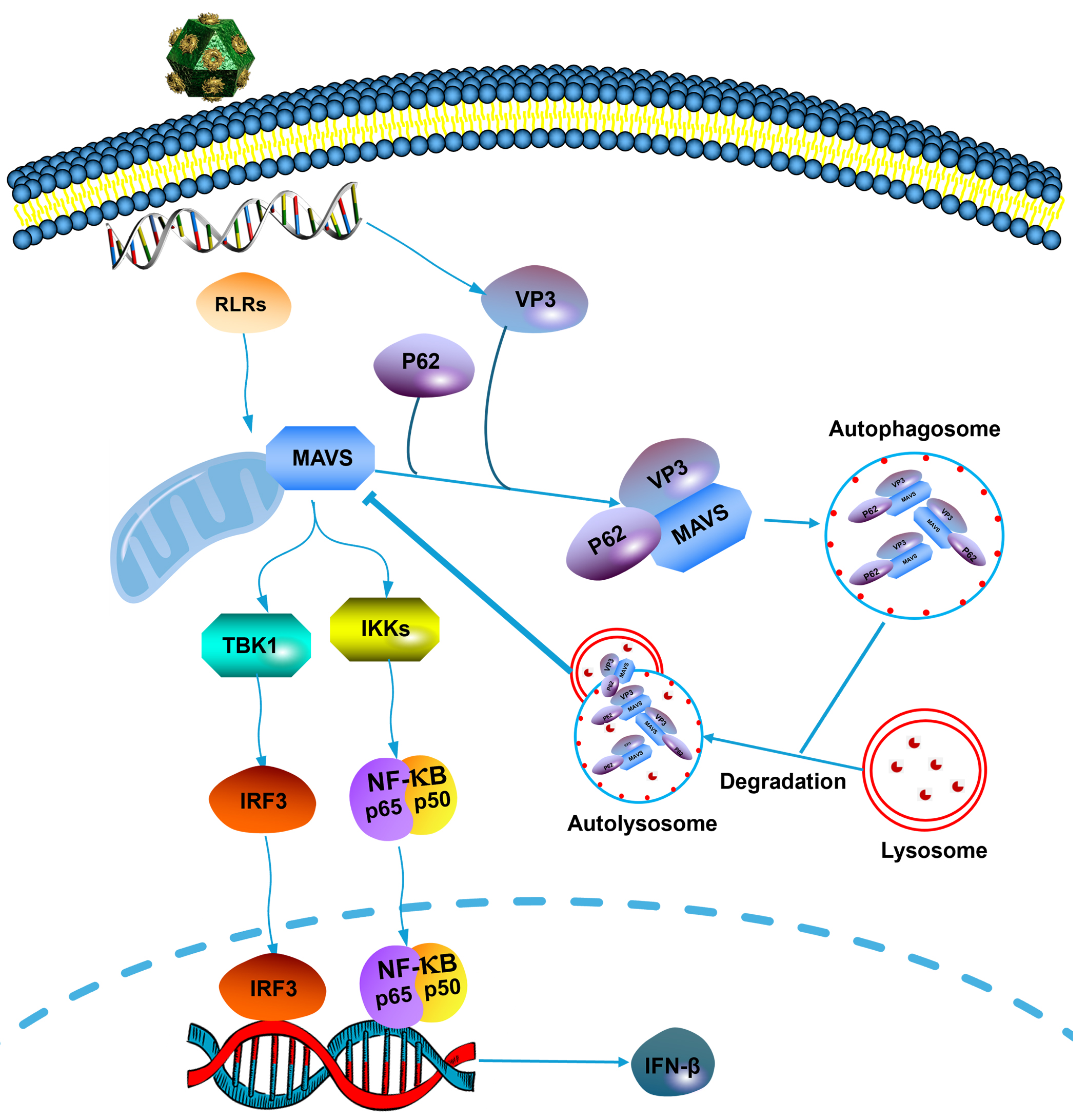 Fig. 7.
Fig. 7.Schematic representation of the role of EMCV VP3 in inhibiting
the type I interferon signaling pathway. Upon EMCV infection, viral protein VP3
can interact with MAVS by recruiting and interacting with p62, then degrades MAVS
via the autophagy–lysosomal pathway to evade the innate antiviral response of
the host. EMCV, encephalomyocarditis virus; RLRs, retinoic acid-inducible gene-I (RIG)-like receptors;
VP3, viral protein 3; MAVS, mitochondrial antiviral signaling protein; TBK1, TANK-binding kinase 1;
IKKs, inhibitor of nuclear factor-
IFNs produced by cells stimulated by pathogens are an important antiviral factor that the RLR pathway regulates. MAVS is an adapter essential for regulating IFN expression in the RLRs signaling pathway [23]. Multiple research studies have demonstrated the importance of the MAVS post-translational modification in initiating downstream signaling pathways. Viral infection of MAVS knockout mice has been shown to inhibit interferon production in mouse embryonic fibroblasts and dendritic cells, leading to increased mortality in mice [24, 25]. Some viral proteins have been found to have negative effects through immune evasion against MAVS molecules; for example, the NS1 protein of duck tambucu virus inhibits the activation of the IFNs signaling pathway by binding to the carboxyl terminus of the host MAVS [26]. Hepatitis B virus (HBV) X protein downregulates IFN production by interacting with the host MAVS protein [27]. The NS3/4A protein of type B GB virus inhibits IFN production by cleaving the host MAVS protein to affect its proper localization [28]. Studies on foot-and-mouth disease virus (FMDV) showed that the structural proteins VP1 and VP3 could block the activation of the host RNA receptors; it may also act as a direct innate immune antagonist within the cytoplasm to antagonize host cell innate immune defense mechanisms [29, 30].
EMCV proteins 3C, 2C, and the leading protein L have been reported as interferon
antagonists [4, 6, 31]. EMCV 2C can block the natural defenses by binding with
MDA5; the V26 amino acid of the EMCV 2C protein displays an indispensable
function in restraining the IFN-
Overall, our findings show that EMCV VP3 promotes virus replication by inhibiting the type I IFN signaling pathway. VP3 specifically binds to MAVS and degrades MAVS depending on the p62-mediated autophagy pathway. The discovery of a new method employed by EMCV VP3 to suppress innate immunity responses enhances our understanding of the immune evasion tactics utilized by EMCV to escape the host’s immunity.
All data generated or analysed during this study are included in this published article. Uncropped western blot images used for analysis are provided as Supplementary Material.
Conceptualization and investigation: XZ, ZH, YZ, DM, ZY, SY, JX, RF; methodology and resources: XZ, ZH, YZ, DM, ZY, SY and JX; writing and original draft preparation: XZ, ZH, YZ, DM, ZY and SY ; critical review and editing: JX and RF. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript.
Not applicable.
We would like to thank Dr. Shasha Li for her invaluable assistance in drafting and reviewing this manuscript.
This research was funded by the College Students Innovative Entrepreneurship Training Program project (state level) of Northwest Minzu University, grant number 202210742022, Fundamental Research Funds for the Central Universities, grant number 31920230162, 31920230160 and 31920240116, and National Natural Science Foundation of China, grant number 32260037.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.