


1 Department of Ophthalmology, The Second Clinical Medical College of Jinan University, 518020 Shenzhen, Guangdong, China
2 Department of Ophthalmology, Shenzhen People’s Hospital, 518020 Shenzhen, Guangdong, China
Abstract
Retinal degeneration (RD) is a group of chronic blinding diseases characterised by progressive retinal cell death. As the disease progresses, vision deteriorates due to retinal cell death and impaired retinal integrity, eventually leading to complete loss of vision. Therefore, the function and environmental homeostasis of the retina have an important impact on the pathogenesis and treatment of RD. Ubiquitination, as a complex post-translational modification process, plays an essential role in maintaining retinal homeostasis and normal function. It covalently combines ubiquitin with protein through a series of enzyme-mediated reactions, and participates in cell processes such as gene transcription, cell cycle process, DNA repair, apoptosis and immune response. At the same time, it plays a central role in protein degradation. There are two major protein degradation systems in eukaryotic cells: the ubiquitin-proteasome system and the autophagy-lysosomal system. The protein degradation pathway maintains retinal protein homeostasis by reducing abnormal protein accumulation in the retina through two modes of degradation. Either dysregulation of ubiquitination or disruption of protein homeostasis may lead to the development of RD. This article aims to comprehensively review recent research progress on ubiquitin-related genes, proteins and protein homeostasis in the pathogenesis of RD, and to summarize the potential targeted therapy strategies for it. The review is expected to provide valuable guidance for further development and application of ubiquitination in RD.
Keywords
- ubiquitination
- ubiquitin-proteasome system
- autophagy
- retinal degeneration
The human visual system is one of the most crucial sensory systems. Deterioration in the quality of vision can have a markedly negative impact on human being, including quality of life, an increase in the number of people with disabilities, the occurrence of accidents, social isolation, and psychological well-being [1]. Visual impairment and loss have become a rapidly growing public health concern across the world. As of 2019, at least 2.2 billion people suffer from visual impairment, and at least 1 billion of them have untreated or preventable visual impairment. Among these 1 billion people, the main diseases that cause distance vision impairment or blindness include cataracts, ametropia, age-related macular degeneration (AMD), glaucoma and diabetic retinopathy (DR), and the number is expected to rise [2]. Several of the diseases are retinal degeneration (RD). RD is a group of chronic blinding diseases characterised by the progressive death of nerve cells such as photoreceptor cells, retinal ganglion cells (RGCs) and (or) retinal pigment epithelial cells (RPE). As the disease progresses, vision deteriorates due to retinal cell death and impaired retinal integrity, eventually lead to complete loss of vision [3]. Depending on the pathogenesis, RD can be divided into two categories: inherited retinal degenerative diseases (IRDs) and complex retinal degeneration. Common types of RD include AMD, DR, retinitis pigmentosa (RP) and Stargardt disease (STGD) [4]. RD is one of the major blinding diseases in the world, and the treatment of RD is also the focus of blindness prevention at this stage. As a result, a lot of research has been done on the pathogenesis and treatment of this disease.
There are many factors that contribute to the development of RD. Aging, oxidative stress, protein aggregation and inflammation are crucial factors in the occurrence of AMD [5]; hyperglycemia, inflammatory and oxidative stress are significant pathogenetic mechanisms in DR [6]. Due to the complexity of the causative factors, there are many unanswered questions about the pathogenesis and treatment of such diseases. It has been found that there are two main reasons for the occurrence of progressive apoptosis in RD. On one hand, genetic mutations cause impaired normal retinal function and retinal cell death [7, 8]. On the other hand, acquired factors can disturb the retinal microenvironment, disrupt the balance of retinal proteins and promote dysfunction and cell death of retinal neurons [9, 10, 11]. Ubiquitination is a process in which ubiquitin binds to a substrate under the co-mediation of enzymes by ubiquitin-activating enzyme (E1), ubiquitin-transferring enzyme (E2) and ubiquitin ligase enzyme (E3). It plays an important role in maintaining the environmental homeostasis and normal function of the retina. Thus when mutations in the genes or environmental factors affect the process of ubiquitination, it may lead to RD. Meanwhile, the protein degradation system plays an important regulatory role in maintaining retinal protein homeostasis [12]. There are two protein degradation systems: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (ALPS) (Fig. 1). The UPS is a system that selectively degrades proteins by ubiquitination modification of the substrate, while the ALPS utilizes lysosome to degrade cellular components in a process called autophagy [13]. In recent years, a growing number of studies have discussed the role of ubiquitination as well as protein degradation systems in RD, but there is a lack of overviews of the most recent research in this area.
