
 , Shuai Wang 2,*
, Shuai Wang 2,*1 Medical School, Nantong University, 226000 Nantong, Jiangsu, China
2 Department of Cardiology, Affiliated Hospital of Nantong University, 226000 Nantong, Jiangsu, China
†These authors contributed equally.
Abstract
Background: Heart failure (HF) is a clinical syndrome that seriously
endangers human health and quality of life as the terminal stage of
cardiovascular diseases. Ferroptosis as a new iron-dependent programmed cell
death mode that is closely related to the occurrence and development of
cardiovascular diseases. Dihydroorotate dehydrogenase (DHODH) has been found to
play a crucial role in inhibiting ferroptosis and improving mitochondrial
function, and its expression can be upregulated by estradiol (E
Graphical Abstract
Keywords
- heart failure
- ferroptosis
- dihydroorotate dehydrogenase
- estradiol
- CoQ
The latest Global Burden of Cardiovascular Disease report indicates that the number of deaths from cardiovascular disease has increased to 19.8 million in 2022 [1], revealing the impact of the current global population growth and aging trends on cardiovascular disease. Heart failure (HF) is the clinical end-stage of most forms of primary cardiovascular disease (CVD), and it is a leading global cause of patient morbidity and mortality. To facilitate the design of more effective targeted cardioprotective interventions, there is a clear need to further explore the pathological mechanisms that underlie cardiomyocyte injury in HF. HF patients tend to present with a range of serious symptoms, high rates of hospitalization and mortality, and a poor overall prognosis. While evidence-based treatment implementation can help protect against mortality among HF patients, rates of patient death remain very high. In patients with stable HF, the annual mortality rate is approximately 6–7%, while these rates can climb to 25% or higher in hospitalized patients with acute HF [2, 3, 4].
Cardiomyocytes are terminally differentiated cells, and they are thus unable to undergo further division or proliferation. Cardiomyocyte death thus inevitably results in a decline in the overall number of these cells, compromising cardiac structural and functional integrity in a manner that can aggravate HF development and progression. There is thus clear potential clinical significance to research focused on the pathways that regulate cardiomyocyte death, with the goal of designing new strategies to inhibit cardiomyocyte death and prevent CVD incidence by preserving overall heart health. The suppression of cardiomyocyte death offers a promising means of preserving heart function and thereby effectively preventing heart disease.
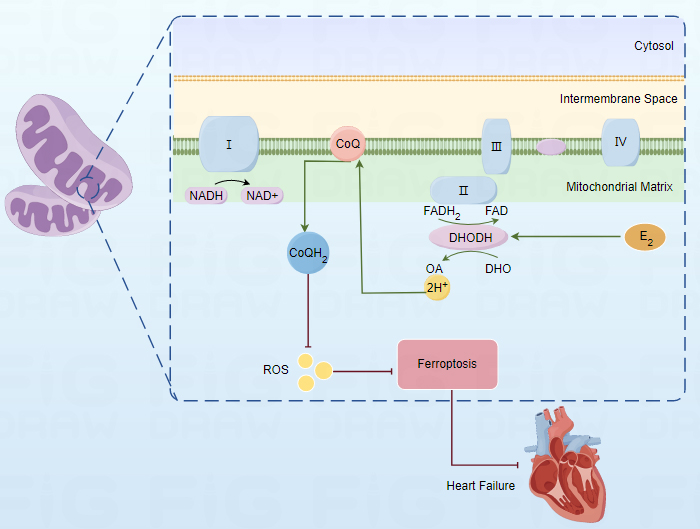
Dihydroorotate dehydrogenase (DHODH) is an iron-containing enzyme that plays an
essential role in the process of ab initio pyrimidine synthesis. DHODH functions
by catalyzing a redox reaction that processes dihydrooxalate into oxalate, which
is then further converted into uridine monophosphate that is used to generate RNA
nucleotides, supporting ribosomal biosynthesis. As it plays key roles in
pyrimidine synthesis and the functionality of mitochondria, inhibitors of DHODH
were first employed for the treatment of rheumatoid arthritis and multiple
sclerosis. DHODH can inhibit mitochondrial inner membrane-related ferroptotic
induction via reducing pan-quinone coenzyme Q (CoQ) to pan-alcoholic CoQH
Ferroptosis was first proposed as a mode of cell death in 2012 by Dixon et al. [15]. This unique cell death pathway is regulated by phospholipid hydroperoxidase (GPX4) signaling pathway and driven through the iron-dependent peroxidation of phospholipids and is subject to regulation by various pathways including mitochondrial activity, iron metabolism, redox homeostasis, and sugar, lipid, and amino acid metabolism pathways. A range of disease-related signaling pathways also regulate ferroptotic activity, which is distinct from other modes of cell death including necrosis, apoptosis, autophagy, and pyroptosis. Ferroptosis primarily occurs through a mechanism related to the dysregulation of iron metabolism, the disruption of amino acid antioxidant systems, and the aggregation of lipid peroxides. The initial disruption of normal homeostatic iron metabolism can result in a rise in the levels of free iron within cells, and divalent iron can then catalyze reactive oxygen species (ROS) generation via the Fenton reaction, further promoting lipid peroxidation such that these peroxides aggregate and trigger ferroptotic death [16]. Ferroptotic cells are characterized by morphological features including reduced mitochondrial volume, increased membrane density, damage to the mitochondrial outer membrane, and the reduction or absence of cristae [17], without any corresponding nuclear changes, chromatin condensation, or destruction of the cell membrane. Iron homeostasis is vital to normal mammalian cell function, particularly for the maintenance of cardiovascular health. HF is a leading cause of death associated with hemochromatosis and thalassemia [18]. Iron deficiency is also a common complication observed in HF patients, serving as a risk factor that is related to poor prognostic outcomes and independent of anemia complications. Under conditions of iron deficiency, myocardial iron content decreases, in turn limiting the oxygen transport capacity of patients and inducing erythropoietin resistance, with these pathological changes potentially culminating in HF. Ferritin serves as a vital mediator of cardiac iron homeostasis and normal cardiac function [19], with ferritin deficiency within myocardial cells contributing to elevated levels of ROS biogenesis. This can give rise to cardiac damage and contribute to greater susceptibility to iron overload-related ferroptosis and cardiomyopathy.
One research team identified DHODH as a ferroptosis-related factor that is
independent of the classical GPX4 signaling pathway [20]. DHODH inactivation can
trigger the extensive peroxidation of mitochondrial lipids and drive ferroptotic
induction in cancer cells expressing low levels of GPX4 while also synergizing
with other ferroptotic inducers to drive these effects in cancer cells expressing
high levels of GPX4. Mechanistically, DHODH thus functions in parallel with
mitochondrial GPX4 while remaining independent of cytoplasmic GPX4 or ferroptosis suppressor protein 1 (FSP1),
inhibiting mitochondrial inner membrane-related ferroptotic activity via reducing
panurea to pan-alcohol, which is an antioxidant with anti-ferroptosis activity.
E
Specific pathogen-free healthy male C57BL/6 mice (8 weeks old, 20–25 g) were
obtained from the Laboratory Animal Center of Nantong University (Nantong,
China). The Animal Ethics Committee of Nantong University approved all animal
studies, which were consistent with the Guide for the Care and Use of Laboratory
Animals. The animal model mice were raised in a standard laboratory environment
that consisted of a 12-hour light/dark cycle, with a constant temperature of 25
°C. The mice had free access to animal feed and tap water. Mice were
randomized into Sham+sesame oil (as a control for E
Information on the materials used in this study can be found in the Supplementary Material.
Measurements of the left ventricular end-diastolic diameter (LVIDd), left ventricular end-systolic diameter (LVIDs), left ventricular end-diastolic interventricular septum thickness (IVSd), and other indices were made along the long axis and short axis sections of the left ventricle. M-mode ultrasound measurements were processed to calculate indicators that reflect left ventricular systolic function, including left ventricular ejection fraction (LVEF) and fractional shortening (FS). Cardiac functional parameters were measured based on the average values from 3–5 cardiac cycles.
