








1 Department of Pathology, College of Medicine, King Saud University (primary affiliation), King Saud University- Medical City (secondary affiliation), 11461 Riyadh, Saudi Arabia
Abstract
Podocytes are epithelial cells lining the outer surface of the renal glomerular capillaries and they play a pivotal role in maintaining the structural and functional integrity of the glomerular filtration barrier. Podocytes react to injury in various ways and any injury to these highly specialized cells can progress to podocyte dysfunction, resulting in a group of proteinuric renal diseases called podocytopathies. Podocytopathies include a wide spectrum of primary and secondary kidney diseases, including minimal change disease, diffuse mesangial sclerosis, focal segmental glomerulosclerosis, collapsing glomerulopathy, diabetic, membranous and lupus nephropathies. Etiologically, they can be idiopathic, genetic or secondary to infections and drugs, metabolic diseases, hemodynamic factors or associated with various immune and non-immune systemic diseases. This manuscript provides a basic understanding of podocyte structure, causes of podocyte injury, response to the injury and the subsequent progression to podocytopathies. The pathogenesis of these diseases is set around podocytes. The clinical and morphological manifestations, the commonality and heterogeneity of these podocytopathies are also discussed. As our knowledge of podocyte biology improves, so will our treatment avenues with a more podocyte-centric personalized approach.
Keywords
- podocytes
- podocytopathies
- minimal change disease
- diffuse mesangial sclerosis
- focal segmental glomerulosclerosis
- collapsing glomerulopathy
- diabetic nephropathy
- membranous nephropathy
- lupus nephropathy
The normal human kidneys filter about 180 liters of blood per day with no significant protein loss in the urine [1]. Glomeruli are the basic filtration units of the kidney and they filter the blood circulating in it and the filtered fluid ultimately becomes urine. A glomerulus is made up of a ball-like tuft of capillaries floating in a urinary space, surrounded by a Bowman’s capsule. Podocytes are one of the four primary cells in a renal glomerulus, lining the outside of the glomerular capillaries. The other cells normally present in a renal glomerulus are endothelial cells, mesangial cells and parietal epithelial cells (PECs). The endothelial cells line the inside of the glomerular capillaries, the mesangial cells are located in the interstitium and the PECs line the inside of the Bowman’s capsule (Fig. 1A). The glomerulus receives its blood supply from afferent arterioles of the renal circulation and drains out into efferent arterioles. The resistance of the efferent arterioles causes high pressure in the glomerular capillaries, leading to the filtration of fluid and certain molecules out of the capillary lumen into the urinary space, termed the ultrafiltrate, which then enters the tubules and collecting ducts to eventually form urine. The remaining non-filtered blood returns into vascular circulation via the efferent arterioles. This glomerular filtration barrier (GFB) controls the filtration of various molecules including proteins from the glomerular capillary lumen into the urinary space and it retains 99% of the plasma proteins. Any abnormality in the GFB leads to proteinuria ranging from mild albuminuria to nephrotic syndrome. The GFB is made up of podocytes, glomerular basement membrane (GBM) and endothelial cells (Fig. 1B–D) [2]. Although the mesangial and PECs are not part of the GFB, they contribute indirectly to infiltration and also the pathogenesis of certain podocytopathic diseases. Notably, mature podocytes have very little regenerative capacity and once significantly injured, they undergo cell death without compensatory regeneration and therefore are terminally differentiated cells. This lack of regenerative capacity of podocytes is a critical point and maintained integrity of these cells is crucial for normal functioning of the kidney. This article reviews, based on world literature, podocyte biology along with causes, mechanisms, consequences and clinical manifestations of podocyte injury. Podocyte-centric renal diseases are termed podocytopathies and they include minimal change disease (MCD), diffuse mesangial sclerosis (DMS), focal and segmental glomerulosclerosis (FSGS), collapsing glomerulopathy (CG), diabetic nephropathy and immune-mediated glomerulopathies. These are also discussed in this review.