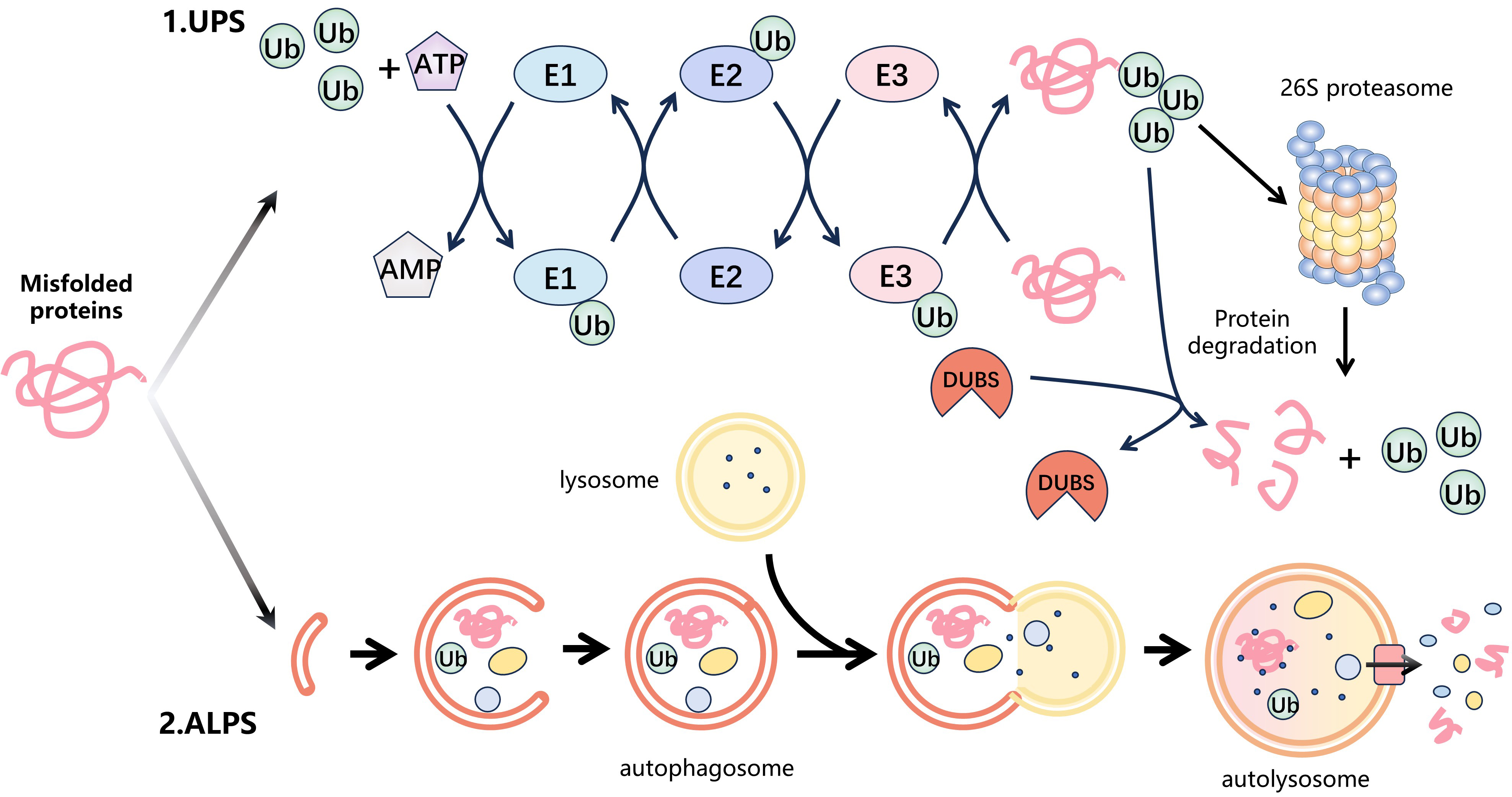 Fig. 1.
Fig. 1.Protein degradation systems. Intracellular misfolded proteins are mainly degraded by two protein degradation systems: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal system (ALPS). (1) The UPS is the major protein degradation pathway. Firstly, the ubiquitin-activating enzyme (E1) catalyzes the activation of ubiquitin (Ub) by utilizing energy from ATP and then transfers the activated Ub to the ubiquitin-conjugating enzyme (E2) to form the E2-Ub complex. The E3 ubiquitin ligase (E3) identifies the E2-Ub complex and transports ubiquitin to the substrate protein. This cycle is repeated, leading to the formation of polyubiquitin chains on the substrate. Finally, the 26S proteasome specifically recognizes the ubiquitinated substrate protein and degrades it. In addition, the process of ubiquitination is reversible. Deubiquitinases (DUBs) remove ubiquitin from the substrate proteins by hydrolysing ester, peptide or isopeptide bonds at the C-terminus of ubiquitin. (2) ALPS is some other significant degradation pathway that utilizes lysosomes to selectively eliminate aging, damaged, or surplus cellular components via a mechanism called autophagy. Firstly, cell receives a signal that triggers autophagy and forms autophagosomes in the cytoplasm to capture the target protein. These autophagosomes then merge with intracellular lysosomes to form autophagic lysosomes. Eventually, the target protein is degraded by the internal hydrolases of the autolysosomes.
This article comprehensively reviews recent research progress on ubiquitin-related genes, proteins and protein homeostasis in the pathogenesis of RD, and summarizes the potential targeted therapy strategies for it. The aim is to guide future studies on the role of ubiquitination in RD and its potential application in future treatments. We hope that it will contribute to enhance understanding of RD and facilitate the development of more efficient treatments.
Ubiquitin, a 76 amino acids small protein, is widely found in eukaryotic cells and highly conserved. Its molecular structure contains an N-terminal methionine residue (M1), a C-terminal glycine residue (G76) and seven lysine residues (K6, K11, K27, K29, K33, K48, and K63). The ubiquitin can acts as a signaling molecule through a cascade of enzyme-catalyzed reactions to covalently modify the lysine residue of the substrate protein, realizing protein ubiquitination. When a single ubiquitin molecule modifies a single lysine residue of a substrate, it is referred to as monoubiquitination, which is mianly involved in receptor internalization, vesicular sorting, transcriptional activation, DNA repair, Ras activation, and gene silencing. When multiple ubiquitin molecules modify multiple lysine residues on the substrate protein, it is called multi-monoubiquitination. Additionally, lysine residues of ubiquitin molecules that have been covalently modified on the substrate protein can undergo further ubiquitination modifications to form various types of homogeneous ubiquitin chains, which is called polyubiquitination. Correspondingly, methionine at the M1 end of the ubiquitin molecule possesses a free amino group, which can tandemly connect the ubiquitin molecule to form a linear ubiquitin chain (also known as the M1 chain). Therefore, monoubiquitination, multi-monoubiquitination, and eight different forms of homogeneous ubiquitin chain modification can exist in the cell, all of which have been extensively studied for their corresponding biological functions [14] (Table 1). Furthermore, if a ubiquitin chain combines different lysine residues of ubiquitin itself to form a mixed or branched ubiquitin chain, it further increases the complexity of the ubiquitin chain assembly and topology. It has been shown that mixed ubiquitin chains and branched ubiquitin chains also play significant roles in biological processes. For instance, the products of the K5 gene from Kaposi’s sarcoma-associated herpes virus modify a cell surface immune receptor, known as major histocompatibility complex (MHC) class I molecules through mixed ubiquitin chains linked by K11 and K63, thereby promoting MHC Ⅰ endocytosis [15]. Ubiquitin is also susceptible to small molecule modifications, including SUMOylation, phosphorylation, acetylation, ribosylation, etc., which can result in changes to its structure. These modifications enrich the diversity of ubiquitin structures, affect ubiquitination and deubiquitination cascade reactions and increase the complexity and diversity of ubiquitin signals regulations [14]. In summary, regardless of the various types of ubiquitination modifications or the modification of ubiquitination by other modifiers, there are numerous potential combinations in the ubiquitination process.
