

1 Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, RI 02912, USA
2 Department of Pediatrics, Fukushima Medical University School of Medicine, 960-1295 Fukushima, Japan
3 Providence Veterans Affairs Medical Center, Providence, RI 02908, USA
4 Department of Medicine, Warren Alpert School of Medicine of Brown University, Providence, RI 02903, USA
5 College of Pharmacy, Jinan University, 510632 Guangzhou, Guangdong, China
6 Department of Pediatrics, Warren Alpert School of Medicine of Brown University, Providence, RI 02903, USA
†These authors contributed equally.
Abstract
Bronchopulmonary dysplasia (BPD) is a chronic lung disease in premature infants characterized by alveolar dysplasia, vascular simplification and dysmorphic vascular development. Supplemental oxygen and mechanical ventilation commonly used as life-saving measures in premature infants may cause BPD. microRNAs (miRNAs), a class of small, non-coding RNAs, regulate target gene expression mainly through post-transcriptional repression. miRNAs play important roles in modulating oxidative stress, proliferation, apoptosis, senescence, inflammatory responses, and angiogenesis. These cellular processes play pivotal roles in the pathogenesis of BPD. Accumulating evidence demonstrates that miRNAs are dysregulated in the lung of premature infants with BPD, and in animal models of this disease, suggesting contributing roles of dysregulated miRNAs in the development of BPD. Therefore, miRNAs are considered promising biomarker candidates and therapeutic agents for this disease. In this review, we discuss how dysregulated miRNAs and their modulation alter cellular processes involved in BPD. We then focus on therapeutic approaches targeting miRNAs for BPD. This review provides an overview of miRNAs as biomarkers, and highlights potential pathogenic roles, and therapeutic strategies for BPD using miRNAs.
Keywords
- bronchopulmonary dysplasia
- microRNA
- hyperoxic exposure
- mechanical ventilation
- therapeutics
Bronchopulmonary dysplasia (BPD) is the most common chronic lung disease in premature infants. At present, a majority of preterm infants are able to survive due to advances in prenatal and neonatal care, including the use of antenatal corticosteroids, effective ventilatory support, and surfactant treatment [1, 2]. However, BPD remains the most common complication associated with prematurity, and is increasing in prevalence, most likely due to the increased survival of extremely low gestational age newborns. The incidence of BPD varies widely among countries from 11.8% to 56% [3, 4]. This disease increases the economic, psychological, and social burdens to families due to prolonged stays in intensive care units, a need for home oxygen therapy at discharge, and repeated hospital admissions due to pulmonary exacerbations [5]. For example, BPD costs an average of ~$377,871 per infant during the first year of life [5, 6, 7].
Most infants born before 28 weeks of gestational age require ventilatory assistance and/or supplemental oxygen. The persistent airway obstruction seen in BPD is due to airway inflammation resulting from mechanical ventilation and oxygen therapy [8]. The risk factors for BPD also includes surfactant deficiency, ventilation, and oxygen toxicity. The pathology of BPD is characterized by alveolar dysplasia, vascular simplification, and dysmorphic vascular development [9]. Currently, steroids, surfactant, caffeine, and vitamin A are used for the treatment of BPD [10]. Unfortunately, these therapies have minimally reduced the prevalence of BPD and associated lung injury [11, 12]. BPD also increases the risk of pulmonary and cardiovascular sequelae as well as adverse neurodevelopmental outcome [13, 14, 15]. Hence, there is an urgent need to develop new therapies to prevent lung injury and associated comorbidities in BPD.
microRNAs (miRNAs) are a class of small, non-coding RNAs with an average 22
nucleotides in length. In 1993, the first miRNA, lin-4, was discovered in
C. elegans [16]. In 2000, the second miRNA, let-7, was characterized,
and this miRNA is conserved in many species [17, 18]. In addition to biogenesis,
miRNAs are controlled by different mechanisms at the transcriptional and
epigenetic levels. They bind to the 3
miRNAs are abundant in tissues and fluids, such as blood and tracheal aspirates. These samples have been employed to evaluate the dysregulation of miRNAs in premature infants with BPD. Tables 1,2 (Ref. [19, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59]) summarize upregulated and downregulated miRNAs in clinical samples from infants with BPD.
| Upregulated miRNA | Samples | References | |
| Human BPD | miR-133b, miR-7 | arterial blood | [24] |
| miR-34a | tracheal aspirates | [25] | |
| miR-1252, miR-1254, miR-130a, miR-20a, miR-20b, miR-378b, miR-876 | tracheal aspirates | [26] | |
| miR-199a | tracheal aspirates | [27] | |
| miR-21 | serum extracellular vesicles | [28] | |
| miR-628, miR-185, miR-545, miR-378 | tracheal aspirates | [29] | |
| miR-219 | tracheal aspirates, lung tissues | [30] | |
| miR-203a | serum | [31] | |
| miR-221, miR-223 | plasma | [32] | |
| miR-184 | tracheal aspirates, lung tissues | [33] | |
| Animal models | miR-34a | mouse | [19] |
| miR-21 | mouse | [28] | |
| miR-219 | mouse | [30] | |
| miR-203a | rat | [31] | |
| (pnd2) miR-20b, miR-106a, miR-128, miR-883b, miR-15b | mouse | [34] | |
| (pnd7) miR-122, miR-30e, miR-365 | |||
| (pnd21) miR-133a, miR-205, miR-379, miR-449a, miR-431, let-7f | |||
| miR-141, miR-21, miR-34a | rat | [35] | |
| miR-411, miR-431, miR-699, miR-29a, miR-29c | mouse | [33] | |
| (pnd3) miR-490, miR-1193 | rat | [36] | |
| (pnd7) miR-3584 | |||
| (pnd14) miR-34c, let-7b, miR-3068, miR-872, miR-183, miR-33, miR-182, miR-322, miR-340, miR-142, miR-141, miR-96, let-7f, miR-15b, miR-449a, miR-22, miR-362, miR-301a and miR-365 | |||
| miR-196b, miR-365, miR-146b, miR-137, miR-132 | mouse | [37] | |
| (pnd1) miR-184, miR-347, miR-181a, miR-204, miR-132, miR-328b | rat | [38] | |
| (pnd3) miR-3559 | |||
| (pnd7) miR-466b, miR-466b | |||
| miR-34a | mouse | [39] | |
| miR-29a | mouse | [40] | |
| miR-421 | mouse | [41] | |
| miR-29a | mouse | [42] | |
| miR-451 | mouse | [43] | |
| miR-154 | mouse | [44] | |
| miR‐125b | rat | [45] | |
| miR-421 | mouse | [46] | |
| miR-194 | mouse | [47] | |
| Hyperoxia-exposed cells | miR-219 | MLE 12, mouse lung primary, fibroblasts | [30] |
| miR-34a, miR-34b, miR-34c | MLg | [39] | |
| miR-451 | MLECs | [43] | |
| miR-421 | MLE12 | [46] |
Abbreviations: miRNA, microRNAs; BPD, bronchopulmonary dysplasia; MLECs, murine lung endothelial cells; MLE12, mouse lung epithelial cells; MLg, mouse lung fibroblast cell line; pnd, postnatal day.