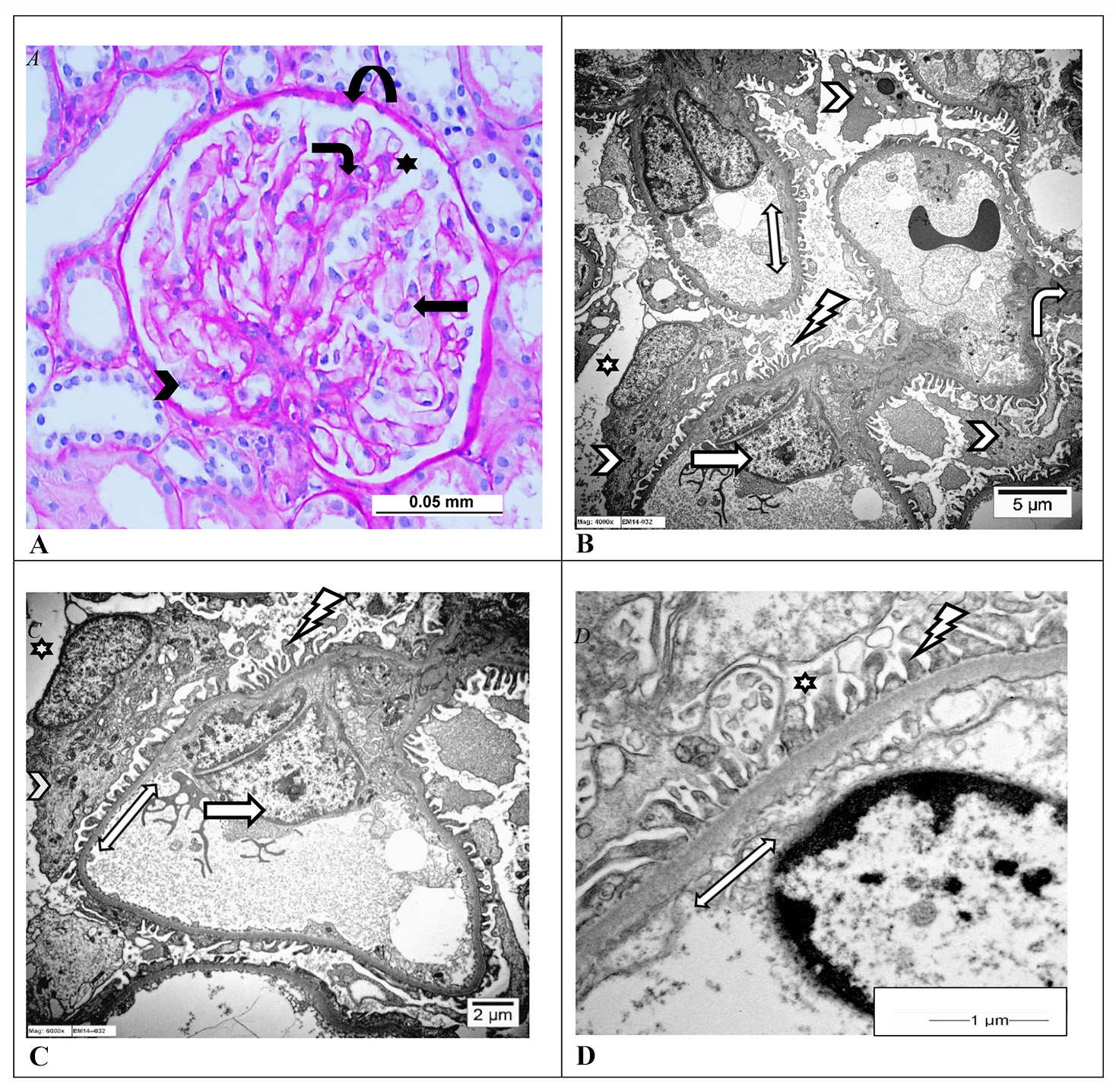 Fig. 1.