| Ubiquitin chain | Biological functions |
| K6 | DNA damage response |
| Mitochondrial homeostasis | |
| K11 | Proteasomal degradation of proteins in mitosis and cell cycle regulation |
| Membrane trafficking | |
| Regulation of Tumor Necrosis Factor (TNF) signalling | |
| K27 | T-cell development |
| Mitochondrial damage response | |
| K29 | Proteasomal degradation |
| Lysosomal degradation | |
| Regulation of Adenosine 5 | |
| K33 | Regulation of AMPK-related protein kinases and T cell receptor (TCR) signalling |
| K48 | Proteasomal degradation |
| K63 | Endocytosis |
| DNA repair | |
| Nuclear factor kappa-B (NF- | |
| Protein complex formation | |
| M1 | NF- |
| Cytokine signalling | |
| Cell death |
UPS is the major pathway for protein degradation in the cell which involved in the degradation of 80–90% of proteins. It consists of ubiquitin, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), E3 ubiquitin ligase (E3), 26S proteasome and deubiquitinases (DUBs).
Ubiquitination is the process in which ubiquitin is covalently attached to proteins through a series of enzyme-mediated reactions. Firstly, E1 activates ubiquitin by utilizing energy from ATP to form a high-energy thioester bond between the carboxyl group of the C-terminal Lys residue of ubiquitin and the sulfhydryl group of its own cysteine (Cys) residue. The E2 then recognizes the E1-ubiquitin (Ub) complex and subsequently conjugates the activated Ub to the Cys residue of its active site via a thioester bond, forming the E2-Ub complex. And E3 identifies the E2-Ub complex and transports ubiquitin to the substrate protein. This process is repeated, leading to the formation of polyubiquitin chains on the substrate. Currently, two E1s (Ubiquitin-activating enzyme, Uba1 and Uba6), about 40 E2s and over 600 E3s have been discovered in human cells [16], collectively regulating the ubiquitination of tens of thousands of substrate proteins within cells. Among them, E3 can be categorized into really interesting new gene finger (RNF), homologous to E6-associated protein C terminus (HECT) and RING-between-RING (RBR) domain family based on structural and functional characteristics. Two distinct mechanisms of Ub transfer to substrate have been identified in E2-Ub complexes under the catalysis of different types of E3. One mechanism entails the RING domain providing action sites for E2 and the substrate, allowing E2 to directly catalyze the transfer of ubiquitin to the substrate. The other mechanism is a two-step process. Firstly, the activated Ubiquitin combines with the conserved cysteine residue of E3 enzyme in the HECT or RBR domain through a thioester bond, and then the E3-Ub complex delivers Ub to the corresponding substrate. For the former mechanism, the formation of the ubiquitin chain is primarily determined by E2. In contrast, in the second mechanism, the formation of the ubiquitin chain is mainly determined by the corresponding HECT/RBR E3. The majority of E3s in cells are RING-type E3s. Therefore, RING-type E3s, in conjunction with E2 plays a key role in ubiquitin chain formation [17]. Finally, the 26S proteasome specifically recognizes and degrades proteins that have been ubiquitinated. At the same time, it dynamically monitors the protein-related biological process through various regulatory mechanisms, ensuring precise control of the degradation of active proteins and preventing ineffective degradation of proteins [18]. In addition, Ubiquitination is a reversible process. DUBs are pivotal enzymes in the deubiquitination process, responsible for the removal of ubiquitin from the substrate proteins by hydrolysing the ester, peptide or isopeptide bonds at the C-terminus of ubiquitin. These enzymes are classified into six families: the ubiquitin C-terminal hydrolases family (UCHs), the ubiquitin-specific proteases family (USPs), the ovarian tumor proteases family (OTUs), the Josephin family, JAMM family and ubiquitin-like proteases family (ULPs) [19]. DUBs also serve as key regulators of the ubiquitin code, managing complex ubiquitin modifications. These processes and enzymes work together to form a complex regulatory network.
Autophagy is a critical intracellular pathway that utilizes lysosomal hydrolases to degrade cellular components in response to various cellular stresses such as energy deficiency, oxidative stress, organelle damage, and DNA damage. Its main role is to recycle cellular components and damaged organelles in order to maintain intracellular homeostasis [20, 21, 22]. Based on their respective mechanisms of action and biological functions, autophagy is classified into three types: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA).