Cardiac tissues were fixed for 24 h using 4% paraformaldehyde, dehydrated with an ethanol gradient (75%, 85%, 95%, 95%, 100%, 100%), treated with xylene, immersed for 3 h in pre-dissolved paraffin, and placed in an embedding box and frozen at –20 °C. A microtome was then used to cut 5 µm sections that were stored at room temperature. For analysis, these sections were immersed twice in xylene (10 min each), rehydrated with an ethanol gradient (100%, 100%, 95%, 85%, and 75%; 5 min each), washed for 5 min with phosphate buffered saline (PBS), stained with hematoxylin, washed for 5 min with distilled water, and immersed in 1% HCl for 30 min for differentiation. Then, samples were incubated for 30 min in 0.1% ammonia in water, rinsed, immersed for 5 min in eosin staining solution, dehydrated using 90%, 95%, and 100% ethanol for 20 s each, incubated twice for 10 min in xylene, sealed using glue, allowed to air dry, and analyzed via microscopy.
Place the sections in the following order in xylene I for 20 minutes, xylene II for 20 minutes, absolute ethanol I for 10 minutes, absolute ethanol II for 10 minutes, 95% alcohol for 5 minutes, 90% alcohol for 5 minutes, 80% alcohol for 5 minutes, 70% alcohol for 5 minutes, and then wash with distilled water. Stain the cell nuclei with hematoxylin: immerse the sections in Weigert’s hematoxylin stain from the Masson staining kit for 5 minutes, wash with tap water, differentiate with 1% hydrochloric acid alcohol for a few seconds, wash with tap water, and rinse with running water for several minutes to return blue. Stain with eosin and phloxine for 5–10 minutes using the Masson staining kit, and quickly wash with distilled water. Treat with phosphomolybdic acid: immerse the sections in the phosphomolybdic acid solution from the Masson staining kit for about 3–5 minutes. Do not wash with water, and directly stain with Fast Blue solution from the Masson staining kit for 5 minutes. Differentiate: treat with 1% acetic acid for 1 minute. Dehydrate and clear the sections by immersing them in 95% alcohol I for 5 minutes, 95% alcohol II for 5 minutes, absolute ethanol I for 5 minutes, and absolute ethanol II for 5 minutes, and then in xylene I and xylene II for 5 minutes. Remove the sections from xylene and allow them to air-dry slightly. Mount the sections with neutral resin and cover them with a cover slip. Examine the sections under a microscope and capture and analyze the images.
Hand-held suckling mice (1–2 days old), scrub the chest and abdomen with 75% alcohol, then use an ophthalmic scissor to cut open the sternum and extrude the heart, put it into a PBS dish and wash it repeatedly, then transfer it into a small glass bottle and fully cut it into a pulpy shape. The cut heart was transferred into a blue-covered wide-mouth bottle containing 10 mL collagenase, gently blow the tissue fragments, and place them into a constant temperature oscillator at 37 °C for 5 minutes. The cell suspension after oscillation was transferred to a 15 mL centrifuge tube, centrifuged at 1000 rpm for 5 minutes, and the rest of the heart tissue was added with 10 mL collagenase, gently blown, and then placed into a constant temperature oscillator at 37 °C for 5 minutes. After centrifugation, 2 mL of complete culture was added to the cells, and the cells were blown evenly and left standing. The suspension after centrifugation was digested again, and the cycle was repeated for 7 times in total to make the tissue completely digested. All the cells collected above were collected and transferred into a new centrifuge tube, and then centrifuged again, and the supernatant was discarded. Then add an appropriate amount of complete medium to the centrifuge tube and gently shake to re-suspend the cells. After filtration with a cell sieve, transfer the cells to a Petri dish. Add Brdu (5-bromo-2-deoxyuridine) to the medium and place it in an incubator for 1.5 hours at a differential speed. The cells that are adherent to the wall are fibroblasts, and the cells that are not adherent to the wall are cardiomyocytes. Take the supernatant and inoculate it in a Petri dish. Change to serum-free medium after 48 hours, and starve for 24 hours before drug addition and transfection.
All cell lines were validated by short tandem repeat
(STR) profiling and tested negative for mycoplasma. Retrieve the cryovial labeled with H9C2 cells and thaw it rapidly in a 37
°C water bath. Cells were all cultured in a humidified incubator at 37
The cells were inoculated into 96-well plates at a concentration of 5
Negative control siRNA (si-NC) and si-CoQ were synthesized by GenePharma (Suzhou, China). Use Lipo3000 transfection reagent to transfect cells according to the manufacturer’s scheme. Add an appropriate amount of Lipo3000 transfection reagent to a certain amount of Opti-Minimal essential medium (Opti-MEM) medium and mix it thoroughly; then use a certain amount of serum-free DMEM and an appropriate amount of si-NC or si-CoQ transfection reagent to mix it thoroughly; shake and mix the two dilutions that have been configured at a ratio of 1:1, and place them at room temperature for 20 min. Add the above mixture to the well plate containing cells one by one with a pipette gun, and then place it in the cell incubator for incubation. After 6 hours, replace it with fresh culture, and then culture for 24–48 hours.
Total RNA was extracted from the above-mentioned mouse heart tissues and cells. The reverse transcription reaction system was prepared by using the reverse transcription kit (G3337), gently mixed and centrifuged, and the reverse transcription program was set to complete the reverse transcription on the PCR instrument. Take 0.1 mL PCR reaction plate, prepare the following reaction system, and prepare 3 tubes for each reverse transcription product. After the sample was added, the PCR sealing plate membrane was used to complete the sealing with the sealing plate instrument, and the microwell plate centrifuge was used to complete the centrifugation. Then the amplification was completed on the fluorescence quantitative PCR instrument.
Total protein was extracted from mouse heart tissues or H9C2 cells. The
bicinchoninic acid (BCA) protein detection kit (ThermoFisher, MI, USA) was used
to measure the protein content. According to the measured protein concentration,
5
Cell samples were homogenized or lysed using PBS or lysis buffer, and the supernatant was collected by centrifugation. Thiobarbituric acid (TBA) storage solution and MDA detection working solution were prepared and dissolved by heating. For the measurement, add the blank control, standard, or sample to the centrifuge tube, then add the MDA detection working solution. Mix well and heat for 15 minutes. Cool to room temperature, centrifuge, and collect the supernatant. Transfer the supernatant to a 96-well plate and measure the absorbance at 532 nm using a microplate reader. Calculate the molar concentration of MDA based on the standard curve.
In accordance with the standard process, the cells were collected and cleavage,
and then the BCA protein analysis kit was used for the accurate determination of
protein concentration. At the same time, the ferrous ion colorimetric test kit
was used for the scientific analysis of ferrous ion content: the probe was loaded
with ferrous ions in the cells, and the optical density value (OD value) of the
generated substance at the strong absorption peak of 593 nm wavelength was
determined, and the corresponding calculation was performed, so as to determine
the content of ferrous ions (Fe
Fresh cell model was measured according to the standard procedure. The cells were washed twice with PBS and centrifuged. The supernatant was discarded, and protein removal reagent was added, followed by full eddy current oscillation. The samples were rapidly frozen-thawed twice with liquid nitrogen and 37 °C water bath, and then placed in a 4 °C refrigerator or ice bath for 5 minutes, and then centrifuged for 10 minutes at 4 °C. The supernatant was used for the determination of GSH. The absorbance was measured at 405 nm with an Enzyme-Linked Tmmunosorbent Assay (ELISA) kit, and compared with the standard curve.