Fig. 1.Normal glomerulus. (A) Light microscopic photomicrographs of a normal glomerulus from a human kidney showing capillary loops with podocytes (arrowhead), mesangial cells (bent arrow), endothelial cells (arrow) and parietal epithelial cells lining the Bowman’s capsule (curved arrow), and the capillary tufts are floating in the urinary space (star). (Periodic acid–Schiff stain, Scale bar = 0.05 mm; uranyl acetate). (B–D) Electron microscopic photomicrographs of capillary loops of a normal glomerulus from a human kidney showing the glomerular basement membrane (GBM) (double-headed arrow) lined on the urinary space (star) side by podocytes (arrowhead). The podocyte shows intact foot processes (lightning arrow) with no significant cytoplasmic vacuolization or microvilli formation and no denudation of the GMB surface. Also seen are normal mesangial cells (bent arrow) and endothelial cells (arrow) facing the luminal side of the GBM. (Uranyl acetate and lead citrate, B: Scale bar = 5 μm; C: Scale bar = 2 μm; D: Scale bar = 1 μm). Images from Pathology Department, King Khalid University Hospital-Medical City, King Saud University, Riyadh, Saudi Arabia.
Podocytes are visceral epithelial cells lining the outer surface of the
glomerular capillary loops exposed to the glomerular urinary space and they,
along with the GBM and endothelial cells, form the GFB. There are approximately
500–600 podocytes per glomerular tuft in the adult human kidney [3].
Architecturally, they have a cell body from which arise multiple branching
primary, secondary and tertiary processes. The tertiary processes have
finger-like terminal ends or pedicles called podocyte foot processes (FPs) (Fig. 1B–D). The podocyte FPs interdigitate with FPs of neighboring podocytes and they
adhere to the outer surface of the GBM of the glomerular capillary loops with the
help of anchor proteins like
They include the following:
The unifying pathology of podocyte-centric proteinuric glomerulopathies, regardless of the causative agent or triggering factor, is podocyte injury. The injury can be caused by intrinsic or extrinsic factors such as genetic mutations, infections, toxins, medications, certain circulating permeability factors, metabolic disorders, maladaptive responses to podocyte stretch and VEGF inhibition [9, 10].
Genetic mutations that cause structural or functional dysregulation of crucial
podocyte proteins result in hereditary podocytopathies. Several genetic
abnormalities have been recognized in association with podocytopathies [11].
Mutations in type IV collagen genes are linked to Alport syndrome, mutations
encoding cytoskeletal proteins
Podocytopathies can be mediated by certain extrarenal soluble circulating pathogenic permeability factors that injure podocytes [15]. The proposed permeability factors include soluble urokinase-type plasminogen activator receptor, cardiotrophin-like cytokine factor 1 and anti-CD40 antibody [16]. IL-4 and IL-13 too have been hypothesized as possible circulating factors in the pathogenesis of podocytopathies [17]. None of these factors have been conclusively recognized and further testing needs to be carried out.
Several infections can trigger cytokine-mediated podocytopathies. They include viral infections like HIV, Hepatitis C Virus (HCV), parvovirus B19, Epstein–Barr virus and cytomegalovirus infection [18, 19]. The severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) infection causing the coronavirus disease 2019 (COVID-19) has been added to the list of viral infections that can induce podocytopathies [20]. Parasitic infections like Borrelia burgdorferi and Plasmodium falciparum have also been implicated [21, 22]. There are many therapeutic medications that are directly toxic to podocytes and trigger podocytopathies, including interferon (alpha, beta, gamma) lithium, bisphosphonates (pamidronate), sirolimus and anthracycline (doxorubicin) and purine antagonists (puromycin aminonucleoside) [23, 24, 25].
A hemodynamic response occurs when there is a decrease in the number of
functioning nephrons. It results in an increased workload on the remaining viable
nephrons, leading to glomerular hyperfiltration, increased intraglomerular
capillary pressure and capillary lumen dilatation. The capillary dilatation
causes disruption of the SD and FPs connection, leaving an empty uncovered and
exposed space on the surface of the GBM. This propels the podocytes to undergo
compensatory hypertrophy and stretch themselves over the exposed portion of the
GBM surface, and in the process, the podocytes become structurally unstable. With
time, this detrimental podocyte shear stress and hypertrophy gives way to
podocyte loss and results in secondary FSGS [26]. In addition, concurrent
downregulation of
Chronic hyperglycemia causes direct injury to podocytes, mesangial cells and
endothelial cells and drives the pathogenesis of diabetic glomerulopathy [28].