Macroautophagy is a widely typical and highly conserved autophagy pathway. In the process of macroautophagy, cargo proteins are captured by signal-activated phagophore to generate a double-membrane autophagosome. Subsequently, the autophagosome merge with the intracellular lysosome to form autolysosome and then the target protein is degraded by the internal hydrolases of the autolysosome [23]. Mitophagy is a subtype of selective macroautophagy that preserves mitochondrial function and cellular homeostasis by specifically eliminating dysfunctional mitochondria from the cytoplasm. This process serves to mitigate the detrimental impact of environmental stressors and decrease cellular apoptosis. In contrast to macroautophagy, microautophagy is a lysosomal degradation process in which substrates are engulfed and degraded through the invagination of the membrane, without the formation of autophagosomes [24]. Macroautophagy and microautophagy can phagocytose large structures through both selective and non-selective mechanisms, whereas CMA is limited to the selective degradation of soluble proteins [25]. In CMA, the soluble substrate protein specifically interacts with chaperone proteins from the heat shock cognate protein 70 (HSC70) family via a pentapeptide motif (KFERQ-like motif) and enters the lysosome for degradation via lysosomal associated membrane protein 2A (LAMP2A) on the lysosomal membrane [26].
In protein degradation, the UPS is responsible for degrading short-lived and soluble misfolded or unfolded proteins, while autophagy is involved in clearing long-lived and insoluble proteins. These two pathways operate independently and have different substrate specificities. Recent research indicates a potential synergistic relationship between UPS and autophagy. Impairment of UPS function may trigger the activation of ALPS as a compensatory mechanism, and inhibition of autophagy can impact proteasome activity, leading to the accumulation of ubiquitination substrates [27].
Additionally, ubiquitination modifications are involved in the recognition of
substrates in both UPS and autophagy. For instance, the proteasome can be
ubiquitinated and transported to the lysosome for degradation under cellular
stress conditions [28]. Hypoxia-inducible factor 1
AMD is a progressive retinal disease that affects the macula and the RPE layer beneath it. It is one of the most common causes of irreversible loss of central vision in the elderly. AMD can be classified into two distinct types according to its clinical manifestations: dry AMD, characterized by macular atrophy, and wet AMD, distinguished by neovascularization, exudation, and hemorrhaging. AMD is affected by a combination of multiple risk factors such as age, obesity, hypertension, excessive light exposure, and smoking. In the pathophysiology of AMD, RPE degradation, oxidative stress, and inflammation are considered to be important pathogenic mechanisms of AMD [29]. These mechanisms are intricately linked to retinal ubiquitination and the maintenance of protein homeostasis [30]. Therefore, it is crucial to explore the role of ubiquitination and protein degradation system in the progression and treatment of AMD.
A comprehensive association analysis of over 3000 neovascular AMD cases and more than 8000 controls from an East Asian cohort in a study by Huang et al. [31] revealed that a missense single nucleotide variant (SNV), rs7739323, which is located in the ubiquitin protein ligase E3D (UBE3D) gene is significantly associated with the development of neovascular AMD. This suggested that UBE3D, a ubiquitination-related genes, may be involved in the pathogenesis of neovascular AMD. Xia et al. [32] further verified the role of UBE3D in the mechanism of neovascular AMD through in vivo and in vitro studies. In zebrafish in vivo experiments, the UBE3D gene was knocked out. Subsequent observations revealed that the eyes of UBE3D morphants exhibited abnormal eye development, an increased number of pigment granule deposits and heightened angiogenesis. In the in vitro experiments, lentiviral gene transfer technology was used to overexpress or knockdown UBE3D expression in hRPE cells. They observed that the UBE3D-up group exhibited enhanced cell proliferation and migration following oxidative damage stimulation, whereas the converse effect was observed in the UBE3D-down group. Simultaneously, their experiments showed that UBE3D was negatively correlated with Cyclin B1, a pivotal modulator of the cell cycle in mitosis. Furthermore, the researchers found that UBE3D and autophagy may act in concert to mitigate oxidative damage while low expression of UBE3D exacerbates oxidative damage and inflammatory responses. Tao et al. [33] also validated the aforementioned research findings. The study observed abnormal accumulation of melanosomes, melanofuscin and lipofuscin, atrophy of photoreceptors, ALPS dysfunction, reduced antioxidant processes, and cell cycle dysregulation in the RPE of UBE3D knockout mice, all of which are associated with cellular senescence. Xu et al. [34] investigated the genetic susceptibility factor UBE3D-v379m SNV in relation to AMD and found that UBE3D is recruited to the double-strand break (DSB) site through a proliferating cell nuclear antigen (PCNA)-interacting protein (PIP) box and binds the Krüppel-associated box domain domain-associated protein 1 (KAP1) via R377R378, thereby mediating the homologous recombination process in heterochromatin regions. The oxidation of individuals carrying the V379M SNV leads to the segregation of UBE3D from KAP1, which impairs the DSB repair process. These findings imply that UBE3D may serve as a pivotal factor in the pathogenesis of AMD, impacting retinal development, oxidative stress, DNA damage repair and autophagy while playing a crucial role in retinal aging and neurodegeneration. Furthermore, these studies suggest that UBE3D deficiency exerts multiple effects on retinal homeostasis and propose that polymorphisms in ubiquitin-related genes may influence retinal susceptibility to age.
In another study, Xing et al. [35] identified differentially expressed
genes in RPE from AMD samples and healthy controls by downloading two datasets,
GSE99248 and GSE125564, from the Gene Expression Omnibus (GEO) database. The
researchers examined the human retinal pigment epithelial cell line ARPE-19 in
AMD samples and found that five (PSMD4, PSMD8, PSMA4,
PSMB5, and PSMB6) of the top 10 key genes in cellular expression levels
were linked to proteasome function. This suggested that proteasome-mediated
degradation may be significantly involved in AMD. The expression levels of
PSMD4, PSMD8 and PSMA4 were upregulated in ARPE-19
cells from AMD samples compared to those from healthy samples. The
PSMD4-encoded protein, proteasome 26S subunit non-ATPase 4, recruits the
phosphorylated and ubiquitinated NF-kappa-B inhibitor alpha (IkB
The above studies demonstrated the influence of ubiquitination and protein degradation systems on AMD from a genetic perspective and reveal the potential correlation between ubiquitination, proteostasis, and AMD mechanism. The regulation of ubiquitination-related genes and proteins impacts the homeostasis of retinal proteins, and conversely, the maintenance of protein homeostasis influences the ubiquitination process, collectively contributing to the pathophysiological progression of AMD.