| Downregulated miRNAs | Samples | References | |
| Human BPD | miR-152, miR-30a | arterial blood | [24] |
| miR-17 | plasma | [48] | |
| miR-489 | lung tissues | [49] | |
| miR-29b | plasma, lung tissues | [50] | |
| miR-876 | tracheal aspirates | [26] | |
| miR-30a | lung tissues | [51] | |
| miR-574 | blood | [52] | |
| miR-3713, miR-3151, miR-1295, miR-1286, miR-380, miR-15a, miR-3175, miR-493, miR-3193, miR-105, miR-4300, miR-631, miR-2116, miR-4304, miR-3125, miR-4303, miR-1908, miR-205, miR-3674, miR-615, miR-4305, let-7i, miR-4330, miR-1255b, miR-125b-1, miR-24-1, miR-646 | tracheal aspirates | [29] | |
| 90 miRNAs | umbilical cord vein blood | [53] | |
| miR150 | plasma | [32] | |
| miR-342 | tracheal aspirates | [54] | |
| Animal models | miR-489 | mouse | [49] |
| miR-876 | mouse (BALF) | [26] | |
| miR-342 | mouse | [54] | |
| (pnd2) miR-299, miR-139p, miR-300, miR-122 | mouse | [34] | |
| (pnd7) miR-335p, miR-714 | |||
| (pnd21) miR-720 | |||
| miR-342, miR-126, miR-335, miR-150, miR-151 | rat | [35] | |
| miR-322, miR-411, miR-431, miR-609, miR-680 | mouse | [33] | |
| (pnd3) miR-377 | rat | [36] | |
| (pnd7) miR-542, miR-99a, miR-139, miR-208a, miR-33, miR-190a, miR-335, miR-708, miR-15b, miR-674, miR-188 | |||
| (pnd14) miR-181c, miR-465, miR-382, miR-208a, miR-351, miR-503, miR-127, miR-664, miR-298, miR-376a, miR-186, miR-134, miR-92a, miR-378a, miR-541, miR-154 | |||
| miR-363, miR-196a | mouse | [37] | |
| miR-342 | mouse | [55] | |
| (pnd1) miR-92a, miR-6215, miR-135a, miR-449c, miR-449a, miR-376b, miR-122, miR-154, miR-543, miR-490 | rat | [38] | |
| (pnd3) miR-338, miR-122, miR-6215, miR-1, miR-133a, miR-133b, miR-208a, miR-490, miR-741, miR-204, miR-466b, miR-466c, miR-490 | |||
| (pnd14) miR-337, miR-344, miR-122, miR-1, miR-208a, miR-19b, miR-154, miR-542, miR-3559, miR-29c, miR-450a, miR-186, miR-3068, miR-29b, miR-34b, miR-500, miR-3068, miR-224, miR-201, miR-344g | |||
| miR-17, miR-18a, miR-19a, miR-19b, miR-20a, miR-92 | mouse | [56] | |
| miR-20b | rat | [57] | |
| miR-214 | rat | [58] | |
| miR-425 | rat | [59] | |
| Hyperoxia-exposed cells | miR-20b | AEC II | [57] |
| miR-425 | RLE-6TN | [59] |
Abbreviations: AEC II, primary type II alveolar epithelia cell; BALF, bronchoalveolar lavage fluid; pnd, postnatal day; RLE-6TN, rat type II alveolar epithelial cell.
Accumulating evidence demonstrates that the expression of miRNAs is altered in
the blood of premature infants with BPD. For example, expression of miR-203a (n =
4), miR-221 (n = 38) and miR-223 (n = 38) was significantly upregulated in the
blood of patients with BPD at 28 days to 3 months of age compared to age-matched
controls (n = 4 or n = 21) [31, 32]. Since BPD is established at this age, these
dyregulated miRNAs could be diagnostic biomarkers of this disease. In contrast,
levels of miR-150 (at 28 days to 3 months of age), miR-574 (at
Blood exosomes are extracellular vesicles secreted by living cells into the circulating blood. Using next-generation sequencing and bioinformatic analysis, 328 miRNAs were upregulated and 90 miRNAs were downregulated in exosomes from umbilical cord vein blood of preterm infants who subsequently developed BPD (12 BPD infants vs 14 non-BPD infants). These miRNAs were those primarily enriched in the PI3K/Akt and angiogenesis-related signaling pathways [53]. Among them, blood levels of miR-200a-3p were increased whereas the expression of miR-103a and miR-185 was most significantly reduced [53]. Levels of miR-21 in serum extracellular vesicles on the 28th day of life were significantly increased in premature infants with severe BPD (n = 2) compared to those without BPD (n = 3) [28]. These findings suggest that alterations of miRNA in exosomes could serve as potential biomarkers of BPD. Further investigation is warranted to determine the mechanisms by which BPD alters these miRNAs or vice versa, and to evaluate whether these changes in the blood correlate with their levels in the lung.
In tracheal aspirates, levels of miR-628, miR-185, miR-545, and miR-378,
detected by miRNA array, were significantly increased in premature infants with
severe BPD (n = 17) compared to those with mild/moderate BPD (n = 8) [29]. This
is in contrast to reduced expression of miR-185 in the blood of infants with BPD
[53]. Expression of miR-1252, miR-1254, miR-130a, miR-20a, miR-20b, miR-378b, and
miR-876 was higher in the tracheal aspirates from patients with severe BPD (n =
25, 23–28 weeks gestation) compared to gestational age-matched full-term
controls (n = 25) [26]. Tracheal aspirates collected in the first postnatal week
from premature infants who developed BPD exhibited significantly increased
miR-34a (n = 5) and miR-199a (n = 10) expression compared to controls [25, 27].
Also, miR-219 expression was markedly increased in tracheal aspirates and lung
tissues of infants with severe BPD compared to post-conception age matched
full-term infants (n = 30) [30]. In contrast, 28 miRNA levels were significantly
decreased in the tracheal aspirates from premature infants with severe BPD
compared to those with mild/moderate BPD [26, 29]. Expression of miR-342 (n =
10), miR-30a (n = 9), miR-489 (n = 4), and miR-29b (n = 4) was decreased in
tracheal aspirate cell pellets from neonatal infants with BPD (n = 10) [49, 51, 54]. Among these miRNAs, miR-34a has been widely studied and is involved in Wnt
signaling, TGF-
Pathway analysis indicated that differentially expressed miRNAs observed in BPD are associated with molecular and cellular functions including cell signaling, DNA replication, cell cycle, cell apoptosis, and inflammatory responses [25, 29]. Further studies are warranted to better understand the role of specific miRNAs in altered cellular functions seen in BPD. Tracheal aspirates contain immune cells, epithelial cells, and mesenchymal stromal cells among others. Defining which cells in the tracheal aspirates show specific alterations in miRNAs that may contribute to BPD will be important.
