






 , Yuan Li 1,†, Yaoyuan Zhang 2,†, Kai Yin 2, Xu Chen 3,*
, Yuan Li 1,†, Yaoyuan Zhang 2,†, Kai Yin 2, Xu Chen 3,* , Xiao Zhu 1,*
, Xiao Zhu 1,*1 Guangxi Key Laboratory of Diabetic Systems Medicine, Guilin Medical University, 541199 Guilin, Guangxi, China
2 Department of General Practice, The Fifth Affiliated Hospital of Southern Medical University, 510900 Guangzhou, Guangdong, China
3 College of Pharmacy, Guilin Medical University, 541199 Guilin, Guangxi, China
†These authors contributed equally.
Abstract
Background: Diabetic nephropathy (DN) is a common microvascular complication of diabetes mellitus (DM). Ferroptosis is an atypical form of iron-dependent, modulated cell death that has been shown to occur in human umbilical vein endothelial cells (HUVECs). Leonurine (LEO) is a single active ingredient extracted from Leonurus japonicus Houtt. It has various biological activities, including anti-inflammatory and anti-cancer effects. However, whether LEO affects ferroptosis in DN has yet to be investigated. Methods: An animal model of DN was established by subjecting C57/BL6 mice to a high-fat diet (HFD) while being induced with Streptozotocin (STZ). A cellular model of DN was established by exposing HUVECs to a high glucose (HG) concentration of 30 mM. Results: LEO was found to improve DN and to attenuate the degree of glomerulosclerosis and tubular atrophy in the mouse model. Additionally, it markedly decreased the levels of ferroptosis markers. Molecular analyses revealed that LEO inhibited HG-induced oxidative stress in HUVECs, thereby decreasing endothelial cell (EC) dysfunction. Furthermore, LEO was found to reduce ferroptosis and reverse EC dysfunction by increasing the expression of glutathione peroxidase 4 (GPX4) and nuclear factor erythroid 2-related factor 2 (Nrf2). The suppression of Nrf2 in HG-induced HUVECs inhibited LEO-GPX4 axis-mediated ferroptosis and increased EC dysfunction. Conclusions: LEO exerts anti-DN effects both in vivo and in vitro by suppressing GPX4-mediated EC ferroptosis. Mechanistically, LEO appears to induce Nrf2-mediated GPX4 expression to inhibit ferroptosis, thereby reducing EC dysfunction. This study provides a new perspective on the treatment of diseases using natural medicines. It involves a novel form of cell death that could potentially lead to better treatment of DN.
Graphical Abstract

Keywords
- leonurine
- diabetic nephropathy
- ferroptosis
- endothelial cell
Diabetic nephropathy (DN) represents one of the most common microvascular complications and a leading causes of end-stage renal disease (ESRD). Approximately 30% to 40% of patients with diabetes show DN [1]. Long-term metabolic disorders and persistent hyperglycemia can lead to progressive kidney impairment, resulting invarious complications [2]. Notably, the combination of hyperglycemia and insulin resistance [3] induces microvascular complications. Hyperglycemia is the main etiological factor in the development of diabetic kidney disease [4]. Although current treatments such as renin-angiotensin system (RAS) blockade, vitamin D receptor activators, incretin-related drugs, and target inflammation therapies of diabetic nephropathy to a certain extent, they are unable to stop the development of end-stage renal disease [5]. Microvascular endothelial cells (ECs) are believed to be the primary target of hyperglycemic damage [4]. During hyperglycemia, damaged ECs impair the vascular endothelium through several pathophysiological mechanisms, including exocytosis, podocyte/fenestrated EC extracellular matrix crosstalk, and inflammatory responses [6]. The pathophysiology of DN is multifactorial, with EC death playing a crucial role in its onset and progression. Although current treatments can delay the development of DN, they do not protect against progression to ESRD. Therefore, there is an imperative for further research into the targeting endothelial cells to improve DN.
Ferroptosis, a newly named mode of programmed cell death in 2012, differs
from existing apoptosis, autophagy, and necrotic apoptosis, among others, in that
ferroptosis is driven by iron-dependent lipid peroxidation of unsaturated fatty
acids, especially phospholipids, on cell membranes. Ferroptosis is regulated by
the system Xc
Traditional Chinese medicine is widely used to treat diabetes and its complications. Leonurine (LEO) is a natural chemical compound extracted from the traditional Chinese herbal medicine Herba Leonuri [12]. The strong anti-inflammatory [13] and antioxidant [14] effects of LEO have been extensively demonstrated. This agent can reduce the damage caused by excessive ROS in many diseases, including cardiovascular diseases [15], ischemic stroke [16], and atherosclerosis [17]. However, the possible effects of LEO in diabetes-induced DN and the underlying mechanisms remain unknown.
In light of the above described data, the goal of this investigation was to examine how LEO affects ferroptosis in DN. To validate our hypothesis, we used high glucose (HG)-induced human umbilical vein endothelial cells (HUVEC) and diabetic mice models for in vivo and in vitro experiments, respectively. Our study found that LEO inhibited ferroptosis in glomerular endothelial cells of DN mice and improved the DN process. And in vitro experiments, LEO protects HUVECs against HG-induced endothelial cell dysfunction and oxidative stress. Additionally LEO inhibited ferroptosis and reduced lipid peroxidation in HUVECs. Mechanistically, we found that the inhibition of ferroptosis in HUVEC by LEO was through nuclear factor erythroid 2-related factor 2 (Nrf2)-GPX4 axis mediated effects. Our findings provided a therapeutic strategy depended on ferroptosis to reduce kidney damage in DN.
The reagents that were utilized were leonurine (20220301, Xinxiang Olan Biotechnology Co., Ltd, Shijiazhuang, Hebei, China), Fer-1 (HY-100579, MedChemExpress, Rahway, NJ, USA), and fetal bovine serum (M1043150S, BBI Solutions, south Wales, UK).
There are three treatment groups overall as a result of the randomized animal
division. Allpro cedures were conducted under protocols approved by the
Institutional Animal Care and Use Committee of Guilin Medical University.