Based on these studies, ubiquitination modification and protein degradation
systems are seen as potential targets for AMD therapy. Li et al. [36] discovered that Smurf1 (Smad ubiquitylation regulatory factor-1, a
C2-WW-HECT structural domain E3 ubiquitin ligase) is upregulated in retinal
injury and degeneration. It was not only involved in epithelial-mesenchymal
transition (EMT) and inflammatory responses, but also established a potential
link between the transforming growth factor-
In conclusion, the dysfunction of ubiquitination and protein degradation systems has multiple impacts on the pathogenesis of AMD. The ubiquitin-related gene UBE3D plays a crucial role in maintaining retinal function and homeostasis. Furthermore, genes PSMD4, PSMD8, and PSMA4, which are associated with proteasome function, have been demonstrated to be strongly correlated with AMD. The current study indicates that inhibition of the E3 ligase Smurf1 can attenuate the inflammatory response, ROS production, and EMT during the progression of AMD. Conversely, activation of autophagy helps sustain the structure and function of the retina, thereby slowing down the advancement of AMD.
DR, a common complication of diabetes, is the primary cause of vision impairment in adults aged 20 to 79. It is estimated that 415 million people aged 20 to 79 had diabetes in 2015 and this number is expected to increase to 642 million by 2040 due to the increasing prevalence of diabetes, an ageing population and longer life expectancy for people with diabetes [39]. According to the severity of the lesion, DR is classified as non-proliferative DR (NPDR) and proliferative DR (PDR). The main fundus manifestations of NPDR are microaneurysms, hemorrhages and exudates. Neovascularization, vitreous proliferation, macular oedema and even retinal detachment are the principal fundus manifestations of PDR. Hyperglycaemic (HG)-induced retinal vascular damage is the major factor contributing to DR. Other factors that contribute to the development of DR include inflammatory response, oxidative stress, apoptosis, and neoangiogenesis.
Ubiquitination is intricately linked to the pathogenic mechanisms outlined
previously. The ovarian-tumor-domain-containing deubiquitinases (OTUDs) are
members of the DUBs that can reverse protein ubiquitination. Zhou et
al. [40] reported that a novel and rare OTUD3 c.863G
The aforementioned studies have examined the correlation between ubiquitination
and DR at the genetic level. In addition, many studies have explored the
relationship between ubiquitination-, deubiquitination-related enzymes, and the
development of DR at the protein level. Hu et al. [42] reported
ubiquitin-specific peptidase 25 (USP25), a DUBs, as an inflammatory regulator
involved in the progression of DR. The researchers found that ubiquitination
related proteins were significantly elevated in DR Tissues compared to normal
tissues, with the greatest difference in USP25 expression. In the HG environment,
USP25 promotes microglia activation by participating in Rho-associated
coiled-coil-containing protein kinase (ROCK) to regulate NF-
Furthermore, the disruption of retinal proteostasis is closely associated with DR. HG disrupt the autophagy pathway in retinal cells, resulting in cells apoptosis, vascular endothelial cell damage and neovascularisation [49]. Recently, more efforts have delved deeper into the potential mechanisms of autophagy in DR, leading to the identification of new therapeutic targets for the treatment of DR.
Feng et al. [50] reported that, under HG-conditions, the
upregulation of high mobility group box 1 (HMGB1) in RPE cells led to increased
lysosomal membrane permeability (LMP) through a cathepsin B (CTSB)-dependent
pathway, ultimately resulting in dysfunction in the ALPS. Liu et
al. [51] also found that in early DR, the vitreous humour secreted
large amounts of the neurodegenerative factor Glial cell maturation
factor-
Tight glycaemic control remains the primary treatment strategy for DR. Retinal laser photocoagulation and anti- vascular endothelial growth factor (VEGF) drugs have significantly reduced the incidence of severe vision loss in DR. However, these interventions are still ineffective in preventing the onset of DR in patients with diabetes. Therefore, there is an urgent necessity to research new pharmaceuticals for the treatment of DR.
Trotta et al. [52] discovered that
In conclusion, numerous ubiquitination-related genes and proteins have been linked to the development of DR, yet there are still many unknowns in the current research landscape. E3 ligases TRIM40 and NEDD4L, as well as DUBs family USP25 and USP48, have been identified to participate in the progression of DR by promoting retinal inflammatory response. E3 ligase TRIM46 and DUBs family USP48 are involved in the progression of DR by promoting ferroptosis in retinal cells. Furthermore, HG environment can disrupt autophagy and promote DR through HMGB1 and GMFB-mediated pathways. It is hoped that regulating autophagy through multiple pathways will lead to the discovery of drugs for DR. For instance, BHB inhibits abnormal cellular autophagy by up-regulating BDNF, NGR1 activates PINK1-dependent mitochondrial autophagy to prevented DR, and adiponectin inhibits angiogenesis caused by autophagy by regulating the PI3K/AKT/mTOR signal pathway. There are still many possibilities for the development of DR therapeutics.
IRDs are a group of rare eye diseases caused by genetic defects that result in progressive retinal degeneration and irreversible visual impairment, affecting approximately 1:2000 people worldwide [55]. The most common of these IRDs is RP, others include Stargardt disease (STGD), Leber hereditary optic neuropathy (LHON), Leber congenital amaurosis (LCA), cone or cone-rod dystrophy (CORD) and X-chromosome-associated cone dystrophy (COD), etc. IRDs are one of the most genetically heterogeneous groups of human diseases. More than 270 related genes have been identified, and research into gene therapy treatments for IRDs is active [7]. However, ubiquitination has been found to be important in the pathogenesis and treatment of some IRDs.