Place the cells in a PBS solution containing 3% to 4% formaldehyde at room temperature for 10 to 30 minutes to fix them. Then, suck out the fixative solution and wash the cells with PBS 2 to 3 times. To quench excess formaldehyde, place the cells in a PBS solution containing 10 mM ethanolamine (or 0.1M glycine) for 5 minutes. Next, add a PBS solution containing 0.1% Triton X-100 to the fixed cells, and incubate them for 3–5 minutes to enhance their permeability. Then, wash the cells with PBS 2–3 times again. Add DAPI solution to the cell culture medium and incubate the cells at 37 °C for about 5 minutes. Wash the cells with PBS 2–3 times again. Then, add a sealing agent to preserve the fluorescence (if using a cover slip, seal the cells with a cover slip). Finally, observe the cells under the excitation/emission wavelength of 493/517 nm.
If the data follows a normal distribution, ANOVA (Analysis of Variance) can be
used, which is a method to compare the differences in means between two or more
groups assuming that the data comes from a normal distribution. If the data does
not follow a normal distribution, Kruskal-Wallis test should be chosen. This is a
non-parametric method that does not depend on the distribution assumption of the
data and is mainly used to compare whether the median of three or more
independent samples is equal. Cell culture and animal experiments were
independently performed at least three times. Data are reported as means
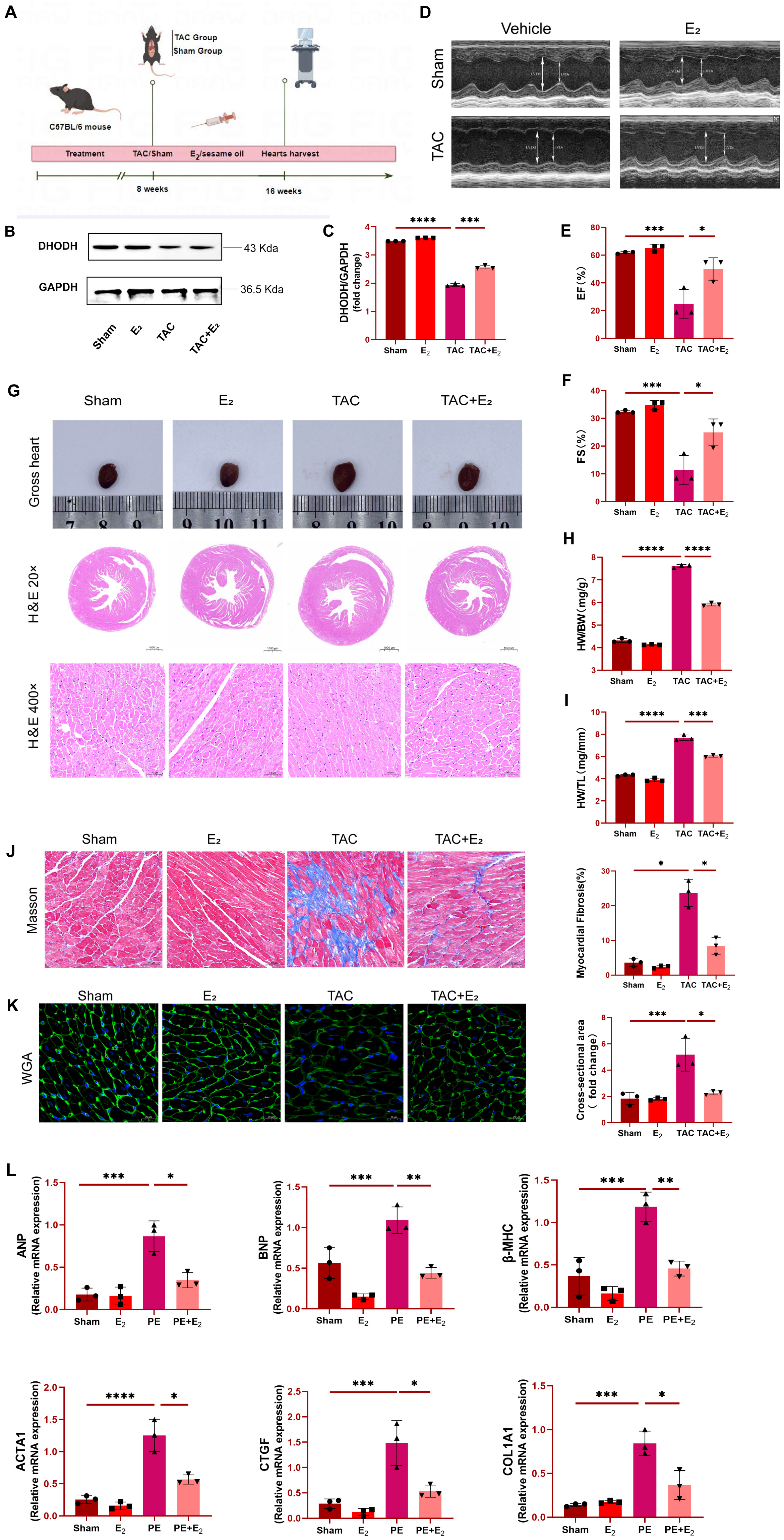
C57BL/6 mice were initially subjected to TAC surgery in order to generate a
murine model of HF (Fig. 1A), after which animals in the experimental group were
injected subcutaneously with E
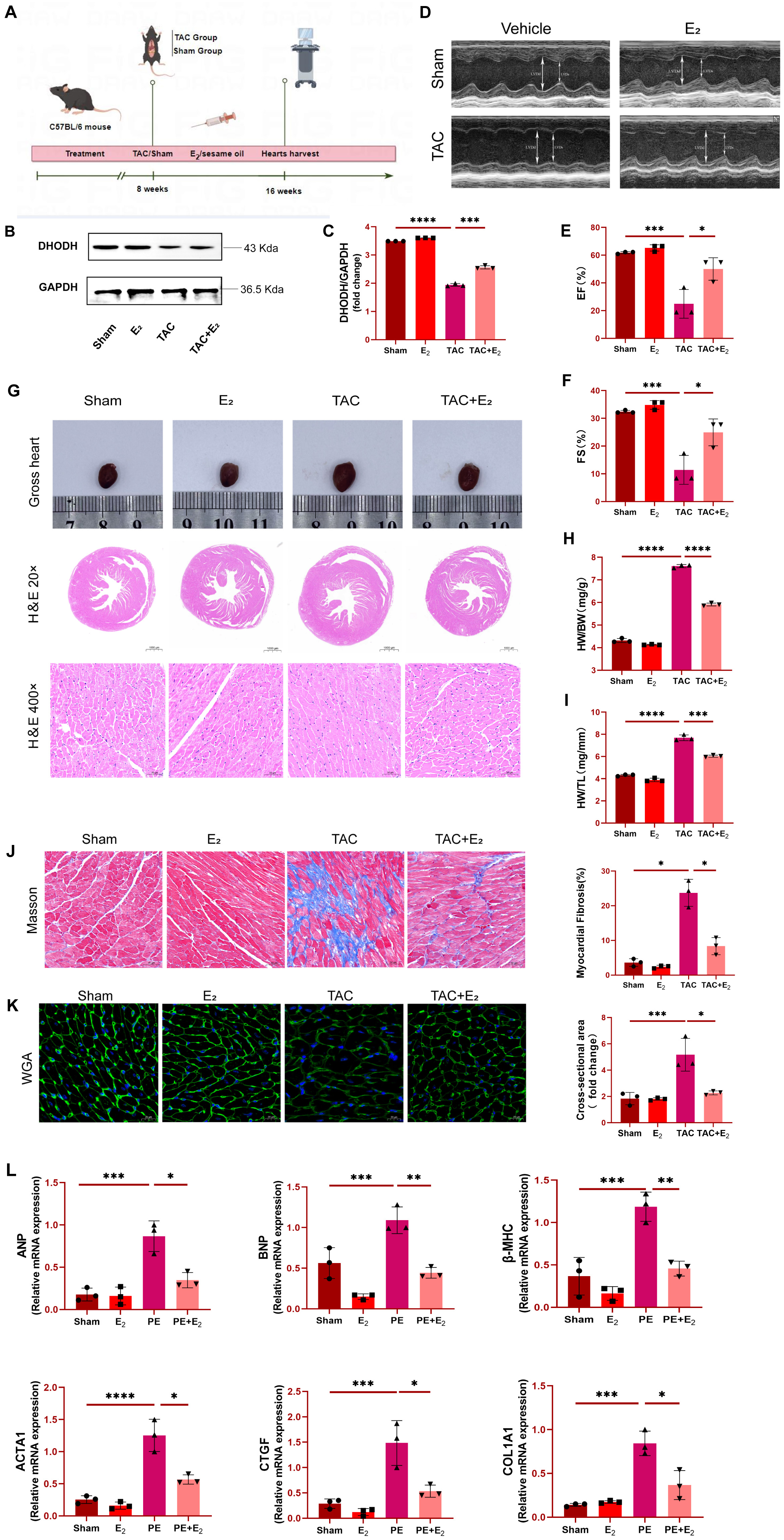 Fig. 1.