Animal models can help us better understand the role of miRNAs in the pathogenesis and potential treatment of BPD. Hyperoxia-exposed neonatal rodents are common animal models used to mimic BPD because hyperoxia alone results in lung injury similar to that seen in BPD. Other rodent models have used an inflammatory injuryto mimic BPD. We summarize the upregulation or downregulation of miRNAs in animal models of BPD (Tables 1,2). It is important to note that miRNA expression is altered differently depending on oxygen concentration, exposure time, animal species and cell types.
Neonatal hyperoxia (60% oxygen for 21 days) resulted in the upregulation of
various miRNAs at pnd2, pnd7 and pnd21 in mouse lung tissues [34], suggesting
dynamic changes of these miRNAs. This is corroborated by the findings in a rat
model showing that neonatal hyperoxia (60–85% oxygen for 14 days) upregulates
miR-490 and miR-1193 at pnd3, miR-3584 at pnd7, and 19 miRNAs including miR-365
at pnd14 [36]. Lung miRNA expression profiling in neonatal mice exposed to
hyperoxia (80% oxygen) or normoxia for either 14 days or 29 days showed several
dynamically regulated miRNAs [33]. These miRNAs include miR-411, miR-431,
miR-699, miR-29a, and miR-29c. In neonatal rats exposed to hyperoxia (80
Lung levels of miR-206 were significantly reduced in mice exposed to hyperoxia (60% oxygen exposure on pnd2, pnd7, pnd21) compared to air-exposed controls [60]. In newborn mouse lungs, miR-342 was significantly downregulated after 21 days of hyperoxic exposure (60% oxygen) compared to room air controls [55]. Interestingly, reduction of miR-299, miR-139p, miR-300, and miR-122 was observed at pnd2, and miR-335p and miR-714 at pnd7, and miR-720 at pnd21 in rodent lungs [34]. These findings suggest that neonatal hyperoxia decreases the expression of certain miRNAs in a dose- and time-dependent manner. This is confirmed by another study using a rat model showing that neonatal hyperoxia (60–85% oxygen for 14 days) reduced miR-377 at pnd3, downregulated 11 miRNAs including miR-139, miR-208a, and miR-188 at pnd7, and 16 miRNAs at pnd14 [36], illustrating the dynamic temporal expression of miRNAs in the lung after neonatal hyperoxia.
Using miRNA expression profiling, Dong et al. [33] identified 4 dynamically regulated miRNAs in the lungs of neonatal mice exposed to hyperoxia (80% oxygen) or room air for either 14 or 29 days. Ruiz-Camp et al. [39] reported that 14 miRNAs, including miR-29c and miR-34a, were dysregulated at pnd5 and pnd14 in neonatal mice exposed to hyperoxia (85% oxygen). In a neonatal hyperoxia exposure model (85% oxygen from 4 to 14 days of age), lung miR-489 expression was reduced. This may serve as a compensatory mechanism, because inhibiting miR-489 improved lung development after hyperoxia whereas miR-489 overexpression inhibited lung development [49]. Levels of miR-876-3p were decreased in the bronchoalveolar lavage fluid of mice exposed to hyperoxia (85% oxygen) from pnd3 to pnd14 [26]. Mu et al. [57] found that miR-20b was downregulated in the lungs of rats exposed to hyperoxia (95% oxygen) for 48 h. Wu et al. [59] demonstrated that miR-425 was downregulated in the lungs of rats with hyperoxia-induced lung injury (90% oxygen for 7 days). Zhang et al. [58] demonstrated that miR-214 expression was lower on pnd3, pnd7, and pnd14 in lungs of neonatal rats after 95% oxygen exposure compared to those from air exposed (21% oxygen) controls. Exposure to 95% oxygen for 10 days (from pnd3 to pnd13) in newborn rats downregulated the levels of 5 miRNAs, such as miR-342, in the lung at pnd13 [35]. Levels of miR-363 and miR-196a were downregulated in the lungs of neonatal mice exposed to hyperoxia (95% oxygen for 3 days) as demonstrated by miRNA arrays [37]. Neonatal hyperoxia (100% oxygen) from pnd1 to pnd4 significantly reduced lung miR-342 expression with a nadir at pnd2, pnd4, pnd7 and recovery at pnd14 [54].
Pathway analysis reveals that downregulated miRNAs are mainly related to immune and inflammatory processes, whereas upregulated miRNAs are associated with extracellular matrix remodeling. Different oxygen levels (60%–100%) and exposure durations (3–14 days) have been used to induce lung injury in rodents. Different durations of air recovery are also commonly used to investigate the long-term effects of neonatal hyperoxia on lung injury. Therefore, further studies are required to investigate the impact of different concentrations of oxygen, different durations of exposure and of air recovery on dysregulation of miRNAs. Additionally, further investigations are warranted to determine cell-specific changes in lung miRNAs after neonatal hyperoxia.
In the pups of pregnant rats endocervically inoculated with an E. coli suspension, levels of lung miR-184, miR-347, miR-181a, miR-204, miR-132, and miR-328b were upregulated, whereas expression of lung miR-122, miR-490 and another 8 miRNAs was downregulated after intrauterine infection compared to controls at pnd1 [38]. At pnd3, lung levels of miR-3559 were upregulated, whereas lung levels of miR-122, miR-490 and another 10 miRNAs were downregulated in this model. Furthermore, at pnd14, lung miR-466b levels were upregulated, while expression of lung miR-122 was most significantly downregulated after intrauterine infection [38]. This is in corroboration with the findings that lung miR-122 was reduced in mice exposed to hyperoxia as neonates [34]. These data suggest that specific miRNAs dynamically participate in the progression of lung injury after intrauterine infection/inflammation, resulting in BPD.
Hyperoxia-exposed cells are commonly used to study mechanisms underlying the
pathogenesis of BPD. We summarize upregulation or downregulation of miRNAs in
cultured cells exposed to hyperoxia (Tables 1,2). The lung contains more than 40
types of cells. Thus, the impact of hyperoxia on miRNA expression in various lung
cell types, such as alveolar epithelial, endothelial, and fibroblast cells,
differs. In lung epithelial cells, expression of miR-219, and miR-421 was
increased with exposure to hyperoxia (85% oxygen) for 6 h to 24 h [30, 46].
Gilfillan et al. [43] demonstrated that miR-451 expression was
significantly increased in murine lung endothelial cells exposed to 100% O
Despite the vast heterogeneity of miRNAs that are altered in different animal
models and in premature infants with BPD, some are more commonly upregulated.