The study by Toulis et al. [56] investigated the potential relevance of the DUB gene USP45 to retinal function. Following the knockout of USP45 in a zebrafish model, their observations indicated that USP45 may be essential for vertebrate retinal development and suggested that genes encoding enzymes of the ubiquitin pathway may be promising candidates for causing IRDs. USP48 may be involved in the regulation and stabilisation of ciliary proteins that are key to photoreceptor function, the regulation of intracellular protein transport, and the transport of cilia to the outer segments of photoreceptors. The impairment of the cilium in retinopathy can give rise to a spectrum of pathologies, including RP, LCA, and CORD, collectively designated as retinal ciliopathies, which constitute approximately a quarter of IRDs [57, 58]. The study of Sánchez-Bellver et al. [59] identified USP48 as a potential new gene candidate for retinal ciliopathies. Furthermore, a ubiquitin-binding gene, DNA topoisomerase 1 Binding Arginine/Serine Rich Protein (TOPORS), is closely related to the normal function of photoreceptor ciliary proteins and centrosomes. Mutations in TOPORS have been identified as a cause of autosomal-dominant RP [60].
RP is the most prevalent class of IRDs. The principal lesions of RP occur on photoreceptors and the RPE. It is characterized by symptoms such as nyctalopia, loss of peripheral vision and a significant reduction or absence of electroretinograms (ERGs). The fundus features of RP are usually identified by the classic triad of retinal pigmentation, arteriolar attenuation and waxy disc pallor. Between 1/7000 and 1/3000 people worldwide have RP, which often starts early and cannot be accurately diagnosed. According to the localization of the causative genes and their expression properties, the identified RPs are broadly classified into autosomal-dominant RP (adRP, 15–25%), autosomal-recessive RP (arRP, 5–20%) and x-linked RP (x-RP, 10–15%), and the remaining 40–50% have different phenotypic characteristics. The rhodopsin (RHO) gene is responsible for approximately 25% of adRP cases, the USH2A gene causes about 20% of arRP cases, and the retinitis pigmentosa GTPase regulator (RPGR) gene accounts for about 70% of x-RP cases. In total, about 30% of RP is caused by mutations in these three genes [61].
In addition to the genes previously mentioned, a number of genes and proteins associated with ubiquitination have been found to be associated with RP. Studies have confirmed that TOPORS causes RP as previously stated. Wang et al. [62] conducted a more in-depth study of TOPORS mutations and found that all pathogenic truncation variants of TOPORS were clustered at residues 807~867. This provided a more precise localization for subsequent studies. Conversely, Czub et al. [63] identified protein chaperone 26S protease regulatory subunit 4 (P26S4/PSMC1) as a protein that interacts with TOPORS. This interaction is thought to be involved in retinal maintenance and retinopathy development. The present study contributes to study the mechanism of RP caused by TOPORS mutations. Furthermore, mutations in the gene for kelch-like protein 7 (KLHL7, a component of the cul3-based Cullin-RING ubiquitin ligase) have been identified as a cause of retinitis pigmentosa (RP), indicating that ubiquitination plays a significant role in retinal health and disease [64]. A study by Kim et al. [65] demonstrated that KLHL7 increases terminal uridylyl transferase 1 (TUT1) ubiquitination, which facilitates the ubiquitination of TUT1 and stimulates the nucleolus stress response. This study suggests that the KLHL7-TUT1 axis may be a critical factor in RP, providing valuable insights into the pathogenesis of RP.
In addition, disruption of the retinal protein environment is a major factor contributing to the progression of RP and a further consequence of the disease. Point mutations in the rhodopsin (RHO) gene are the most common genetic factors for adRP. The most commonly occurring RHO mutation is a mutation that replaces a proline with a histidine at amino acid residue 23 in rhodopsin (called P23H). The change of P23H causes the accumulation of misfolded retinal proteins in the cells. This leads to increased endoplasmic reticulum stress (ERS), which ultimately results in retinal degeneration [66, 67]. Qiu et al. [66] discovered that misfolded retinal proteins in photoreceptor cells in RP lead to abnormal activation of autophagy, which counteracts the protective effect provided by the complementary UPS mechanism. It was found that the ratio of autophagy to proteasome activation (referred to as the A:P ratio) in the retina of P23H mice was nearly 50% higher than in normal control mice. Thus, it is crucial to maintain retinal protein homeostasis to delay RP progression.
There is considerable interest in the potential role of ubiquitination and
protein degradation systems in the treatment of RP. Park et al. [68]
identified a novel function of the Drosophila ubiquitin-specific protease 14
(USP14) in endoplasmic reticulum stress. The overexpression of USP14 in a
Drosophila model demonstrated a protective effect against endoplasmic reticulum
stress while maintaining proteasome activity. Furthermore, it was observed that
the level of retinoschisin-1 (Rh-1) protein was restored in a Drosophila model of
adRP, and retinal degeneration was inhibited. These findings suggest that
manipulation of USP14 may serve as a potential therapeutic strategy for RP. Other
studies indicate that the E3 ubiquitin ligases
SORRD1 and SORRD2 increase degradation of
misfolded retinal RH1 (Rh1G69D) by ubiquitination, preventing Rh1G69D-induced
photoreceptor dysfunction and retinal degeneration [69]. In addition, the study
by Varner et al. [70] identified that the deubiquitinase
ovarian tumor domain-containing 7B (Otud7b)/Cezanne is highly expressed in retinal photoreceptor cells. In
photoreceptor retinal degeneration mice and RP model mice, deficiency of Otud7b
activated the NF-
COD and CORD are two genetic diseases that cause progressive apoptosis of
retinal photoreceptor cells. 32 genes have been identified, most of which are
common to both diseases. COD is specifically characterised by apoptosis of the
central retinal cone cells. In the early stages of the disease, patients
experience day blindness, loss of central visual field and colour discrimination.