Fig. 1.Estradiol (E
As the above results support the successful establishment of the murine TAC
model system, analyses of iron levels in murine cardiomyocytes were next
conducted. A significant increase in these iron levels was observed in mice from
the TAC group, whereas iron concentrations in mice from the TAC+E
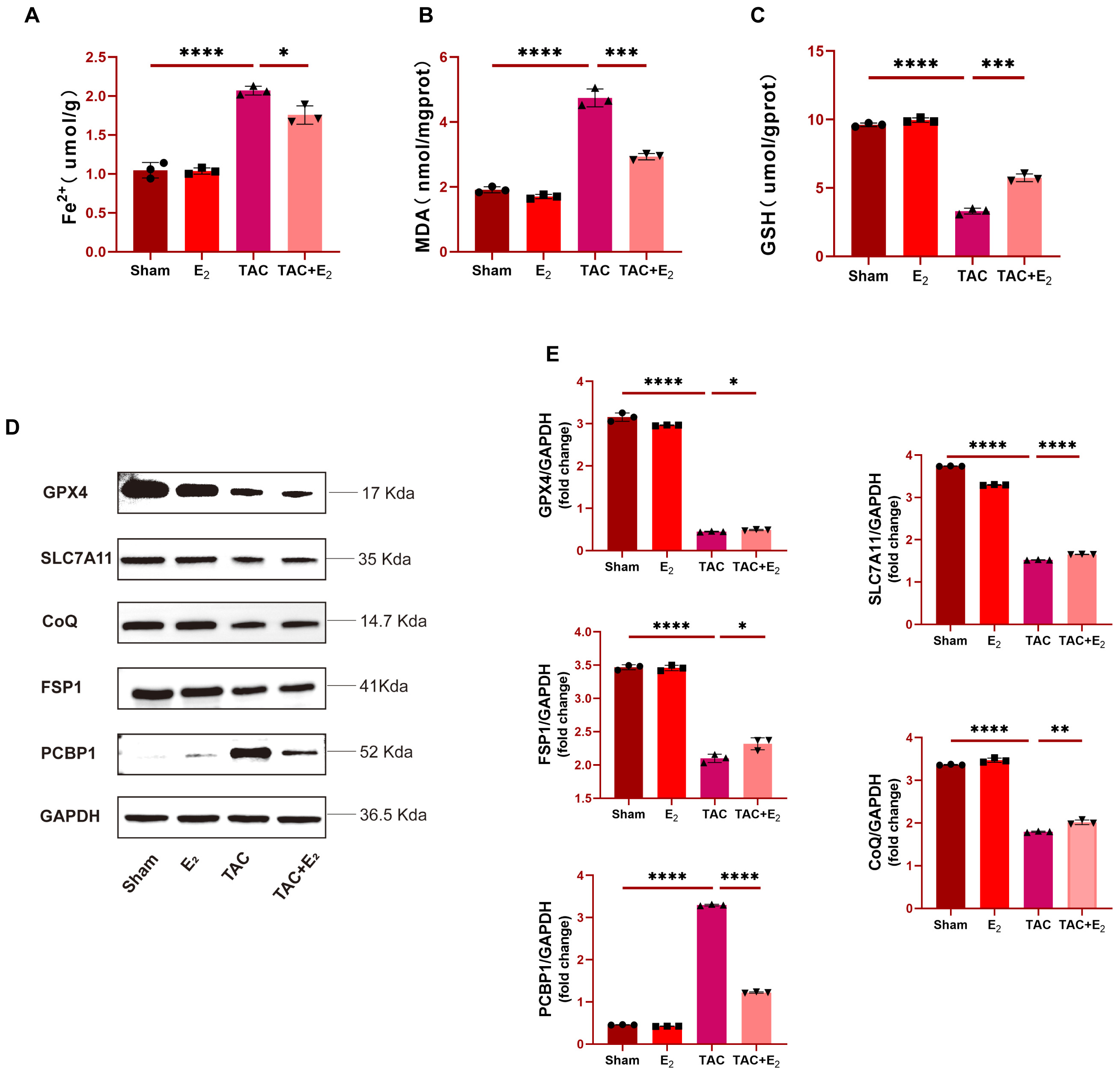
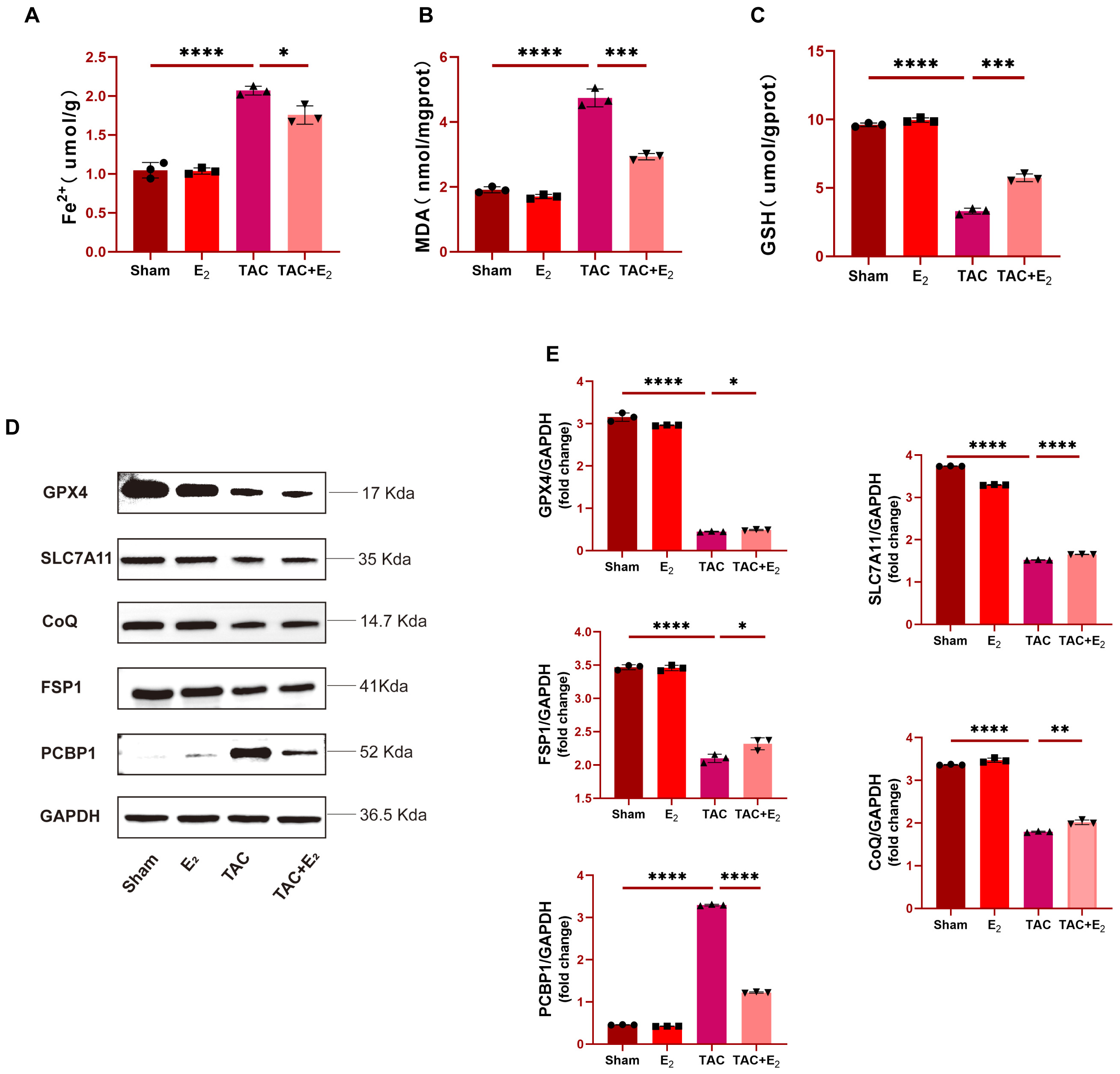 Fig. 2.