(Approved No. GLMC202105049). C57BL/6J mice, aged four weeks, were procured from
Cavens Biotechnology Co., Ltd. (Nanjing, China) (SCXK2021-0013) and housed in a
dedicated pathogen-free facility with a relative humidity of 55
Human HUVECs (Chinese Academy of Sciences, Shanghai, China) were grown in
DMEM supplemented with 10% fetal bovine serum (FBS), and penicillin/streptomycin
(100 U/mL). We established cellular model of DN by treating HUVECs with HG at a
concentration of 30 mM for 24 h in a 37 °C humidified incubator. Cells
were co-treated with LEO at a concentration of 40 µM and HG at 30 mM as a
treatment group. The cells were incubated in a cell culture incubator with 5%
CO
Cell viability was evaluated using the Cell Counting Kit-8 assay kit (Beyotime, Songjiang, Shanghai, China) as recommended by the manufacturer. Cells were grown in 96-well plates for 24 h. They were then treated with HG at concentrations of 10, 20, 30, 40, 50, or 60 µM, and subsequently with LEO at doses of 5, 10, 20, 40, 80, or 100 µM. Following the appropriate treatment, cell counting kit-8 (CCK8) detection reagent was added and allowed to incubate for two hours before reading the absorbance at 450 nm with a microplate reader.
Using 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylimidacarbocyanine (JC-1) fluorochrome (C2006, Beyotime), to detect mitochondrial membrane potential (MMP) in HUVEC under
different treatment conditions. HUVECs were seeded at 10
Protein concentrations were measured using a bicinchoninic acid assay (BCA) protein assay kit (CWBIO, Beijing, China). Proteins (20 µg per lane) were loaded onto an 8% sodium dodecyl sulfate (SDS)-polyacrylamide electrophoresis gel (Solarbio Co., Beijing, China) and separated by running at 80 V for 30 min and then 120 V for 1 h. The proteins were subsequently transferred onto a 0.45 µM polyvinylidene fluoride membrane (PVDF, Merck Millipore, Darmstadt, Germany). The PVDF membranes were blocked using QuickBlock™ (P0252, Beyotime Biotechnology) for 30 min at room temperature, and then incubated overnight at 4 °C with gentle shaking with a 1:1000-dilution of specific primary antibody. The antibodies used were as follows: anti-GAPDH (60004, Proteintech, Wuhan, Hubei, China), anti-GPX4 (6773-1-lg, Proteintech, Wuhan, Hubei, China), anti-FTH (DF6278, Affinity, Liyang, Jiangsu, China), anti-FTL (10727-1-AP, Proteintech, Wuhan, Hubei, China), anti-CD31 (AF3628, R&D, Minneapolis, MN, USA), and anti-VE-cadherin (sc-9989, Santacruz, Pudong New Area, Shanghai, China).
Total RNA was isolated from aortic tissues or cells using TRIzol reagent
(15596026, Invitrogen, Carlsbad, CA, USA) and reverse-transcribed into cDNA with
StarScript II RT Mix and gDNA Remover (A224-10, Genstar, Changping, Beijing,
China). Real-time PCR analysis was performed using 2
| Gene | Species | Sequence for primers |
| TGF- |
Human | Forward: GGCCAGATCCTGTCCAAGC |
| Reverse: GTGGGTTTCCACCATTAGCAC | ||
| CD31 | Human | Forward: AACAGTGTTGACATGAAGAGCC |
| Reverse: TGTAAAACAGCACGTCATCCTT | ||
| CD44 | Human | Forward: CTGCCGCTTTGCAGGTGTA |
| Reverse: CATTGTGGGCAAGGTGCTATT | ||
| TNF- |
Human | Forward: CCTCTCTCTAATCAGCCCTCTG |
| Reverse: GAGGACCTGGGAGTAGATGAG | ||
| IL-6 | Human | Forward: ACTCACCTCTTCAGAACGAATTG |
| Reverse: CCATCTTTGGAAGGTTCAGGTTG | ||
| MCP-1 | Human | Forward: CAGCCAGATGCAATCAATGCC |
| Reverse: TGGAATCCTGAACCCACTTCT | ||
| GPX4 | Human | Forward: GAGGCAAGACCGAAGTAAACTAC |
| Reverse: CCGAACTGGTTACACGGGAA | ||
| FTH-1 | Human | Forward: CCCCCATTTGTGTGACTTCAT |
| Reverse: GCCCGAGGCTTAGCTTTCATT | ||
| SLC40A1 | Human | Forward: CTACTTGGGGAGATCGGATGT |
| Reverse: CTGGGCCACTTTAAGTCTAGC | ||
| SLC11A2 | Human | Forward: TGGAGATCATGGGGAGTCTG |
| Reverse: AAGAAAACCTGGTCCGGTGAA | ||
| GAPDH | Human | Forward: GCACAGTCAAGGCCGAGAAT |
| Reverse: GCCTTCTCCATGGTGGTGAA | ||
| Gpx4 | Mouse | Forward: TGTGCATCCCGCGATGATT |
| Reverse: CCCTGTACTTATCCAGGCAGA | ||
| Tgf- |
Mouse | Forward: TGGAGCAACATGTGGAACTC |
| Reverse: CGTCAAAAGACAGCCACTCA | ||
| Tnf- |
Mouse | Forward: CAGGCGGTGCCTATGTCTC |
| Reverse: CGATCACCCCGAAGTTCAGTAG | ||
| Cd31 | Mouse | Forward: ACGCTGGTGCTCTATGCAAG |
| Reverse: TCAGTTGCTGCCCATTCATCA | ||
| Cd44 | Mouse | Forward: TCGATTTGAATGTAACCTGCCG |
| Reverse: CAGTCCGGGAGATACTGTAGC | ||
| Slc39a14 | Mouse | Forward: GAGTGGGCCGGGATAATGTTT |
| Reverse: GAGATCGCTCGCTCAAGTTGT | ||
| Slc40a1 | Mouse | Forward: TGGAACTCTATGGAAACAGCCT |
| Reverse: TGGCATTCTTATCCACCCAGT | ||
| Fth-1 | Mouse | Forward: CAAGTGCGCCAGAACTACCA |
| Reverse: ACAGATAGACGTAGGAGGCATAC | ||
| Gapdh | Mouse | Forward: AGGTCGGTGTGAACGGATTTG |
| Reverse: GGGGTCGTTGATGGCAACA |
PCR, Polymerase chain reaction; TGF-
The kidneys from mice were dissected and fixed in formalin. Mouse kidneys were dissected and embedded in optimal cutting temperature compound (Sakura Tissue-Tek, Torrance, CA, USA). Sections (5 µM thickness) of mouse glomerulus were cut using a Leica CM3050 S Research Cryostat (Leica, Weizler, Hesse, Germany). Mouse kidneys underwent Masson staining to identify the area and extent of the lesion respectively, while hematoxylin-eosin staining (HE) staining was used to identify glomerulosclerosis. En face images of the glomerulus were captured using a digital camera and analyzed using ImageJ software (win64, National Institutes of Health, Bethesda, MD, USA).