As the disease progresses, patients develop peripheral visual field defects. Han
et al. [72] conducted a study involving eight unrelated families from
Hungary, the USA, Israel and the Netherlands. All members of these families
exhibited a rod-cone or cone/cone-rod dystrophy phenotype. The researchers
identified a double allele loss-of-function variant of UBAP1L (HGNC:
40028) in all of the patients. Ubiquitin associated protein 1 like (UBAP1L) is a
member of the UBA structural domain family, which encodes ubiquitin-related
protein 1-like. One of the pathogenic variants, intron NM
001163692.2:c.910-7G
LCA is the earliest and most severe form of inherited retinal disease (IRD). It is characterised by severe vision loss in childhood, usually with complete loss of cone and rod cell function in both eyes within the first year of life, leading to congenital blindness in infants. Yi et al. [74] sequenced and analysed two individuals with LCA, as well as 3011 internal controls with other inherited eye diseases, and identified two rare USP45-pure mutations in two unrelated families. Immunohistochemistry of USP45 in human and zebrafish retinal sections showed enriched expression in the photoreceptor inner segments, a key site where most forms of LCA are affected. In zebrafish, knockdown of USP45 transcripts resulted in abnormal retinal development, including effects on the photoreceptors; however, wild-type USP45 mRNA was able to rescue the retina. In addition, targeted knockdown of USP45 in mice caused an abnormal electroretinographic response, similar to that of LCA patients. These findings suggest that USP45 double allele mutations are associated with LCA development. The most prevalent causative mechanisms of LCA are mutations that impair the membrane-association and co-localisation of RPE65, a membrane-related protein found in abundance in the RPE, and lecithin-retinol acyltransferase (LRAT), resulting in a disruption of 11-cis-retinal synthesis. Luxturna is a direct gene therapy approved to treat RPE65-LCA (LCA2), caused by mutations in certain genes. The therapy initially restores vision. However, the visual benefits diminish over three years and the photoreceptors continue to deteriorate. Therefore, some studies have focused on the degradation process of key proteins by UPS in order to develop effective gene-based therapeutic strategies. Li et al. [75] showed that systemic administration of sodium 4-phenylbutyrate (PBA), a chemical chaperone, improved the correct folding of RPE65, reduced proteasomal degradation of mutant RPE65, and increased protein stability, enzymatic activity, membrane association and co-localisation of RPE65 and LRAT.
LHON is a disease associated with mitochondrial DNA mutations that cause blindness due to damage or apoptosis of retinal ganglion cell (RGC). Sharma et al. [76] showed that cells with LHON-specific mitochondrial DNA (mtDNA) mutations have impaired mitophagy, leading to accumulation of dysfunctional mitochondria and cell death. Conversely, activating autophagy can repair mitochondrial defects and improve cell survival. The findings suggest that modulation of autophagy may be a potential therapeutic target for the treatment of LHON. Danese et al. [77] demonstrated that autophagy and mitophagy were pathologically increased in LHON-affected patients, indicating the activation of the apoptotic response due to the mitochondrial stress phenotype. However, this phenomenon was not observed in LHON carriers. Therapeutic strategies that balance autophagy and mitophagy levels and mitochondrial homeostasis can be used to reverse the pathological phenotype of LHON-affected cells.
Prom1 is a transmembrane glycoprotein predominantly found in the cytoplasm of the RPE. Mutations in the Prom1 gene disrupt photoreceptor disc morphogenesis, leading to autosomal dominant Stargardt-like macular dystrophy (STGD4). Prom1 may play a key role in the regulation of autophagy through upstream inhibition of mTOR signalling, regulating autophagosome maturation and transport [78]. However, it is unclear whether this is the pathogenic mechanism of STGD.
Overall, current researches on IRDs are focused on the pathogenesis and gene therapy of such diseases. The ubiquitin-related genes TOPORS and KLHL7 are associated with RP, while UBAP1L and USP45 are linked to the development of CORD and LCA, respectively. A significant number of studies have been conducted to refine and enrich the pathogenic mechanism of mutated genes on this basis. Nevertheless, it is important to note that protein homeostasis is also a significant factor in the progression of RP, and both UPS and autophagy play crucial roles in the survival of photoreceptor cells and the maintenance of functional protein activity. Regulation of ubiquitination-related genes/proteins and retinal protein homeostasis represents a promising therapeutic avenue for RP treatment.
Current pharmacological treatments for RD can delay disease progression and improve vision to some extent, but their therapeutic effects are still quite limited. Ubiquitination is an important protein labelling mechanism, and ubiquitination-related genes/proteins not only affect the normal development and function of the retina, but also affect the occurrence of retinal diseases. The protein degradation pathway plays an extremely significant role in regulating the number and activity of retinal proteins, protein-protein interactions and subcellular localisation of proteins. Currently, targeted ubiquitin-related gene/protein therapy has become a focus of development. However, the study of ubiquitination-based drugs remains a relatively unexplored area in the treatment of RD. This review provides a comprehensive analysis of ubiquitin-related genes in RD (Table 2, Ref. [31, 40, 41, 42, 44, 45, 46, 47, 48, 59, 60, 63, 64, 65, 74]) and discusses potential therapeutic strategies for these diseases.