Fig. 2.E
Western blotting was next used to analyze the expression of
ferroptosis-associated factors in tissue samples from these mice including DHODH,
GPX4, SLC7A11, FSP1, CoQ and PCBP1, with the goal of further confirming the
anti-ferroptotic effects of E
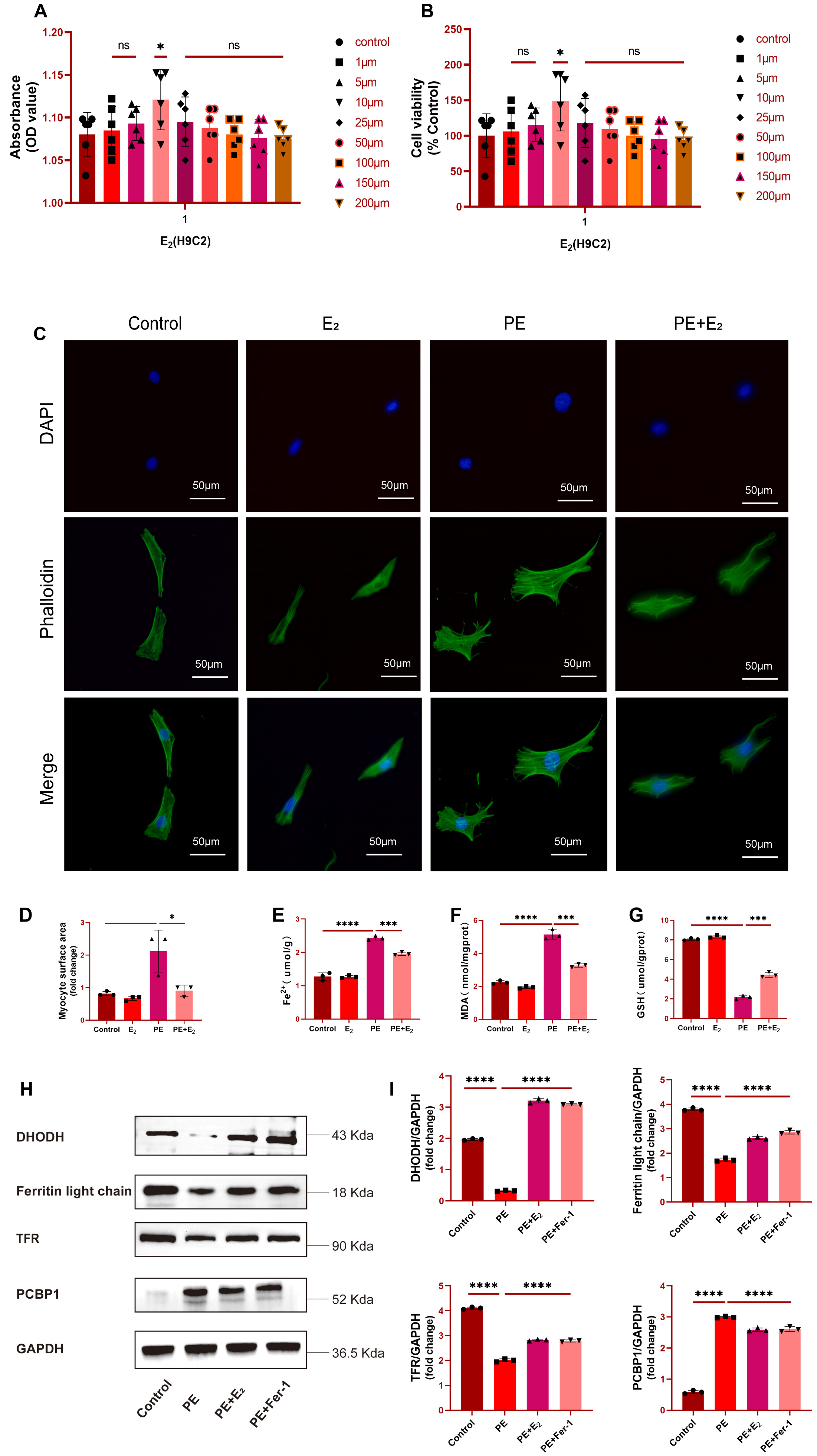
To probe the mechanistic role of DHODH in cardiomyocytes, a series of in
vitro experiments was next conducted. Initially, H9C2 cardiomyocytes were
treated for 24 h with a range of E
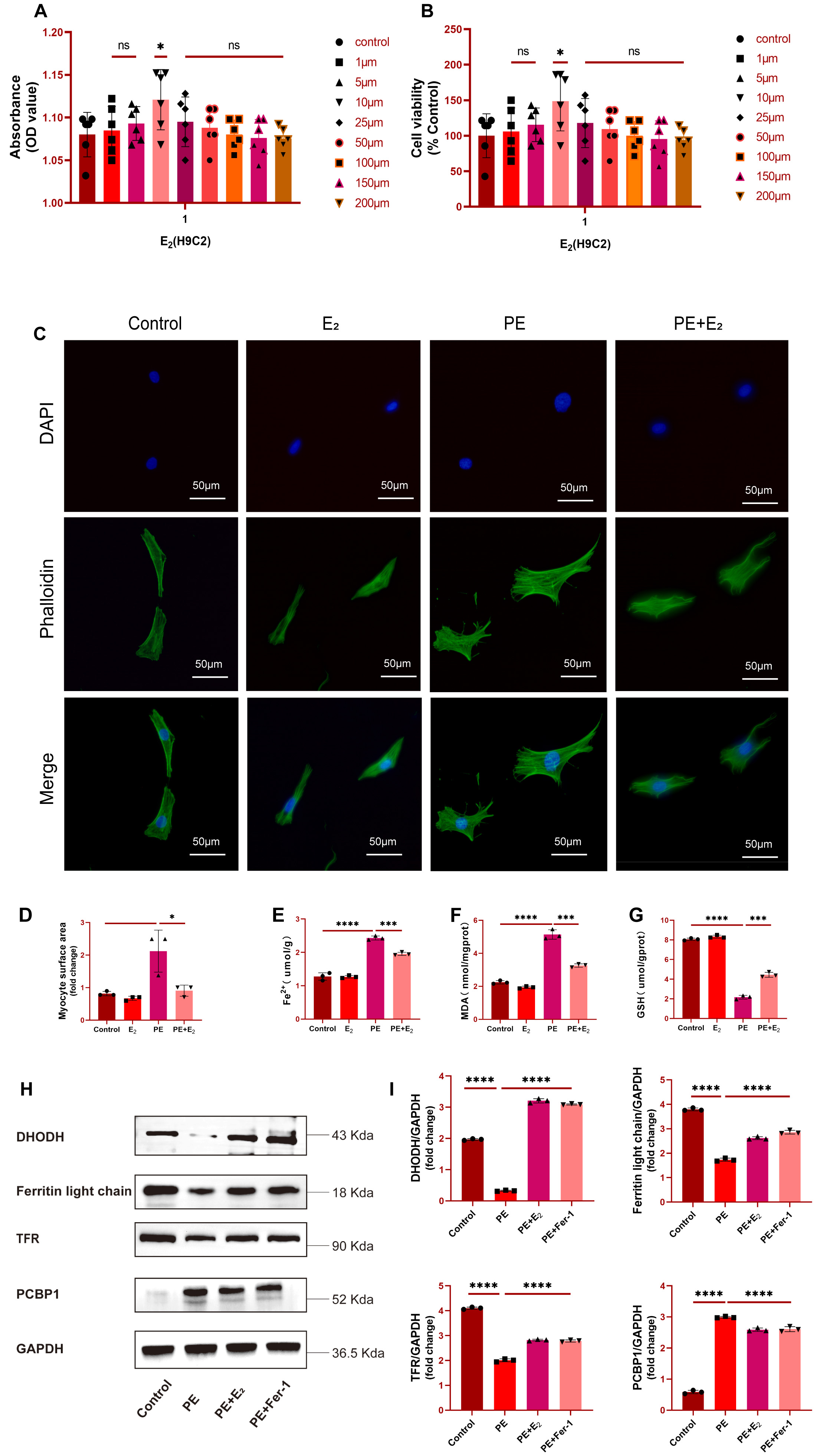 Fig. 3.