The diabetic milieu of high glucose leads to the downregulation of nephrin
protein with resultant disruption of SD and FP injury. In addition, there is
downregulation of
Deposition of immune complexes in glomeruli can directly injure podocytes. A classic example is membranous nephropathy (MN) in which the deposition of sub-epithelial electron-dense immune complexes activates the lectin complement pathway and the membrane attack complex C5b-9 via the alternate pathway, to ultimately bring about podocyte damage and podocytopathic disease [35]. Other immune-mediated conditions in which podocyte damage is well documented include lupus nephropathies and IgA disease [9]. Recent literature has reported the identification of autoantibodies against the SD protein nephrin in some cases of MCD, along with increased serum levels of the anti-nephrin antibody, suggesting an autoimmune etiology in a subset of MCDs [36]. Significant elevation of complements and urinary complement fragments have been documented in patients with FSGS [37, 38].
Podocytes and endothelial cells crosstalk and help stabilize the slit diaphragm and maintain the structural and functional integrity of the GFB. Podocyte-sourced angiogenic VEGF plays a major role in this crosstalk. Notably, VEGF increase and VEGF decrease, both adversely affect the podocyte-endothelial crosstalk and optimal glomerular functions. While reduced VEGF leads to podocyte loss, thickening of the GBM and endothelial cell injury, elevated VEGF causes abnormal neovascularization [29]. The inhibition of VEGF in pregnancy-related hypertension during pre-eclampsia can lead to podocytopathies like MCD and FSGS [39]. Systemic diseases such as mixed connective tissue disorders, certain lymphomas and leukemia can also trigger podocyte injury [40, 41, 42]. Several risk factors are also documented in association with podocyptopathies and they include advancing age, high salt diet, low birth weight and prematurity [18]. Calcium is essential for podocyte actin cytoskeleton structural stability and function and any depletion of intracellular calcium predisposes to podocyte injury. The previously mentioned SOC and TRPC6 channels are instrumental in preserving podocyte integrity [43]. They regulate the entry of intracellular calcium and thereby help maintain calcium homeostasis and any aberrant expression or abnormality in the signaling of these channels contributes towards podocytopathic glomerular diseases like familial FSGS and diabetic nephropathy [34, 44, 45].
Podocytes respond to stressful injurious stimuli in various ways:
(a) Foot process effacement (FPE): The initial response involves remodeling of the podocyte cytoskeletal apparatus and alterations in the SD network, resulting in the simplification, retraction and loss of the FP (Fig. 2A,B) [46]. This phenomenon is the most characteristic response to podocyte stress and it adversely affects the normal function of the GFB. FPE is reversible in non-progressive glomerulopathy like MCD upon treatment with corticosteroids, while in chronic progressive glomerular disease like FSGS, the FPE can be refractory to treatment and tends to be irreversible.