After fixing the cells and frozen sections with 4% paraformaldehyde, the antigen was extracted using Quick Antigen Retrieval Solution for Frozen Sections (P0090, Beyotime). The cells were then blocked with 5% QuickBlockTM Blocking Buffer (P0260, Beyotime) at room temperature for an hour, and primary antibodies were incubated at 4 °C for an overnight period. Following a PBS wash, the samples were incubated for two hours at room temperature in the dark with Alexa Flour 488 or 594 secondary antibodies. The stain used on nuclei was DAPI (28718-90-3, Solarbio Co.). Finally, an anti-fluorescence quencher (S2100, Solarbio Co.) was used to seal and mount the sections.
The levels of iron, MDA and GSH in the glomerulus of mice were determined using the corresponding kits (Abcam, Pudong New Area, Shanghai, China) according to the manufacturer’s instructions. The iron, GSH, and MDA levels in HUVECs were determined using colorimetry. GSH and MDA in the glomeruli of mice and in HUVECs were reacted with kit reagents to determine the absorbance of the sample. The sample levels were then determined by comparison with the standard.
In 6-well plates, 5
The samples were embedded in paraffin and treated with 4% paraformaldehyde. The sections had a thickness of 9 µM. Using Lillie staining (Solarbio Co.), variations in ferrous iron levels in tissues were identified. Following deparaffinization, the tissue sections were soaked for 27 min in Lillie staining solution and then 5 min in deionized water. Then nuclear fast red stained solution for 7 min, finally dehydrated and transparented with anhydrous ethanol and sealed with neutral gum. Images were captured using a fluorescence microscope (BS.3153-PLi, Handelsen, Hamburg, Germany).
As directed by the manufacturer, DCFH-DA (Beyotime Biotechnology, Shanghai, China) examined the reactive oxygen species levels of HUVECs. Following the seeding of HUVECs cells into 24-well plates, fluorescent probes were added and diluted 1:1000 using serum-free media. The cells were then incubated for 20 min at 37 °C in a cell culture incubator. After three rounds of serum-free DMEM washing, the cells were examined under a fluorescence microscope. The Image J program was used to quantify the intensity of the cellular fluorescence.
Results are presented as the mean
The therapeutic effect of LEO on DN was investigated in a mouse model of type
2 diabetes mellitus (T2DM) established by IP injection of STZ (50 mg/kg). Blood
glucose levels were higher in both STZ and STZ+LEO (60 mg/kg) mice than in the
control group (no STZ injection). Oral glucose tolerance tests (OGTT) revealed
that mice administered STZ or STZ+LEO had reduced glucose tolerance, as evidenced
by a faster rise in blood glucose, higher 30-min blood glucose values, and a
slower decrease in blood glucose compared to the control group. These results
indicate successful establishment of a mouse model of T2DM (Fig. 1A). After 12
weeks, STZ-induced DN mice showed significantly increased albuminuria, but this
was attenuated by treatment with LEO (Fig. 1B). Serum creatinine and blood urea nitrogen (BUN) levels
were markedly decreased by LEO treatment (Fig. 1C,D). HE staining revealed that
LEO mitigated the extent of glomerulosclerosis and tubular atrophy in STZ-induced
DN mice (Fig. 1C). Masson’s trichrome staining also showed that LEO alleviated
renal interstitial fibrosis in these mice (Fig. 1E). Expression of the
inflammatory factor tumor necrosis factor-
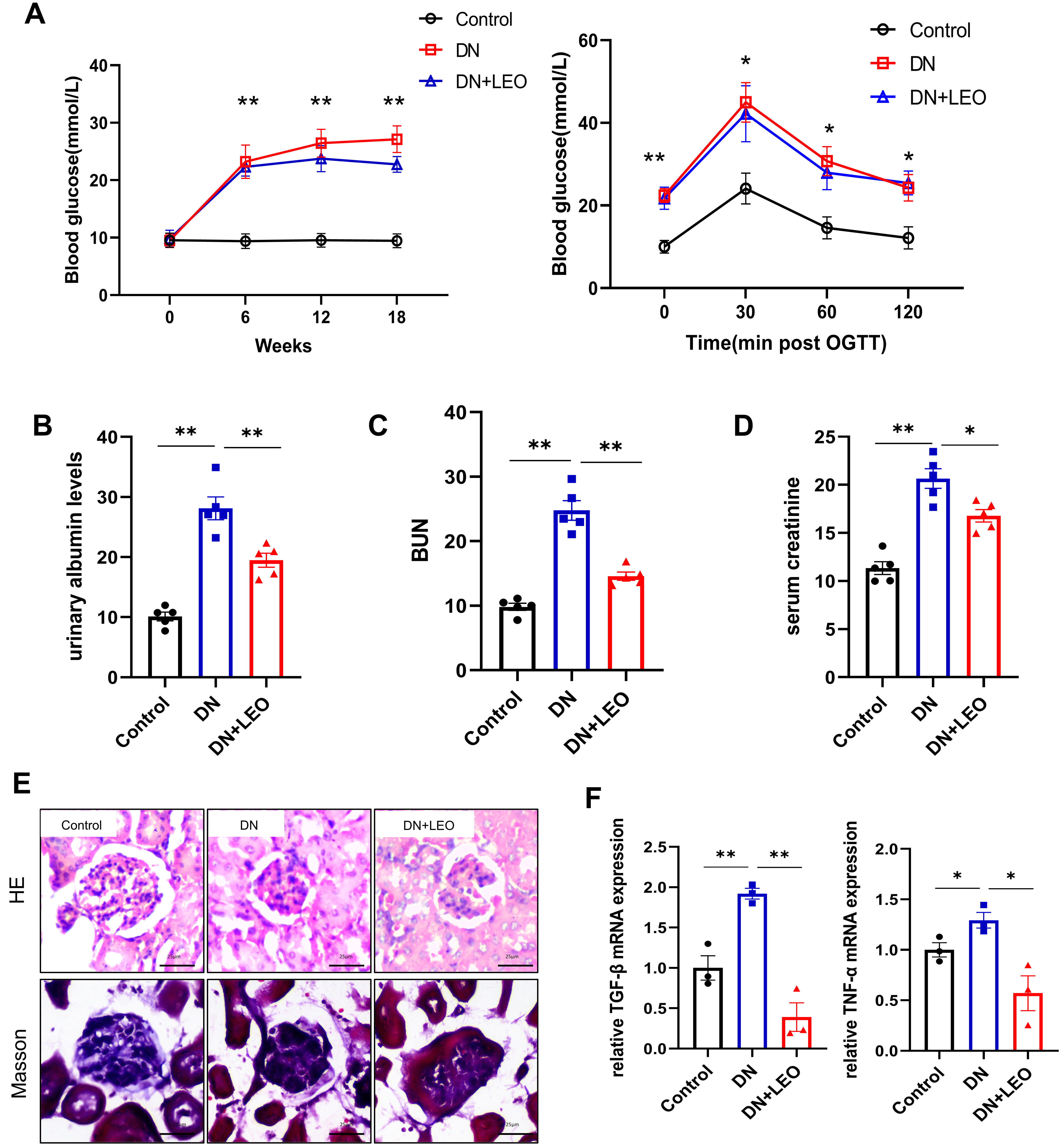 Fig. 1.