Fig. 3.DHODH upregulation protects against phenylephrine (PE)-induced cardiomyocytes
with failure. (A,B) Effects of different concentrations of E
The impact of E
Further analyses of Fe
Subsequently, a ferroptosis-specific inhibitor Ferrostatin-1 (Fer-1) was added
as a positive control. Analysis of protein levels of DHODH, Ferritin light chain
and transferrin receptor (TFR), which are downregulated in ferroptosis, and
ferroptosis marker PCBP1 further showed that these three proteins were
significantly upregulated in PE-induced primary cardiomyocytes with failure, but
returned to baseline levels after E
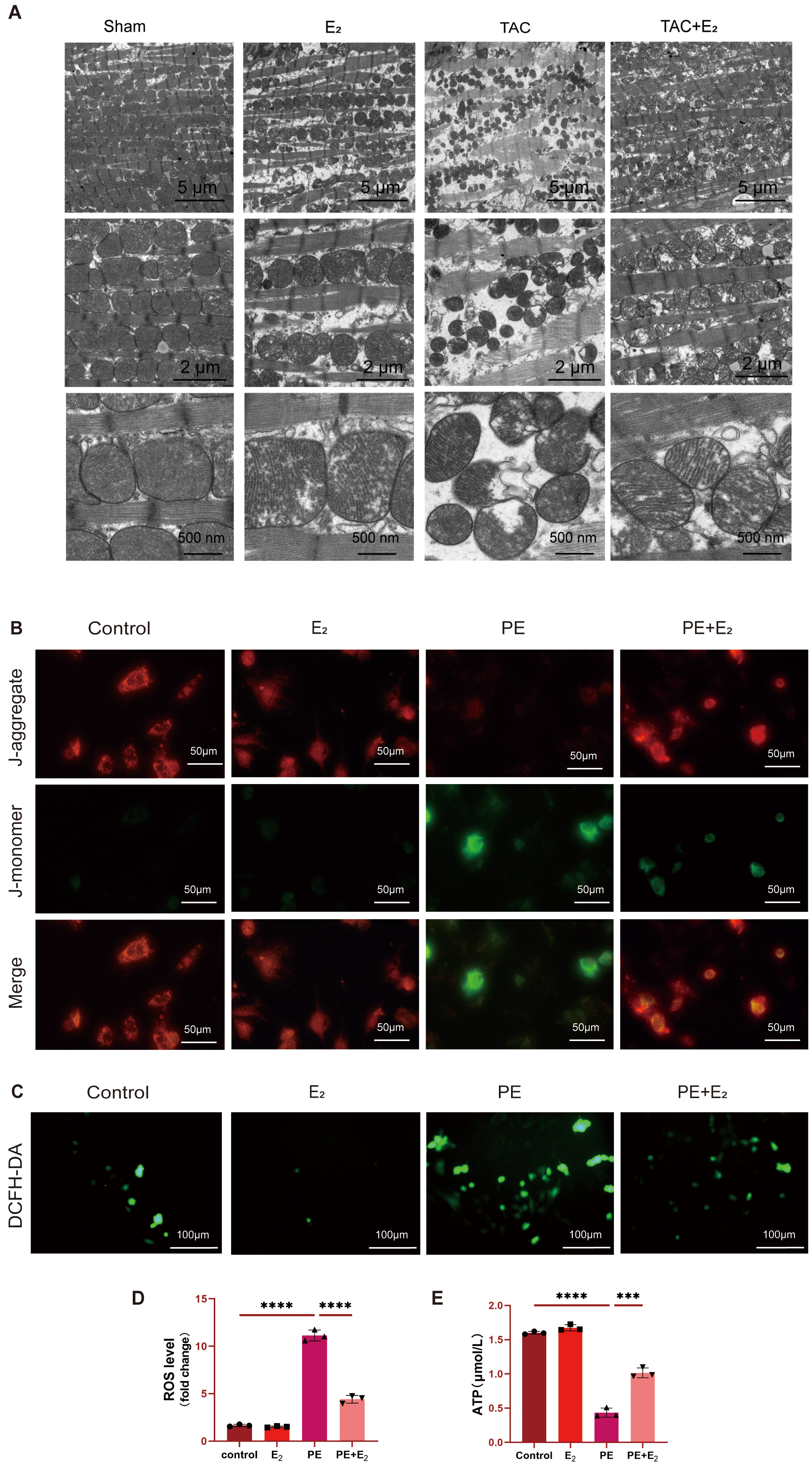
To further interrogate the effects that DHODH has on mitochondria, transmission
electron microscopy was employed to assess mitochondrial changes in the
cardiomyocytes of TAC model mice. These analyses revealed significantly reduced
mitochondria in TAC model mice, with the remaining mitochondria exhibiting
disorderly arrangement, outer membrane rupture, and the partial or total loss of
cristae. In contrast, TAC+E
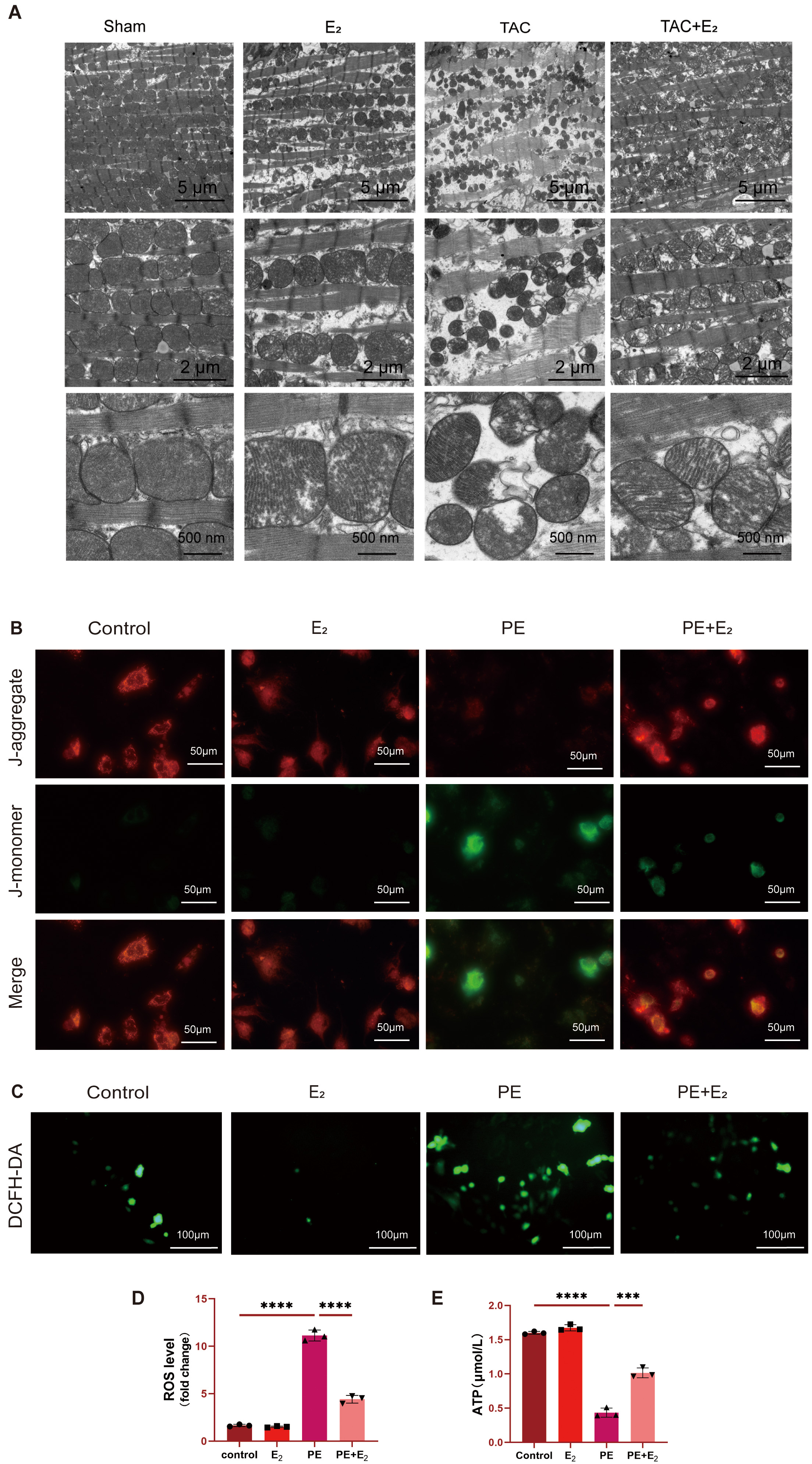 Fig. 4.