These include miRNA-34a, miR-219 and miR-421. Among them, miR-34a has been the
most thoroughly studied. It is upregulated in the tracheal aspirates of premature
infants with BPD and in animal models of this disease. This miRNA targets many
genes, including Wnt1, Snail, cdk4,
SIRT1, Dll-4, and modulates multiple pathways, such as Wnt,
TGF-
Several miRNAs, including miR-133, miR-20b and miR-185, are differentially altered between human and animal samples in BPD. For example, miR-133b expression was upregulated in blood collected during the first 2 weeks of life in 15 subjects with BPD compared to 15 sex-matched control subjects without BPD [33]. In contrast, lung miR-133b was downregulated in a rat model of BPD induced by intrauterine infection/inflammation at pnd3 [56]. Further study is warranted to determine whether miR-133b is secreted from lung tissues through exocytosis and transported into blood during the development of BPD. Compared to gestational age-matched full-term controls, miR-20a expression was increased in the tracheal aspirates of patients with severe BPD [26]. However, miR-20b expression was downregulated in the lungs of rats exposed to hyperoxia as neonates [57]. These discrepancies may be due to altered expression of miR-20b in different lung cells during the development of BPD. Similarly, miR-185 expression was significantly increased in the tracheal aspirates of premature infants with severe BPD compared to those with mild/moderate BPD [29]. In contrast, miR-185 was reduced in the blood of infants who develop BPD compared to controls who do not [53]. Whether miR-185 is reduced in endothelial cells and increased in lung epithelial cells during lung injury observed in BPD remains to be determined [64].
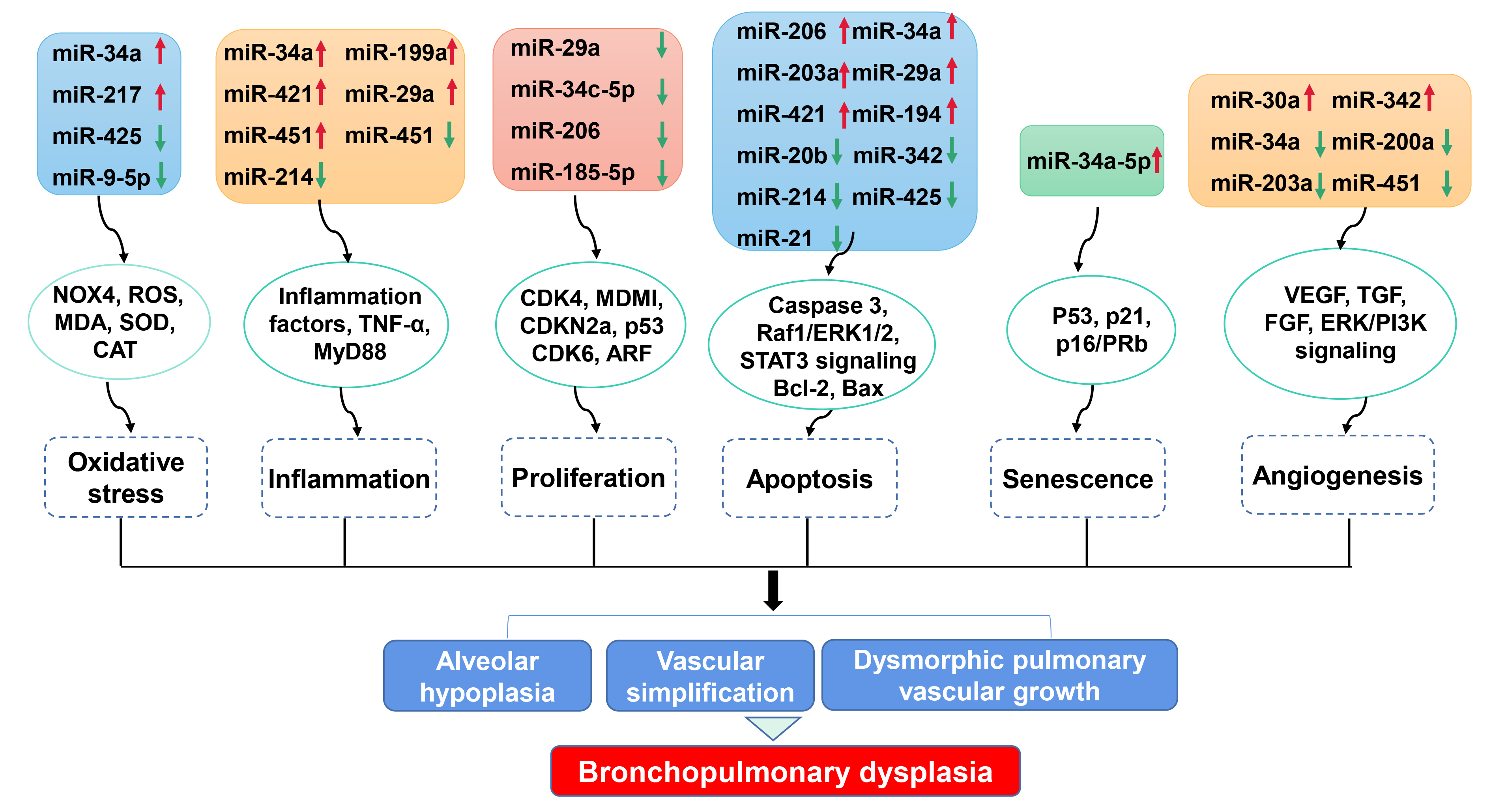
Bioinformatic analyses have identified miRNAs as direct targets of specific cellular processes. Here we summarize the link between dysregulated miRNAs and cellular processes involved in BPD (Fig. 1, Table 3 (Ref. [19, 25, 26, 27, 31, 40, 41, 42, 43, 46, 47, 51, 53, 54, 57, 58, 59, 60, 65, 66])).