Fig. 1.LEO treatment exerts protective effects against renal damage
in DN mice. (A) Fasting blood glucose and OGTT. (B–D) Levels of blood urea
nitrogen (BUN), serum creatinine and urinary albumin in renal tissues were
evaluated using the corresponding kits. (E) HE and Masson staining of kidney
sections. Scale bar: 25 µm. (F) The mRNA expression of TNF-
Ferroptosis plays a crucial role in the onset and progression of DN, with GPX4, ferritin light chain (FTL) and and recombinant ferritin, heavy polypeptide (FTH) being central regulators of this process. Treatment with LEO significantly restored GPX4, FTL and FTH protein levels in ECs (Fig. 2A). Additionally, the mRNA levels of GPX4, FTH-1 and SLC40A1 were lower in mice with DN, while that of SLC39A14 was increased. However, these levels were restored by LEO (Fig. 2B). We next determined the iron content in kidney tissues using the corresponding kit. As shown in Fig. 2C, the iron level was notably increased in the DN group, but decreased in the LEO group. The GSH content was lower in the DN group. However, after LEO administration, the MDA content was markedly reduced and the GSH level increased, indicating the suppression of lipid peroxidation by LEO in DN mice (Fig. 2C–E). To further validate the inhibitory effect of LEO on ferroptosis in the DN model, another index of ferroptosis, Lillie’s ferrous iron stain, was also investigated. Thisrevealed that STZ-induced DN resulted in kidney iron deposition, and that LEO could reverse this iron accumulation in the glomerulus (Fig. 2F). Furthermore, LEO treatment increased the expression of endothelial and ferroptosis markers in DN. Decreased expression of GPX4, FTL and FTH was observed in glomerular vascular ECs (Fig. 2G). LEO treatment promoted VE-cadherin and CD31 protein expression tomarkedly higher levels than observed in the STZ-induced DM group (Fig. 2I). RT-qPCR also showed that LEO treatment significantly restored the expressionof Cd31 and Cd44 (Fig. 2H). Collectively, these results indicate that LEO exerts a protective effect by inhibiting ferroptosis in mice with DN. LEO therefore appears to protect against HG-induced dysfunction and oxidative stress in ECs.
 Fig. 2.
Fig. 2.LEO treatment reduces ferroptosis and endothelial dysfunction
in DN mice. (A) Protein expression of GPX4, ferritin light chain (FTL) and recombinant ferritin, heavy polypeptide (FTH) was determined by
Western blot. (B) mRNA expression of Fth-1, Gpx4, Slc39a14 and Slc40a1 was
determined by RT-qPCR. (C–E) Iron, glutathione (GSH) and malondialdehyde (MDA) levels in renal tissue were
determined using the appropriate assay kit. (F) Results of Prussian blue (ferric)
and Lillie dye (ferrous) staining in the mouse renal glomerulus in each group.
The arrow points to stained ferrous iron. Scale bar: 25 µm. (G) Sections of
renal glomerulus were co-stained for cluster of
differentiation 31 (Cd31) (green) and Gpx4 (red), FTH (red) or
FTL (red). Scale bar: 20 µm. (H) The mRNA expression of Cd31 and Cd44 molecule (Indian blood group) was
determined by RT-qPCR. (I) Protein expression of CD31 and VE-cadherin was
determined by Western blot. Values shown are the mean
To further explore protective role of LEO in ECs and to elucidate the underlying mechanism, the effects of varying doses of high hyperglycemia (HG) on HUVEC were assessed by measuring cell viability using the CCK8 assay. The CCK8 data demonstrated a significant decrease in cell viability at HG concentrations greater than 30 mM (Fig. 3A). After that, HUVEC were exposed to escalating LEO doses, and the viability of the cells was evaluated. Cell viability was significantly decreased at 80 µM of LEO and higher, as shown in (Fig. 3B). Subsequent experiments were therefore performed using 40 µM LEO. To investigate the protective effect of 40 µM LEO on HUVECs, MDA and GSH levels were evaluated to determine lipid peroxidation. LEO inhibited MDA production and increased the level of antioxidant GSH in HUVECs under oxidative stress (Fig. 3C,D). Intracellular ROS generation was evaluated using the 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) probe. Exposure to 30 mM HG led to excessive accumulation of ROS, whereas LEO treatment reduced ROS levels (Fig. 3E). Hence, LEO appears to exert a protective effect against HG-induced oxidative stress in HUVECs. In addition, Western blot results showed that HG reduced the expression levels of cluster of differentiation 31 (CD31) and VE-cadherin in HUVECs, whereas LEO restored their expression (Fig. 3F). Furthermore, the scratch assay showed that HG reduced cell migration, but this was enhanced by treatment with LEO (Fig. 3G). The above results suggest that HG increases EC damage and oxidative stress, whereas LEO protects HUVEC endothelial function.