Fig. 4.DHODH protects against PE-induced ferroptosis in cardiomyocytes
via enhancing mitochondrial function. (A) Representative image of mitochondrial
ultrastructure morphology in mouse cardiomyocytes. Scale bar: 5 µm,
2 µm, 500 nm. (B) Representative JC-1 green/red fluorescence image. Scale
bar: 50 µm. (C,D) Detection of intracellular reactive oxygen species (ROS) levels in H9C2 cells using
DCFH-DA fluorescence probe. Scale bar: 100 µm. (E) Detection of ATP
concentration in H9C2 cells using mouse adenosine triphosphate (ATP) ELISA kit.
Data are shown as mean
The impact of E
To explore the in vitro inhibition of ferroptotic cell death by
E
Mitochondrial function was analyzed by detecting ATP concentrations in H9C2
cells via ELISA. A significant decline in ATP levels was detected in PE-treated
H9C2 cells, while treatment with both PE and E
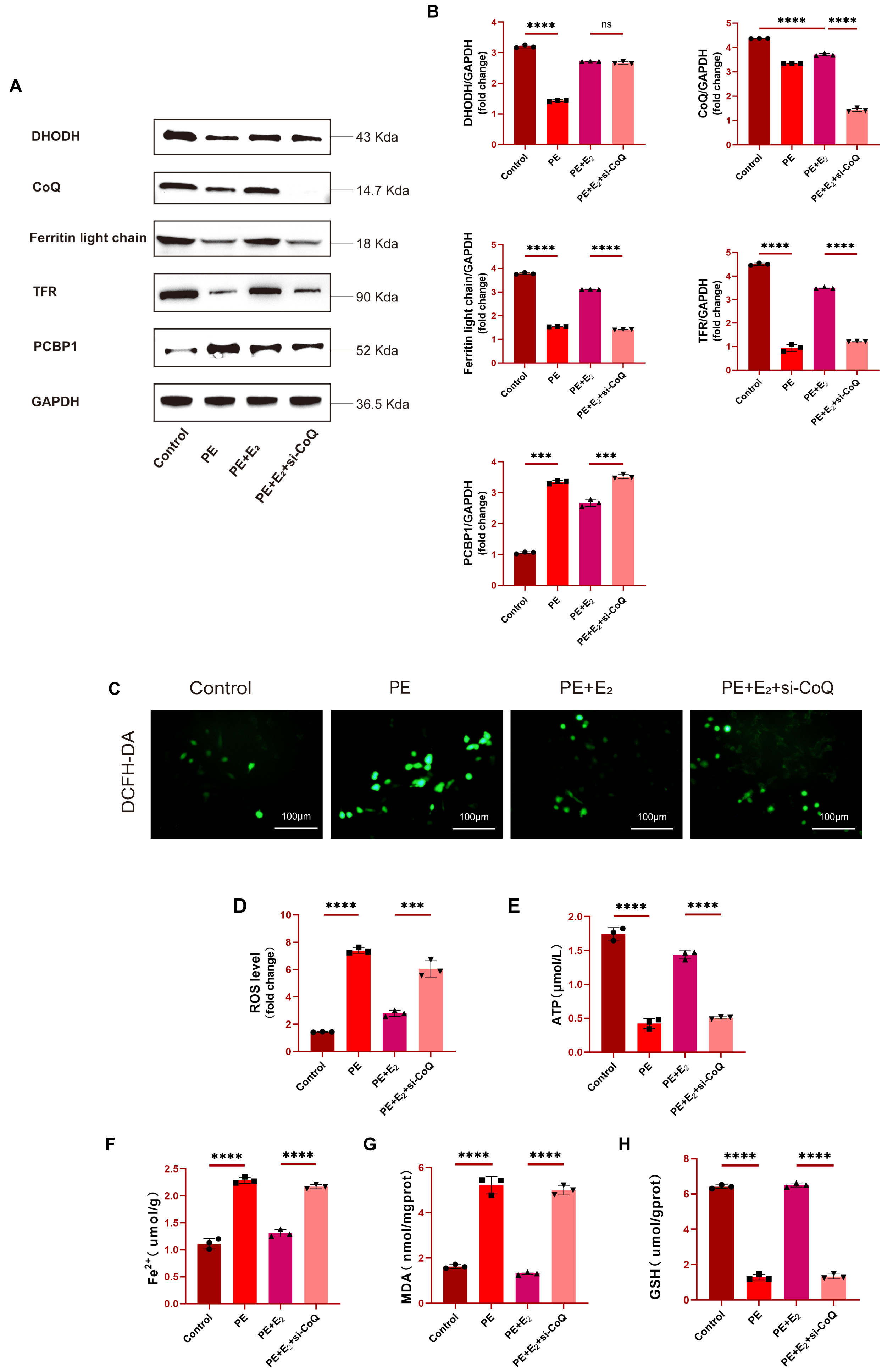
To confirm the functional role that CoQ plays as a regulator of the
anti-ferroptotic effects of DHODH, the knockdown of CoQ in H9C2 cells was
performed prior to PE treatment, with Western blotting being used to confirm
successful CoQ knockdown following si-CoQ transfection. Cells treated with PE
also exhibited lower levels of ferroptosis-related proteins including DHODH,
Ferritin light chain, and TFR as compared to control levels, with the expression
of these proteins being significantly elevated in the PE+E
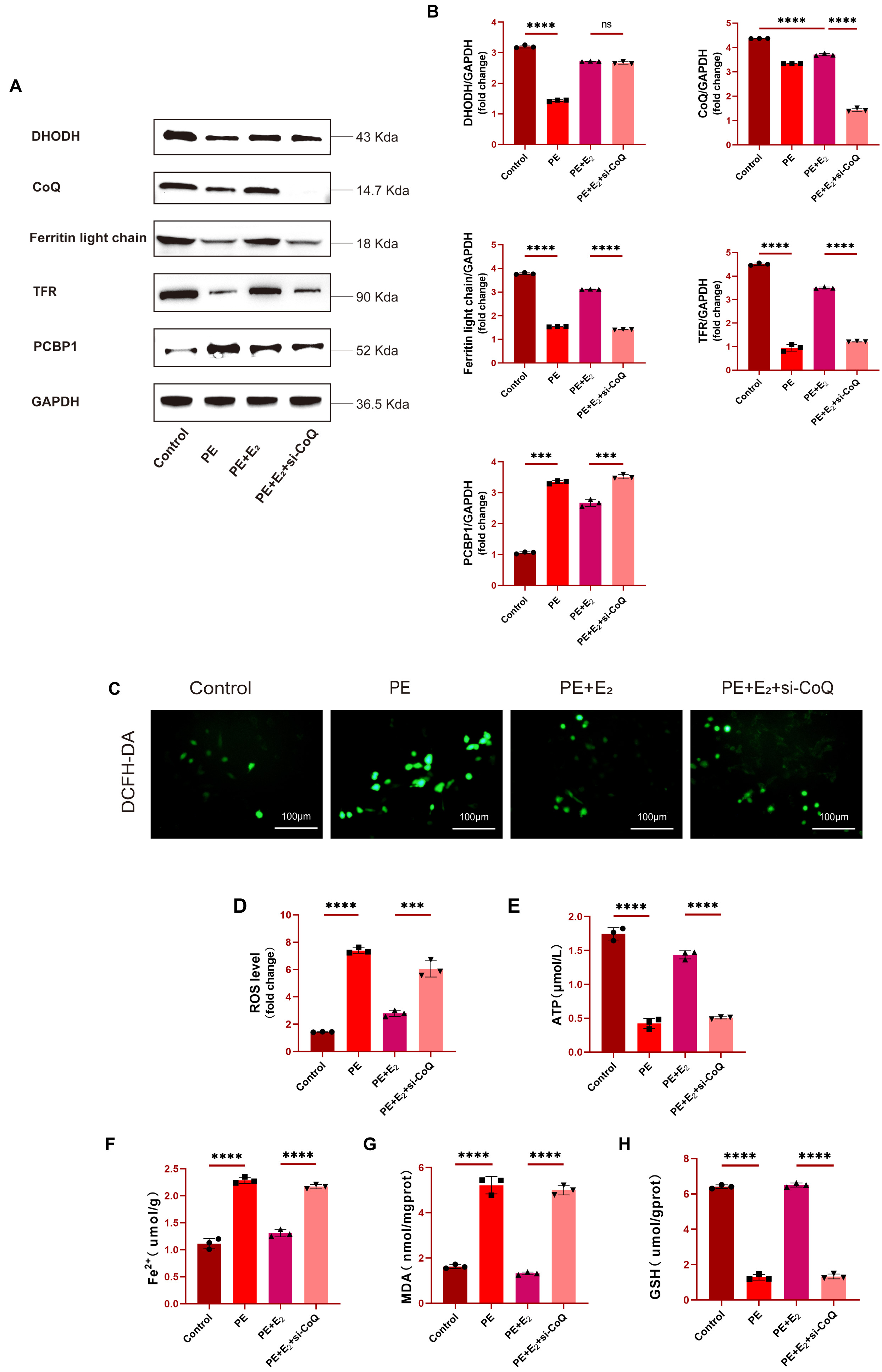 Fig. 5.
Fig. 5.DHODH regulates CoQ to exert cardioprotective effects and
prevent ferroptotic induction. (A,B) Western blotting and quantitative analysis
of DHODH, CoQ, Ferritin light chain and TFR expression in H9C2 cells. (C,D)
Determination of ROS levels in H9C2 cells using DCFH-DA fluorescent probe. Scale bar: 100 μm. (E)
Detection of ATP concentration in H9C2 cells using mouse adenosine triphosphate
(ATP) ELISA kit. (F–H) Determination of Fe
Fe
DHODH is a rate-limiting enzyme located in the mitochondria, responsible for de
novo synthesis of pyrimidine nucleotides, which can associate pyrimidine
nucleotide synthesis with the electron transport chain (Electron transport chain,
ETC) of mitochondrial complex III through CoQ, playing an important role in
cellular energy metabolism [24]. A recent study has found that [5] inhibiting
DHODH gene expression helps promote ferroptosis in tumor cells, and
DHODH-related research has shown significant clinical significance in the
treatment of blood system diseases, liver tumors, and small cell lung cancer
[25, 26, 27]. However, the role of DHODH in cardiovascular diseases has not yet been
widely recognized. Some studies have shown that DHODH-specific inhibitor
Teriflunomide can exacerbate rat cardiac ischemia-reperfusion injury [28]. In
this study, we selected E
The mitochondria, as the cell’s energy center, are involved in various
physiological processes, including programmed cell death, apoptosis, autophagy,
metabolism, calcium flux, and innate immunity [32, 33, 34, 35]. Because of its high energy
demand, the heart is the organ with the most mitochondria [36]. In the
development of the heart, mitochondria play a crucial role, especially in the
development stage of the fetal heart, the differentiation process of cardiac
muscle cells, and maintaining the contractile function of the heart [37, 38, 39]. The
role of mitochondria is indispensable. Mitochondrial damage can break the
metabolic balance and further produce excessive ROS, thereby triggering more
severe cell damage and death [40]. A study has found that [41] mitochondrial