| Disease | Ubiquitin-related genes/proteins | Types of corresponding ubiquitin-related enzymes | Physiological/pathological effects |
| AMD | UBE3D | E3 | Involve in eye development, DNA repair |
| Synergises with autophagy to reduce oxidative damage [31] | |||
| DR | OTUDs | DUBs | Involve in glucose and fatty acid metabolism [40, 41] |
| USP25 | DUBs | Regulating NF- | |
| Promotes microglia activation and the secretion of inflammatory factors [42] | |||
| TRIM40 | E3 | Down-regulation increased DAB1 and promoted inflammatory responses [44] | |
| NEDD4L | E3 | Up-regulation promoted IκB | |
| USP48 | DUBs | Down-regulation induce oxidative stress and ferroptosis and enhances TNF- | |
| TRIM46 | E3 | Promote HG-induced ferroptosis and inhibiting cell proliferation [48] | |
| RP | USP48 | DUBs | Involved in the regulation and stabilisation of ciliary proteins [59] |
| TOPORS | E3 | Interact with P26S4, involve in retinal maintenance and retinopathy development [60, 63] | |
| KLHL7 | E3 | Facilitates the ubiquitination of TUT1 and stimulates the nucleolus stress response [64, 65] | |
| CORD | UBAP1L | Ubiquitin-associated protein 1-like | |
| LCA | USP45 | DUBs | Down-regulation resulted in abnormal retinal development [74] |
RD, retinal degeneration; AMD, age-related macular degeneration; UBE3D,
ubiquitin protein ligase E3D; DR, diabetic retinopathy; OTUDs,
ovarian-tumor-domain-containing deubiquitinases; DUBs, deubiquitinases; USP,
ubiquitin-specific peptidase; TRIM40, tripartite motif-containing 40; DAB1,
Disabled1; NEDD4L, neural precursor cell expressed developmentally downregulated
4; IkB
UPS is widely distributed in cells and its ubiquitination modification has recently emerged as an interesting target for drug therapy. Proteasome inhibitors such as bortezomib (VELCADE) and carfilzomib (KYPROLIS) have been used in the treatment of multiple myeloma [79, 80, 81]. In addition, bendamustine is an E3 ligase inhibitor that targets the E3 ligase linear ubiquitin assembly complex (LUBAC) and has been approved by the U.S. Food and Drug Administration for the treatment of chronic lymphocytic leukaemia, multiple myeloma, non-Hodgkin’s lymphoma, ovarian cancer and other diseases [82]. Although these proteasome inhibitors are mainly used in oncological therapy, no efficacy has been reported in ophthalmology. However, the ubiquitination degradation mechanism has been applied to the treatment of RD. For example, research shows that injecting Smurf1 inhibitor A01 into the vitreous cavity of mice can reverse some of the retinal damage caused by ROS and inflammatory responses, thus slowing the progression of AMD [36].
Several natural plant and drug products are now known to promote UPS function and autophagy, thereby alleviating environment-related retinal damage. For instance, polyphenol quercetin, a natural substance found in fruits and vegetables, has been shown to enhance retinal cell degradation and self-renewal by inhibiting mTOR [83]. NGR1, extracted from Panax notoginseng, has been shown to inhibit inflammatory response and neovascularisation through pink1-dependent enhancement of mitophagy [53]. Both curcumin and nanocurcumin were found to increase proteasome activity in RPE cells in a study by Ramos de Carvalho et al. [84], with nanocurcumin showing less cytotoxic effects. Fung et al. [85] found that the antioxidant luteolin inhibited autophagosome formation, reducing oxidative stress, inflammation and apoptosis in retinal cells. All of these pharmacological studies are designed to minimize retinal cell death and maintain the homeostasis of the retinal environment, with nutritional support serving as a prerequisite for therapeutic success.
Due to the complexity of the proteins involved in the protein degradation system, drugs targeting this system have historically lacked selectivity. Additionally, toxicity, resistance, and efficacy issues of certain drugs have posed challenges, further hindering the development of proteasomal inhibitors. De Cesare et al. [86] have carried out a high-throughput, label-free screening of ubiquitin E2 enzymes and E3 ligases based on Matrix-Assisted Laser Desorption/Ionization Time of Flight (MALDI-TOF) mass spectrometry. This method is a versatile tool for drug discovery in the ubiquitin pathway, which can detect the ubiquitin transfer activity of E2 enzymes and E3 ligases, identifiy E2/E3 active pairs, assess the potency and specificity of inhibitors, and screen compound libraries in vitro without the need for chemical or fluorescent probes. It also reduces the risk of false positives and negatives by using non-physiological E2/E3 ligase substrates. Researchers are shifting from the traditional occupancy-driven approach of enzyme inhibitors to a mechanism that targets event-driven processes. This is achieved through proteolysis targeting chimera (PROTAC) and molecular glue technology, which provides highly selective control of targeting inhibition. PROTACs are small heterobifunctional molecules that consist of a module that binds to the target protein and a module that recruits the E3 ligase. Molecular glue is a low molecular weight molecule that binds to a specific protein and adds the desired interaction partner, such as E3 ligase, to its interactome. This creates a new binding surface on the protein, providing greater specificity, activity and duration of action than conventional occupancy-based small-molecule inhibitors [87, 88]. To broaden the range of potential substrates, several types of PROTACs are also under development, including autophagy-targeted chimeric (AUTACs) [89], lysosome-targeted chimeric (LYTACs) [90] and several PROTACs that have been used to treat breast and prostate cancer by targeting the estrogen and androgen receptors respectively [91]. However, PROTACs are not currently being used to treat RD. It is a feasible concept to develop highly selective PROTACs or molecular glues to regulate retinal cell death.
This review summarizes the involvement of ubiquitination and protein degradation systems in RDs and suggests potential therapeutic strategies for these diseases. However, there are still many uncharted areas regarding the role of ubiquitination modifications in the pathogenesis of RD. Appropriate stimulation of the protein degradation system may help maintain retinal cell homeostasis, but excessive ubiquitination may accelerate disease progression. The next research goal is the identification of targeted ubiquitination regulators for specific retinal diseases and the validation of cellular and animal assays to evaluate their therapeutic potential. The aim of this review is to provide a basis for future research in this area.
All authors JW, XC, YX and YG contributed significantly to searching the literature and writing the original manuscript. JW contributed to literature search, writing and editing of all sections of the review paper. XC, YX, and YG made significant contributions to the conception and design of the manuscript. JW created Tables 1,2 and Fig. 1. XC, YX, and YG undertook a critical review of the entire intellectual content of the manuscript and ultimately approved the version for publication. YG was responsible for ensuring that the descriptions are accurate and agreed by all authors. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.