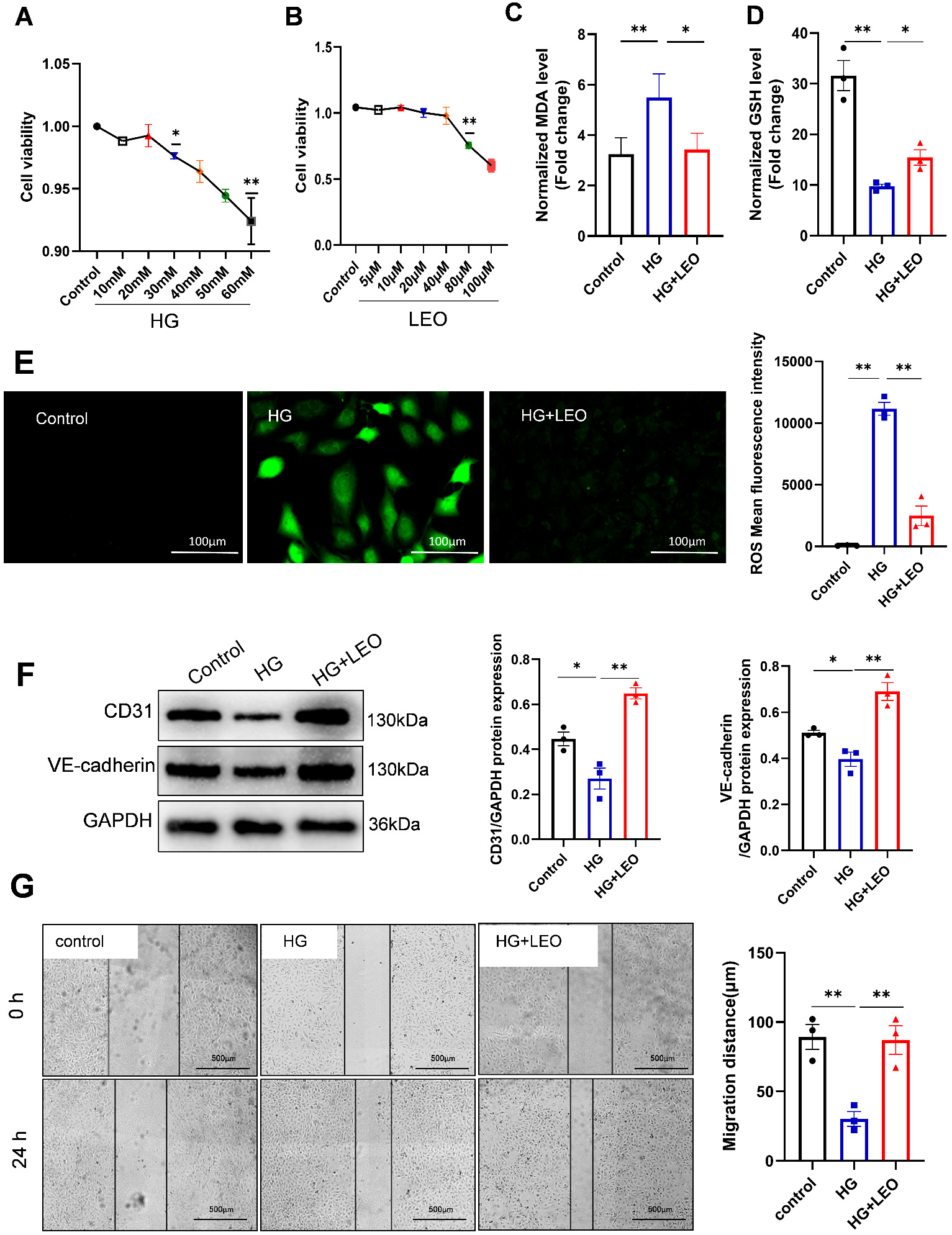 Fig. 3.
Fig. 3.LEO protects human umbilical vein endothelial cells (HUVECs) against HG-induced toxicity and
dysfunction. (A,B) The cell viability of HUVEC cells treated with different high glucose (HG)
or LEO concentrations was determined using the cell counting kit-8 (CCK8) assay. (C,D) The level of GSH
and MDA in HUVECs was determined using the corresponding kit. (E) Reactive oxygen species (ROS) staining
and fluorescence intensity in the control, HG, and HG+LEO groups. Scale bar: 100
µm. (F) Protein expression of CD31 and vascular endothelial (VE)-cadherin was determined by
Western blot. (G) The migration of HUVECs was evaluated with the scratch assay.
Scale bar: 500 µm. Values shown are the mean
We next examined whether LEO could restore EC function by reducing HG-induced
deposition of ferritin. Fer-1 is a typical ferroptosis inhibitor and has been
used to inhibit ferroptosis by attenuating lipid peroxidation and endothelial
dysfunction in ECs [18]. HUVECs were treated for 24 h with HG (30 mM), HG+LEO (40
µM), or ferroptosis inhibitor HG+Fer-1 (2 µM). Similar to Fer-1, LEO
inhibited HG-mediated ferroptosis. Representative microscopy and Western blot
images show the rescue of GPX4, FTL and FTH protein expression by LEO and Fer-1
(Fig. 4A,B). Following induction by HG, iron accumulation was also observed in
HUVECs (Fig. 4C). However, this HG-induced deposition of iron was significantly
reduced by LEO. Ferroptosis is often accompanied by excessive intracellular lipid
peroxidation, with MDA being the end product of lipid peroxidation. The MDA level
was significantly higher in the HG group than in the control group, indicating
that HG increased the cellular level of lipid ROS (Fig. 4D). The production of
MDA induced by HG was significantly reduced by treatment with LEO and Fer-1. GSH
is a crucial defence mechanism against lipid peroxidation and ferroptosis, and
was also found to be lower in the HG group (Fig. 4E). However, the GSH level was
effectively restored by treatment with LEO and Fer-1, and ferroptosis was
inhibited (Fig. 4D). Moreover, LEO and Fer-1 reversed the decrease in GPX4, FTH1
and SLC40A1 expression induced by HG, and increased SLC11A2 expression (Fig. 4F).
In actuality, ferroptosis is linked to serious impairments in the metabolism,
bioenergetics, and morphology of the mitochondria. In the present study, HG
treatment of HUVECs decreased the mitochondrial membrane potential (MMP) (Fig. 4G), leading to mitochondrial damage.