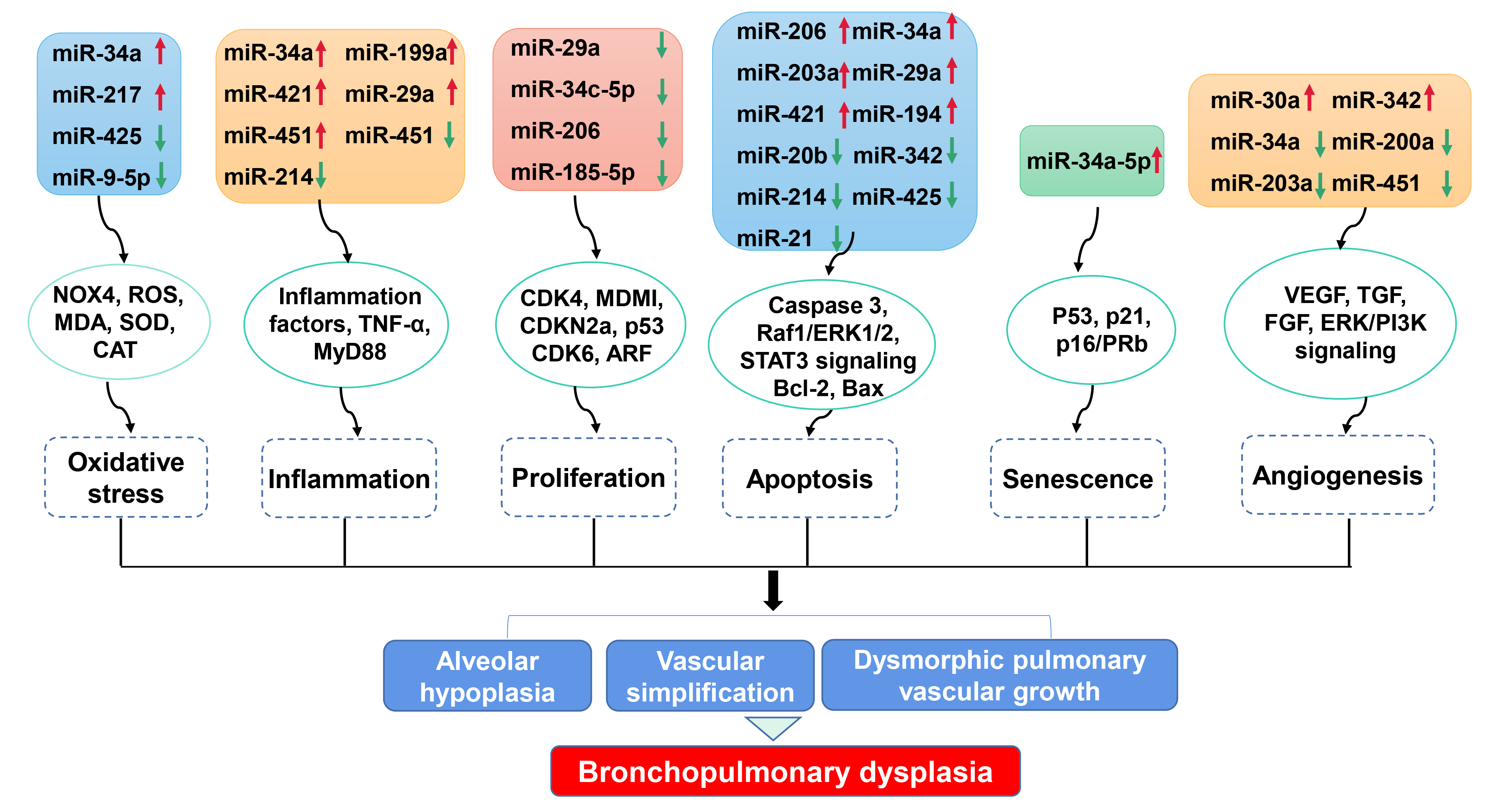 Fig. 1.
Fig. 1.Impact of miRNA dysregulation on cellular processes involved in BPD. Dysregulated miRNAs modulate numerous cellular processes, including oxidative stress, inflammation response, proliferation, apoptosis, senescence and angiogenesis, via various targeted genes. These cellular processes participate in alveolar hypoplasia, vascular simplification and dysmorphic vascular growth, which are key pathological features of BPD. NOX4, NADPH oxidase 4; ROS, Reactive oxygen species; MDA, Malondialdehyde; SOD, Superoxide dismutase; CAT, Catalase; TNF, Tumor necrosis factor; CDK, Cyclin-dependent kinase; MDM1, Mouse double-minute 1; ARF, ADP-ribosylation factor; Raf1, Raf-1 proto-oncogene, serine/threonine kinase; ERK1/2, Extracellular signal-regulated protein kinase 1/2; STAT3, Signal transducer and activator of transcription 3; Bcl-2, B-cell lymphoma 2; VEGF, Vascular endothelial growth factor; TGF, Transforming growth factor; FGF, Fibroblast growth factor; ERK/PI3K, extracellular signal-regulated protein kinase/phosphoinositide 3-kinase.
| Cell process | Change vs controls | miRNAs | Samples | References |
| Oxidative stress | ↓ | miR-425 | RLE-6TN | [59] |
| Inflammation | ↑ | miR-34a | mouse lung tissues | [25] |
| ↑ | miR199a | MLECs | [27] | |
| ↑ | miR-29a | MLE12 | [42] | |
| ↑ | miR-451 | mouse lung tissues | [43] | |
| ↑ | miR-421 | MLE12 | [46] | |
| ↓ | miR-214 | rat lung tissues | [58] | |
| ↓ | miR-876 | mouse lung tissues | [26] | |
| Proliferation | ↓ | miR-206 | A549, H441 | [60] |
| ↓ | miR-29a | MLE12 | [40] | |
| Apoptosis | ↑ | miR-206 | A549, H441 | [60] |
| ↑ | miR-34a | MLE12 | [25] | |
| ↑ | miR-203a | RLE-6TN | [31] | |
| ↓ | miR-342 | MLE12 | [54] | |
| ↑ | miR-421 | mouse lung tissues, MLE12 | [40, 41, 46] | |
| ↑ | miR-29a | MLE12 | [42] | |
| ↑ | miR-421 | mouse lung tissues | [41] | |
| ↑ | miR-194 | mouse lung tissues, BEAS-2B | [47] | |
| ↓ | miR-20b | rat lung tissues, AEC II | [57] | |
| ↓ | miR-214 | rat primary embryonic type II alveolar epithelial cells | [58] | |
| ↓ | miR-425 | RLE-6TN | [59] | |
| ↓ | miR‑21 | AEC II | [65] | |
| Senescence | ↑ | miR-34a | MLE12, SAEC | [66] |
| Angiogenesis | ↓ | miR-34a | MLE12 | [19] |
| ↑ | miR-30a | HPMECs | [51] | |
| ↓ | miR-200a | HUVECs | [53] | |
| ↓ | miR-203a | rat lung tissues, RLE-6TN | [31] | |
| ↑ | miR-342 | mouse lung tissues | [54] | |
| ↓ | miR-451 | mouse lung tissues, MLECs | [43] |
Abbreviations: A549, Human lung adenocarcinoma epithelial cell line,
metastatic cells; AEC II, rat primary type II alveolar epithelia cell; BALF,
bronchoalveolar lavage fluid; BEAS-2B, human pulmonary bronchial epithelial
cells; H441, Human lung adenocarcinoma epithelial cell line, nonmetastatic cells;
HPMECs, neonatal human pulmonary microvascular endothelial cells; HUVECs, Human
umbilical vein endothelial cells; MLE12, mouse lung epithelial cells; MLECs,
murine lung endothelial cells; RLE-6TN, rat type II alveolar epithelial cell; SAEC, human small
airway epithelial cells.