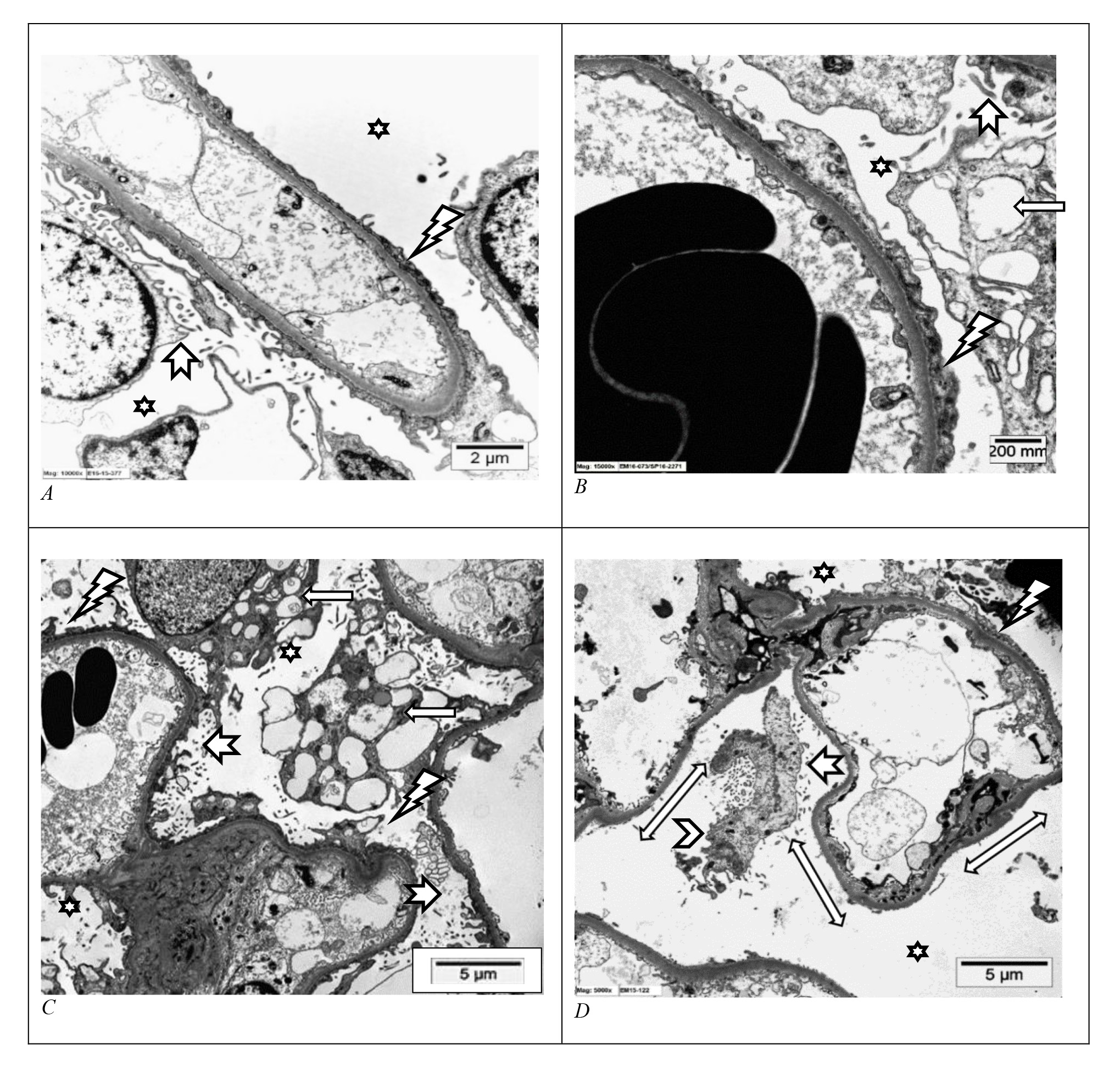 Fig. 2.
Fig. 2.Injured podocytes. (A–D) Electron microscopic photomicrographs from a human kidney of glomerular capillary loops floating in the urinary space (star) showing features of podocyte injury. There is podocyte foot process effacement (lightning arrows), prominent podocyte cytoplasmic vacuolization (arrows), microvilli formation (notched arrows) and areas of denudation of the glomerular basement membrane surface (double-headed arrows) signifying podocyte loss, as well as a detached damaged podocyte (arrowhead). (Uranyl acetate and lead citrate, A: Scale bar = 2 μm; B: Scale bar = 200 mm (millimicron/mμ); C: Scale bar = 5 μm; D: Scale bar = 5 μm). Images from Pathology Department, King Khalid University Hospital-Medical City, King Saud University, Riyadh, Saudi Arabia.
(b) Podocyte vacuolization and microvilli formation: Injured podocytes exhibit many cytoplasmic surface microprojections that protrude into the urinary space termed microvilli. They also show vacuolar degenerative changes in their cytoplasm along with abnormalities in the cellular organelles (Fig. 2C) [47]. Although these are non-specific findings, they are commonly noted in damaged podocytes.