targeted therapy significantly reduces the cardiac damage induced by ferroptosis.
Therefore, maintaining mitochondrial homeosta is of vital importance to cells,
and these regulatory mechanisms are collectively referred to as mitochondrial
quality control (MQC) [42]. Studies have shown that [43] knocking down DHODH
increases the production of reactive oxygen species and lowers the mitochondrial
membrane potential. We have proven by transmission electron microscopy
observation of mitochondrial morphology in mouse cardiomyocytes, JC-1 staining to
detect the mitochondrial membrane potential of cardiomyocytes, ELISA to detect
ATP concentration, ROS fluorescence detection, and other methods that
upregulating DHODH can improve mitochondrial function in cardiomyocytes. Our
study also found that the expression of CoQ was significantly downregulated in
TAC-induced heart failure mouse models and PE-induced primary mouse cardiomyocyte
models. To further investigate the relationship between CoQ expression and
ferroptosis, we downregulated CoQ expression in H9C2 cells using si-CoQ. The
results showed that the downregulation of CoQ partially counteracted the benefits
brought by the upregulation of DHODH using E
There are certain limitations to this experiment. We will continuously improve and refine our experimental protocol to obtain more accurate results. We only evaluated mice 8 weeks after TAC surgery and did not compare with earlier time points, which is a limitation of this study. The sample size for animal experiments is inadequate, and it will be expanded in subsequent experiments. Hemodynamic measurements were not conducted in the various groups of mice in this study and will be addressed in future research. In addition, this study only validated the relationship between DHODH and CoQ, but did not explore the specific action pathways in more depth. This still needs to be further explored and studied in subsequent experiments.
This study found that dihydroorotate dehydrogenase (DHODH) plays a crucial role
in alleviating heart failure through a CoQ10 (CoQ)-related ferroptosis inhibition
mechanism. Key findings include: upregulation of DHODH significantly improves
myocardial hypertrophy and fibrosis caused by transverse aortic constriction
(TAC) surgery, while inhibiting cardiomyocyte ferroptosis and improving
mitochondrial function. Estradiol (E
The original data supporting the conclusions of this paper will be provided by the authors without reservation upon reasonable request.
CW, CC, JZ, QL and SW selected the topic, prepared the initial manuscript draft, searched the literature, generated all figures and revised the manuscript critically for important intellectual content. JS and HS contributed to collection and assembly of data. JL, PG and XW performed statistical analysis of all data. All authors contributed to editorial changes in the manuscript. The final manuscript has been approved by all corresponding author, who agreed to be accountable for all aspects of the work.
All experiments involving animals were approved by the Animal Ethics Committee of Nantong University (Permit Number: S20220310-010).
Not applicable.
This work was supported by the grants from the Multicenter Clinical Collaborative Research Project of Affiliated Hospital of Nantong University (No. LCYJ-A04), Affiliated Hospital of Nantong University Research Physician Development Fund (No. YJXYY202204-YSB59) and Postgraduate Research & Practice Innovation Program of Jiangsu Province in 2022 (No. SJCX22_1622).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.