Moreover, the HG-induced reduction in MMP was attenuated by LEO and Fer-1,
indicating they have protective functions in mitochondria. HUVECs were treated
with LEO and Fer-1 to further explore whether LEO repairs endothelial dysfunction
via ferroptosis. As expected, this restored the mRNA levels of CD31, CD44,
VE-cadherin and TGF-
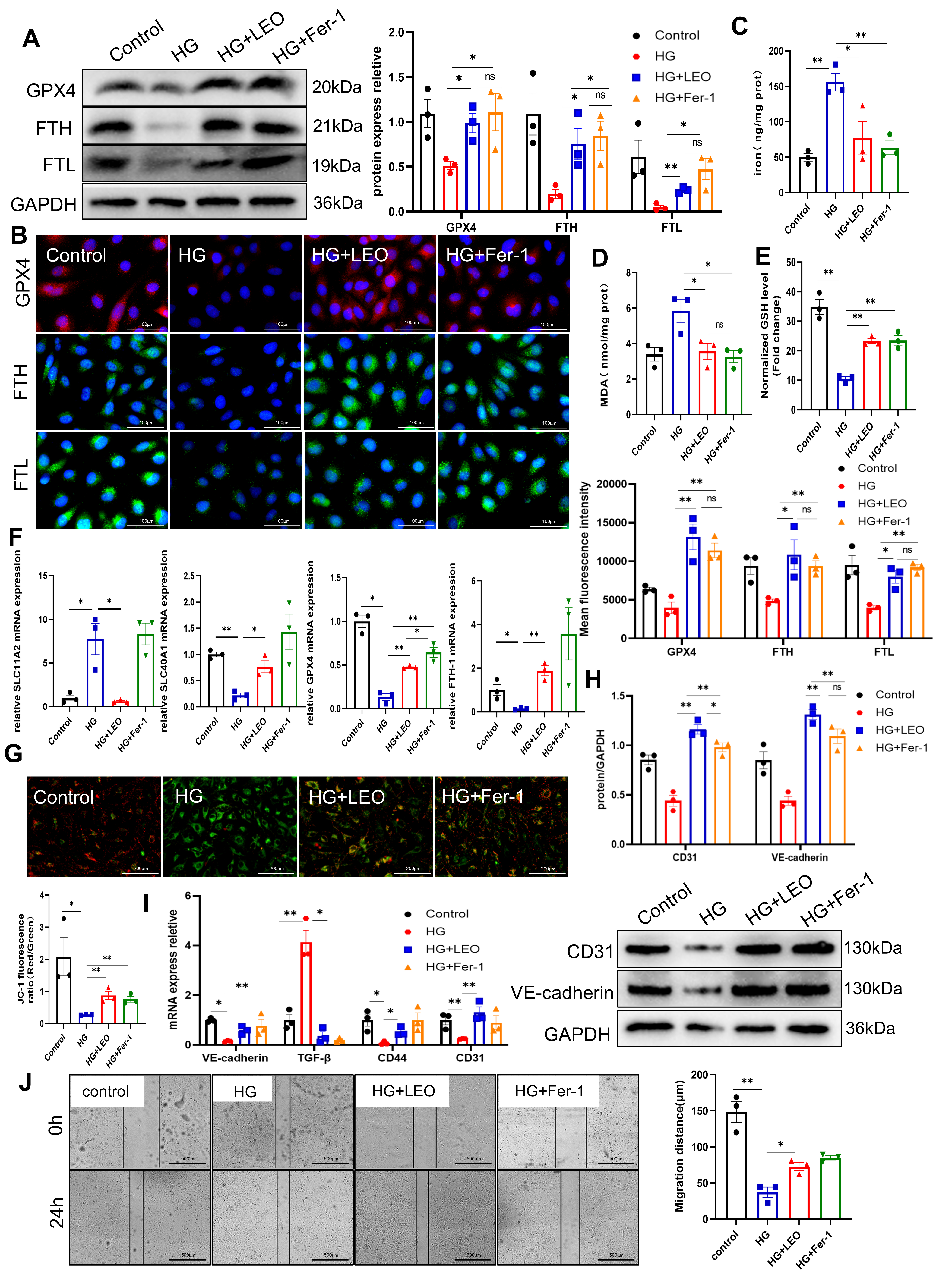 Fig. 4.
Fig. 4.LEO suppresses HG-induced ferroptosis in HUVECs. (A) Protein
expression of GPX4, FTH and FTL was determined by Western blot. (B) Expression of
GPX4 (red), FTH (green) and FTL (green) proteins was detected by immunofluorescence (IF). Nuclei were
stained with DAPI (blue) in HG-induced HUVECs treated with or without LEO. Scale
bar: 100 µm. (C–E) The level of iron, GSH and MDA in HUVECs was determined
using the corresponding kit. (F) The mRNA expression of FTH-1, GPX4, SLC11A2 and
SLC40A1 was determined by RT-qPCR. (G) JC-1 staining and the fluorescence ratio
of mitochondrial membrane potential (MMP) in the control, HG, and HG+LEO groups. Scale bar: 200 µm. (H)
Protein expression of CD31 and VE-cadherin was determined by Western blot. (I)
The mRNA expression of CD31, VE-cadherin, TGF-
Lipid peroxide is produced in significant quantities when GPX4 activity is
suppressed, and this is thought to be an important factor in preventing
ferroptosis. We next studied the mechanism by which LEO protects HG-induced
HUVECs against ferroptosis. HUVECs were first transfected with specific GPX4
siRNA. The knockdown of GPX4 expression was demonstrated by immunofluorescence
(IF) (Fig. 5A). Intriguingly, silencing of GPX4 blocked the inhibitory effect of
LEO on HG-induced ferroptosis, decreased the generation of GSH, and increased the
iron content in HUVECs (Fig 5B,C). LEO reduced the HG-induced accumulation of
lipid peroxide MDA, but this was abolished by transfection with si-GPX4 (Fig. 5D). GPX4 silencing also partially abrogated the effects of LEO on the protein
expression of markers for vascular endothelial dysfunction (CD31 and VE-cadherin)
(Fig. 5E). Knockdown of GPX4 also abrogated the increase in MMP caused by LEO