Expression of miRNAs can be altered by stresses such as exposure to hypoxia. Changes in miRNA expression under oxidative stress could regulate enzymes involved in miRNAs processing. Hypoxia inhibits the expression of DROSHA and DICER1, which could result in incomplete miRNA biogenesis [67]. Oxidative stress and radiation-induced DNA damage can activate p53 which affects the expression of several miRNAs [68]. Furthermore, miRNAs regulate the expression of redox markers and antioxidants, including Cu/Zn SOD, catalase and glutathione peroxidase. Syed et al. [25] showed that hyperoxia-exposed mice have increased myeloperoxidase activity, which was significantly decreased in the lungs of global and epithelial cell-specific miR-34a knockout mice. Hyperoxic exposure increased miR-185 expression in cultured lung epithelial cells, which promoted DNA damage [69]. Therefore, there is a vicious cycle between dysregulated miRNAs and oxidative stress, which could further drive the progression of BPD.
Hyperoxia-exposed wild type mice had an increase in lung neutrophil
infiltration, which was significantly decreased in the lung of global and type II
epithelial cell-specific miR-34a knockout mice [25]. Inhibiting miR-199a
expression attenuated hyperoxia-induced inflammatory responses including
increased interleukin-6 (IL-6), tumor necrosis factor-
GRB2-associated-binding protein 1 (GAB1) is a target gene of miR-29a. Inhibition of miR-29a promoted proliferation of MLE12 cells exposed to hyperoxia, and also protected against neonatal hyperoxia-induced lung injury through GAB1 upregulation [40]. Extracellular vesicle miR-34c-5p derived from bone mesenchymal stem cells enhanced proliferation and migration in human pulmonary microvascular endothelial cells, and inhibited hyperoxic lung injury [70]. Overexpression of miR-103a-3p and miR-185-5p significantly enhanced the proliferation and migration of normal human umbilical vein endothelial cells, whereas overexpressing miR-200a-3p inhibited these responses [53]. A better understanding of the roles of these dysregulated miRNAs in modulating lung injury observed in BPD is needed.
Certain miRNAs directly target key molecules involved in apoptotic pathways, including caspase and Bcl-2 family members [71]. For instance, miR-34a, miR-203a, miR-421, miR-29a, and miR-194 are pro-apoptotic, whereas miR-342, miR-214, miR-20b, and miR-425 inhibit apoptosis in cultured lung epithelial cells and mouse lungs exposed to hyperoxia. Transfection of miR-34a mimics further increased hyperoxia-induced apoptosis in type II alveolar epithelial cells, and these effects were decreased by an miR-34a inhibitor or genetic disruption [25]. In RLE-6TN cells, miR-203a transfection caused apoptosis [31]. Inhibition of miR-29a and miR-421 reduced neonatal hyperoxia-induced apoptosis in the lung by upregulating GAB1 and Fgf10, respectively. Consequently, inhibition of these miRNAs protected against neonatal hyperoxia-induced lung injury in mice [40, 41, 42, 46]. Overexpression of lncRNA CASC2 inhibited hyperoxia-induced apoptosis in pulmonary bronchial epithelial cells and lung injury by inhibiting miR-194 [47]. Upregulating miR-194 blocked these effects.
In contrast, miR-342 overexpression decreased type II alveolar epithelial cell apoptosis under hyperoxic conditions. This was associated with inhibition of Spred3, and the pro-survival Raf1/ERK1/2 signaling pathway [54]. Overexpression of miR-214 inhibited apoptosis in rat bronchial embryonic lung epithelial cells by downregulating the STAT3 pathway, and subsequently protected against neonatal hyperoxia-induced lung injury in rodents [58]. miR-20b overexpression attenuated hyperoxia-induced mitochondrial dysfunction-mediated apoptosis by targeting Mfn1 and Mfn2 [57]. This also inhibited hyperoxia-induced acute lung injury in adult mice. Therefore, miRNAs play a significant role in regulating apoptosis during hyperoxic lung injury.
Cellular senescence refers to the irreversible arrest of cell proliferation, which is characterized by a senescence-associated secretory phenotype and resistance to apoptosis. We reported that early programmed senescence orchestrates postnatal lung development whereas later hyperoxia-induced senescence causes lung injury [72]. miRNAs play a role in modulating senescence by potentially targeting genes on the p53, p21 and p16/pRb pathways. Furthermore, miRNAs also can regulate the actin cytoskeleton structure that contributes to the enlarged and flattened cell morphology, a characteristics of the senescence phenotype [73]. We reported that miR-34a mediates hyperoxia-induced senescence in cultured lung epithelial cells by upregulating the KLF4/p21 signaling pathway [66]. Expression of miR-34a-5p was increased in cultured lung epithelial cells and in the lungs of newborn mice exposed to hyperoxia, as well as in the lung of premature infants requiring mechanical ventilation. Further studies are warranted to understand the contribution of miR-34a to lung injury in BPD using larger animal models.
Aberrant angiogenesis is a key feature of the lung injury observed in BPD. Treatment with an miR-34a inhibitor protected against neonatal hyperoxia-induced vascular simplification in mice, and this was partially mediated via the Ang1/Tie signaling pathway [25]. Inhibiting miR-451 improved pulmonary vascular growth and alveolar simplification in neonatal hyperoxia-exposed mice. This was associated with sustained expression of macrophage migration inhibitory factor and increased expression of vascular endothelial growth factor A (VEGFA), Ang1, Ang2, and the Ang receptor Tie2 [43]. Knockdown of miR-203a protected against neonatal hyperoxia-induced alveolar simplification in rats by increasing VEGFA expression [31]. In contrast, overexpression of miR-342 increased pulmonary vessel density in neonatal mice exposed to hyperoxia [54]. The miR-30a mimic increased angiogenic sprouting in cultured human pulmonary microvascular endothelial cells by inhibiting delta-like ligand 4 (Dll4). Interestingly, these effects were observed in female but not male cells. Deletion of miR-30a expression eliminated the female resilience to neonatal hyperoxic lung injury, suggesting important roles of miRNAs in driving the sexual dimorphism observed in BPD [51, 74]. Overall, these findings suggest that miRNAs could be therapeutic targets to prevent lung injury seen in BPD through modulation of angiogenesis.