treatment of HG-treated HUVECs (Fig. 5F). The mRNA levels of CD31 and CD44 showed
the same trend. si-GPX4 also reversed the decrease in the levels of
inflammation-related factors (interleukin-6 (IL-6), monocyte chemotactic protein 1 (MCP-1), TNF-
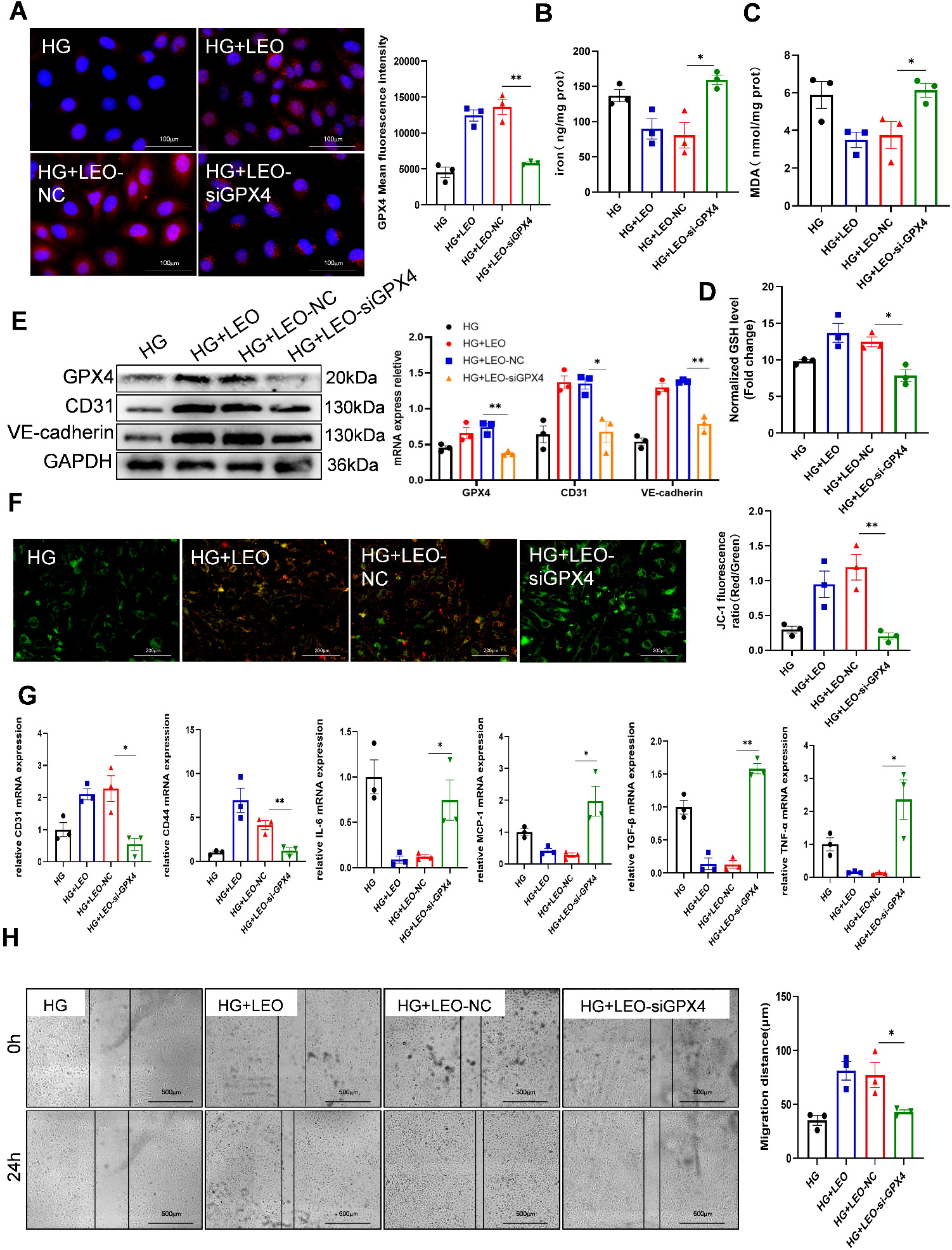 Fig. 5.
Fig. 5.GPX4 is involved in the anti-ferroptosis effect of LEO in
HG-induced HUVECs. (A) Expression of GPX4 (red) protein was detected by IF, and
nuclei were stained with DAPI (blue) in HUVECs treated with LEO and siGPX4 or NC.
Scale bar: 100 µm. (B–D) The levels of iron, GSH and MDA in HUVECs were
determined using the corresponding kit. (E) Protein expression of GPX4, CD31 and
VE-cadherin was determined by Western blot. (F) JC-1 staining and the
fluorescence ratio of MMP in the presence or absence of siGPX4. Scale bar: 200
µm. (G) mRNA expression of CD31, CD44, IL-6, MCP-1, TGF-
Nuclear factor-E2 related factor 2 (Nrf2) is a transcription factor that can regulate GPX4. To further investigate whether LEO affects GPX4 through Nrf2 to subsequently inhibit ferroptosis, we evaluated the expression of Nrf2 following LEO treatment. HG treatment (30 mM) reduced Nrf2 expression, but this increased after the administration of LEO (40 µM) (Fig. 6A). The Nrf2 inhibitor ML385 (5 µM) significantly inhibited GPX4 expression compared to LEO treatment (Fig. 6B,C). ML385 also significantly attenuated the LEO-induced reduction in iron and MDA content, while significantly decreasing the GSH content (Fig. 6D–F). To further examine whether the inhibition of Nrf2 could prevent LEO inhibited EC dysfunction, western blot was used to evaluate CD31 and VE-cadherin expression. ML385 partially abrogated the expression of CD31 and VE-cadherin compared to LEO alone (Fig. 6G). Furthermore, the increase in MMP following LEO treatment of HG-treated HUVECs was abrogated by ML385 (Fig. 6H). Cell scratch experiments also showed that ML385 inhibited HUVEC migration compared with LEO treatment alone (Fig. 6I). These results suggest that LEO inhibits ferroptosis in HG-induced HUVEC by activating the Nrf2/GPX4 signaling pathway.
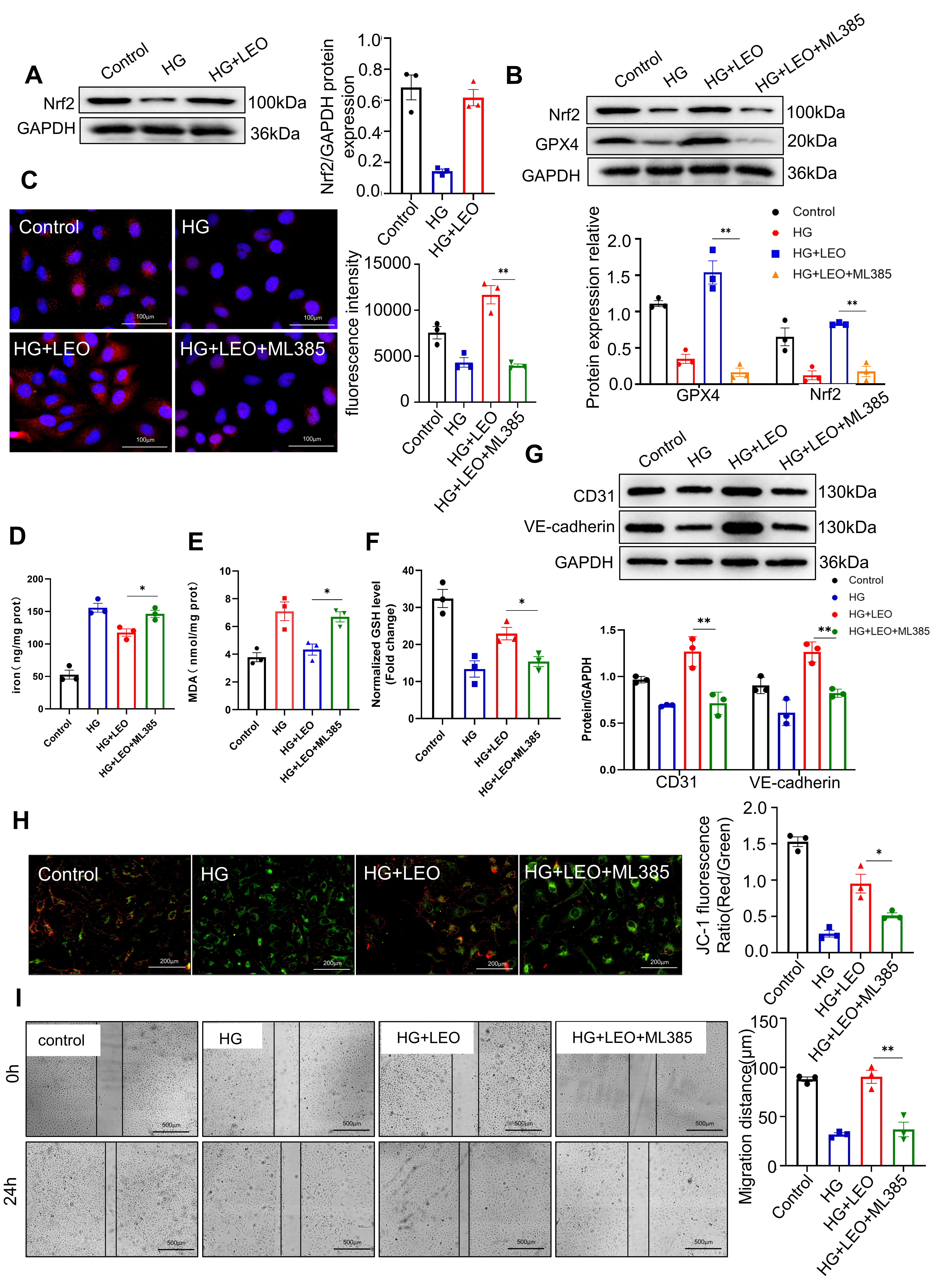 Fig. 6.