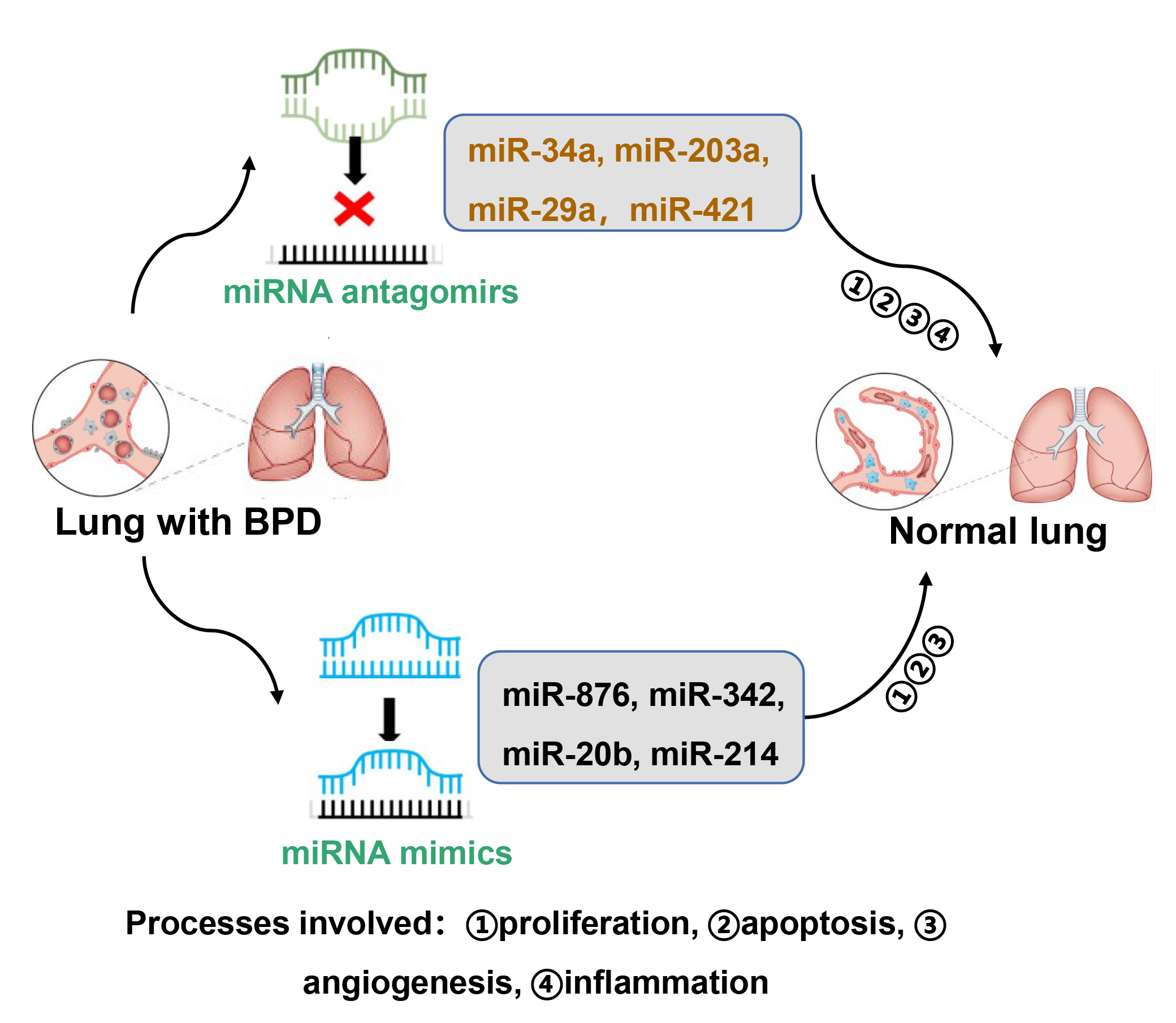
Although many miRNAs are used as biomarkers in clinical medicine and as potential therapeutic agents in treating cancer [75], there are no clinical studies on the use of miRNA modulators for preventing or treating BPD. Here, we summarize the miRNA inhibitors and miRNA mimics that have been used in preclinical animal studies for preventing BPD (Fig. 2).
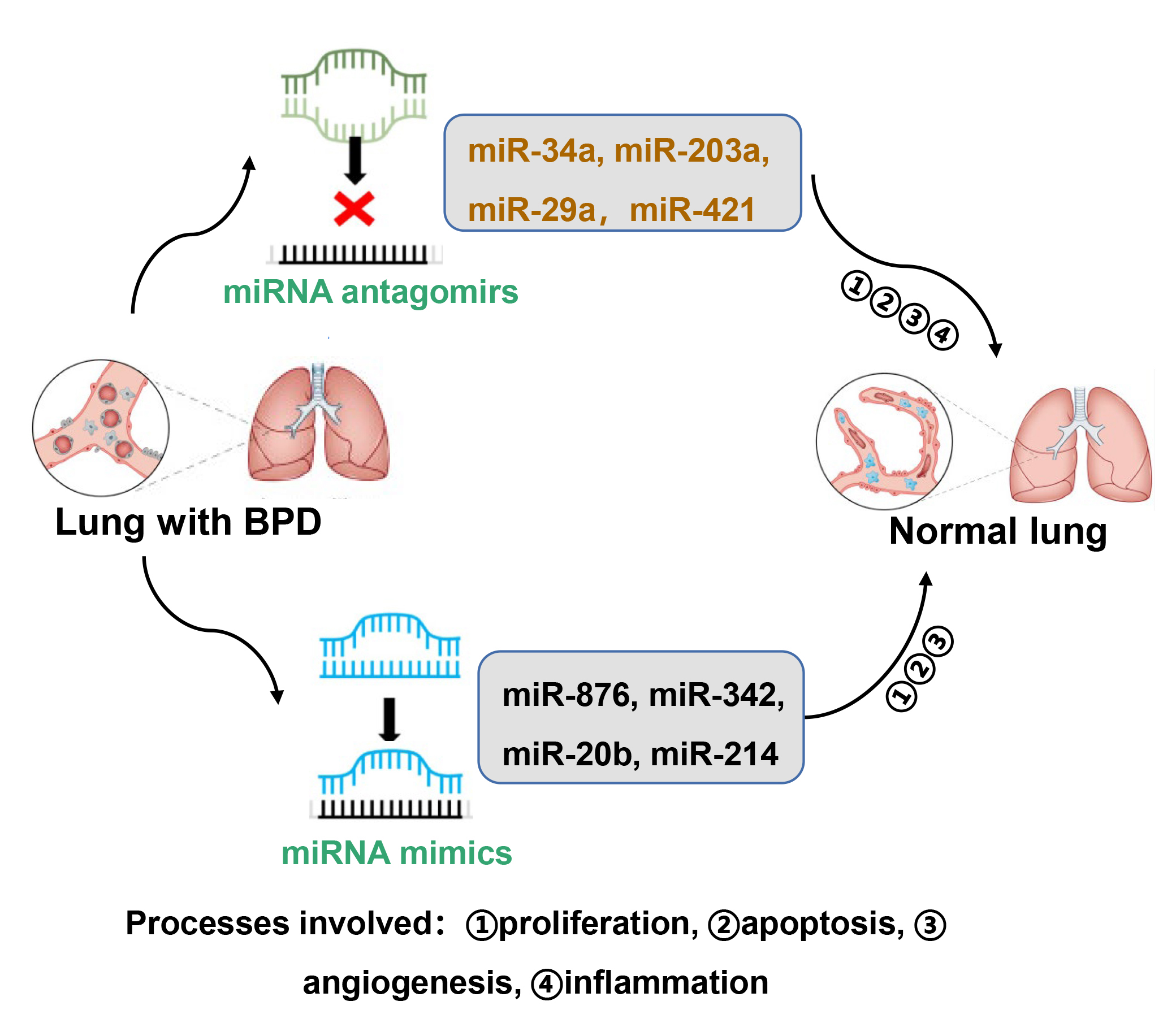 Fig. 2.
Fig. 2.Modulators of miRNAs as potential therapeutic agents to treat BPD. Certain miRNA inhibitors and mimics have been used in animal models of BPD. These include miRNA antagomirs of miR-34a, miR-203a, miR-29a, and miR-421, as well as miRNA mimics of miR-876, miR342, miR-20b, and miR-214. These miRNAs impact alveolar and vascular simplification by modulating proliferation, apoptosis, inflammation and/or angiogenesis.
Intranasal administration of miR-489, miR-34a, miR-421, miR-451 or miR-203a inhibitors during hyperoxic exposure improved alveolar development as demonstrated by increased secondary septation and radial alveolar counts, as well as reduced mean linear intercepts in rodents [25, 31, 39, 41, 43, 49]. Mechanistically, miR-34a and miR-451 inhibitors promote angiogenic activity and blood vessel maturation as well as reduce cellular senescence in the lung after neonatal hyperoxia. Hu et al. [40] subcutaneously injected adenovirus overexpressed-GAB1 or a miR-29a antagomir into mice prior to hyperoxia. Injection of the miR-29a antagomir further inhibited GAB1 overexpression-induced protection against alveolar simplification seen in neonatal hyperoxia. This may be due to reduced inflammatory responses and apoptosis as well as increased proliferation and angiogenesis with the mi-R29a antagomir. Intravenous injection of an miR-134-5p inhibitor ameliorated neonatal hyperoxia-induced lung injury by suppressing ferroptosis [76].
Several miRNA mimics are shown to inhibit lung injury in animal models of BPD via modulation of apoptosis, angiogenesis and inflammation. Lal et al. [26] administered miR-876 mimics intranasally in a single-hit (hyperoxia alone) and the double-hit (hyperoxia plus LPS) model of BPD. Injection of miR-876 mimics attenuated alveolar hypoplasia in both models. Intranasal administration of an miR-342 mimic or venous injection of miR-20b mimics significantly improved chord length and septal thickness in mice exposed to hyperoxia as neonates [54]. This may be explained by the fact that miR-342 mimic administration reduced neonatal hyperoxia-induced apoptosis and endothelial-mesenchymal transition [54]. Injection of hyperoxia exposed rats with miR-20b mimics or an miR-214 agomir alleviated lung injury, as reflected by increased number of alveoli and reduced ratio of alveolar area/pulmonary septal area [57, 58]. Furthermore, miR-20b mimics suppressed hyperoxia-induced mitochondrial dysfunction by directly regulating mitochondrial dynamics. MicroRNA-214 overexpression inhibited hyperoxia-induced lung epithelial cell apoptosis via the PlGF-dependent STAT3 pathway.
It is interesting to note that above treatments with miRNA inhibitors and mimics were prophylactic. Further studies are warranted to investigate the therapeutic effects of these miRNA inhibitors and mimics on lung injury in models of BPD.
miRNAs play important roles in regulating both physiological and pathological processes, including oxidative stress, inflammatory responses, proliferation, apoptosis, senescence, and angiogenesis. All these processes contribute to the development of BPD. The dysregulation of miRNAs has been observed in the blood and tracheal aspirates of premature infants with BPD. Larger scale clinical studies are warranted to identify whether these dysregulated miRNAs are predicttors for developing BPD or biomarkers for diagnosing this disease. Preclinical studies using mice and rats suggest that manipulation of specific miRNAs could serve as potential therapeutic strategies to prevent or ameliorate lung injury in infants with BPD.
Identifying the cell-specific expression of dysregulated miRNAs in the lung of infants with BPD could be accomplished by fluorescent in situ hybridization staining. However, this technique cannot measure multiple miRNAs in different cell types simultaneously. Single-cell microRNA sequencing, as well as spatially resolved and multiplexed miRNA quantification are cutting-edge technologies that could measure miRNAs with spatial resolution, while multiplexing directly from lung samples [77, 78] could help quantify miRNA heterogeneities in tissue samples. This will lead to informed, biomarker-based diagnostics for BPD and a better understanding of the pathogenesis of this disease.
Rodents exposed to hyperoxia in the first days of life are the most commonly used models to study human BPD [79, 80, 81]. Nevertheless, these models cannot recapitulate all the characteristics of BPD. The preterm lamb models the clinical setting of preterm birth and respiratory failure requiring prolonged ventilatory support for days or weeks with oxygen-rich gas [82, 83, 84]. Perhaps, studies using larger animals will increase our understanding of the translational significance of miRNAs in the pathogenesis of BPD and allow us to use these agents in therapies for BPD.
Challenges of miRNA-based therapy include limited cellular uptake resulting in low delivery efficiency, multiple targets leading to off-target effects and toxicity [85]. Targeting miRNAs using the CRISPR/Cas system allows for precise and permanent targeting of mutations and provides an opportunity to target dysregulated miRNAs in BPD [85]. Extracellular vesicles, including exosomes and micro-vesicles, are considered novel tools for intercellular communication because miRNAs are packaged into them and are detectable in body fluids. miRNAs in extracellular vesicles are transferred to target cells to regulate gene expression due to their resistance to RNase digestion and high stability in the serum and various body fluids. Another promising therapeutic strategy could be to use surfactant as a delivery vehicle for miRNA mimics amd/or inhibitors in the lung since surfactant is widely used clinically.
In summary, miRNAs are often modulated in the lungs and other tissues of infants with BPD and in animal and cell models of this disease. Further defining which of the myriad of miRNAs could be targeted therapeutically will be an important goal. Innovations in detection and delivery of miRNAs will allow us to refine our approaches to mitigating BPD and its devastating consequences.
Conception and Design: HY, and PAD. Data acquisition and interpretation: HM, XL and HG. Drafting of the manuscript: HM, XL and HG. Revision of the manuscript: HM, XL, HG, PAD, and HY. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by an NIH R01 R01HL166327, an Institutional Development Award (IDeA) from the NIGMS of NIH under grant #P30GM149398, the Rhode Island Foundation grant #14699_20231340 (to HY), and the Warren Alpert Foundation of Brown University (to PAD).
The authors declare no conflict of interest. Given the role as Editorial Board Member, Hongwei Yao had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.