Fig. 6.LEO attenuates endothelial impairment via the nuclear factor erythroid 2-related factor 2 (Nrf2)-GPX4 axis
in HG-induced HUVECs. (A) Protein expression of Nrf2 was determined by Western
blot. (B) Protein expression of Nrf2 and GPX4 was determined by Western blot. (C)
Expression of GPX4 (red) protein was detected by IF, and nuclei were stained with
DAPI (blue) in HUVECs treated with LEO and ML385 or not. Scale bar: 100 µm.
(D–F) The levels of iron, GSH and MDA in HUVECs were determined using the
corresponding kit. (G) Protein expression of CD31 and VE-cadherin was determined
by Western blot. (H) JC-1 staining and the fluorescence ratio of MMP in the
presence or absence of ML385. Scale bar: 200 µm. (I) The migration of
HUVECs was determined with the scratch assay. Scale bar: 500 µm. Values
shown are the mean
LEO has shown good efficacy in the treatment of cardiovascular diseases such as myocardial infarction and atherosclerosis. There is also increasing evidence supporting the clinical value of LEO in the treatment of diabetes [19] and kidney injury [20]. DN is the most common microvascular complication of diabetes and contributes to renal failure and high mortality rates. Injury to the ECs plays a key role in the early development and progression of DN. Endothelial dysfunction leads to vasoconstriction, vascular inflammation, and several other vascular abnormalities. LEO can reduce histopathological inflammation and act as an anti-oxidant [20], anti-inflammatory [21], and angiogenesis-promoting agent [22]. However, it is not known whether LEO can also exert anti-inflammatory and anti-oxidant effects to slow the progression of DN. The present study therefore investigated whether LEO is involved in the progression of HG/STZ-induced DN.
We first assessed whether LEO inhibits ferroptosis in STZ-induced mice.
Interestingly, our results showed that LEO significantly reduced glomerular
injury and renal fibrosis in STZ-induced T2DM mice (Fig. 1). Additionally, LEO
significantly decreased the levels of BUN, serum creatinine (SCr) and urinary albumin (ALB) compared to
control STZ-induced mice. In particular, LEO alleviateed glomerulosclerosis and
renal tubular atrophy. LEO was also observed to suppress the in vitro
and in vivo expression of the fibrosis-related factor TGF-
Ferroptosis was recently implicated in the progression and therapeutic
response of DN. Previous studies reported that increased levels of Fe
Previous studies have reported the physiological functions of ECs in DN, with
VE-cadherin and CD31 being well-known markers of EC dysfunction [31, 32]. In DN,
the downregulation of VE-cadherin and CD31 in the glomeruli is related to
endothelial dysfunction [33, 34]. The current study found that LEO increased the
expression of GPX4, FTL, and FTH in CD31-positive cells (Fig. 2), indicating that
LEO-mediated ferroptosis might affect EC activity and function in the kidney
during the initiation and development of DN. The viability of HUVECs was impaired
by glucose concentrations as low as 30 mM, but this was attenuated by LEO. Under
normal conditions, ECs form a barrier to prevent leukocytes such as monocytes
from infiltrating the vascular wall and stopping the entry of glucose [4].
However, endothelial dysfunction can generate biomolecules such as inflammatory
factors that contribute to the development of DN. GPX4 can reduce ferroptosis in
DN through the SIRT3 pathway [35]. The present results showed that siGPX4 reduced
CD31 and VE-cadherin expression in HG-treated HUVECs, but this was reversed by
LEO. We also assessed the effects of LEO-mediated ferroptosis on EC dysfunction
using the scratch test and siGPX4 (Fig. 5). During the development of DN,
endothelial dysfunction of ECs can be triggered by oxidized lipids and ROS,
leading to increased expression of several inflammatory factors, such as IL-6,
TNF-
In conclusion, the present results from STZ-induced mice showed that ferroptosis occurs in DN, and that inhibition of ferroptosis by LEO can protect against lipid peroxidation and further aggravation of DN. In addition, the inhibition of ferroptosis by LEO suppressed HG-induced lipid peroxidation and endothelial dysfunction in HUVECs in vitro. GPX4-mediated inhibition of ferroptosis by LEO can therefore attenuate DN by suppressing lipid peroxidation and endothelial dysfunction in HUVECs. Moreover, LEO was found to regulate Nrf2, thereby activating GPX4 to protect against endothelial dysfunction in HUVECs. These results contribute further to our understanding of DN pathogenesis and suggest a therapeutic target for this condition.
All data points generated or analyzed during this study are included in this article and there are no further underlying data necessary to reproduce the results.
XY, KY and YL conducted the experiments. XY, XC and YZ analyzed the data. XY and XZ designed the experiments and wrote the paper. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be ac-countable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
Institutional Animal Care and Use Committee of Guilin Medical University (Approved No. GLMC202105049).
Not applicable.
This work was supported by the grants from the Natural Science Foundation of Guangxi Zhuang Autonomous Region Nos. GuikeAD20238024 and Nos. 2021GXNSFBA196042).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.