(c) Podocyte hypertrophy: It typically results from the hemodynamic insult (glomerular hyperfiltration, increased intraglomerular pressure, shear stress, podocyte mechanical stretch and hypertrophy).
(d) Podocyte detachment and loss: The most common mechanism of podocyte loss is
death via apoptosis [48, 49]. Since these cells are terminally differentiated
cells with limited capacity to regenerate, the loss results in a reduction in
podocyte numbers that cannot be compensated and it renders a portion of the GBM
exposed (Fig. 2D). Podocyte loss is termed podocytopenia and it is the hallmark
of chronic progressive diseases like FSGS. The detached and lost podocytes appear
in the urine as podocyturia [47]. There is associated downregulation of
(e) Activation of PECs: Podocytopenia stimulates the PECs to migrate towards and adhere to the exposed part of the GBM where they proliferate and lay down ECM, resulting in glomerulosclerosis [52]. Podocyte loss of about less than 20% per glomerulus is compensated by hypertrophy and stretching of the remaining viable podocytes, over the exposed denuded portion of the GBM to cover the deficit. In podocyte loss of more than 20% per glomerulus, the area of the exposed GBM is more and can no longer be accommodated by simple podocyte hypertrophy, and it results in synechiae formation between the GBM and the Bowman’s capsule, the hallmark of FSGS. Podocytopenia of over 40% is associated with progressive, more advanced FSGS, and more than 60% loss is associated with global glomerulosclerosis [53]. In non-progressive glomerulopathies like MCD, the podocyte numbers are relatively well preserved.
(f) Proliferation of PECs: PECs are the progenitor cells capable of regeneration and differentiation into podocytes, especially in children under 5 years of age [54]. In certain conditions, the podocytopenia propels the PECs to proliferate and differentiate towards the podocyte phenotype as a reparative response in an attempt to replace the lost podocytes. This type of podocyte loss followed by mild PEC hyperplasia and attempted generation of podocytes is seen in the pathogenesis of DMS. In cases of severe acute podocyte injury, there is significant podocyte loss triggering marked aberrant PEC hyperplasia with incomplete differentiation towards the podocyte phenotype, and histologically, this collection of dysregulated PECs appears as pseudocrescents obliterating in the urinary space [55]. This phenomenon forms the basis of pathogenesis in cases of CG.
(g) Alteration in GBM structure and excess production of ECM: Podocytes are instrumental in the synthesis of the GBM so, podocyte injury can affect the structure and thickness of GBM and also promote abnormal production and deposition of ECM [31].
Podocytes are crucial in maintaining the integrity of the GFB and upon injury, there is a disturbance in the podocyte cytoskeletal machinery, the SD and FPs functions, subsequent FPE with disruption of the GFB and alteration in the permeability of the GMB [56]. Clinically, FPE translates into proteinuria. All podocytopathies present with proteinuria and proteinuria-related clinical features. The proteinuria may be nephrotic (more than 3.5 gm/day) or sub-nephrotic (between 0.2 gm/day to less than 3.5 gm/day). The patients with nephrotic range proteinuria present with nephrotic syndrome, classically defined as proteinuria of more than 3.5 gm/day with resultant hypoalbuminemia, hyperlipidemia and edema. The edema is usually due to sodium retention and hypoalbuminemia. Acute tubular injury can also occur in some cases of marked proteinuria. Microscopic hematuria may also be noted. In chronic progressive podocytopathies, patients can develop hypertension and renal insufficiency. Urine analysis, with the exception of MCD, tends to show both viable and apoptotic podocytes (podocyturia) [51].
Podocytopathies are glomerular diseases driven and unified by podocyte injury, FPE and proteinuria. A feature universal to all podocytopathies is the loss or “simplification” of the 3-dimensional architecture of the tertiary finger-like interdigitating FPs, and it is caused by alteration of the actin cytoskeleton with consequent disruption of the SD. Kidney biopsy is the mainstay of diagnosis for all types of renal disease including podocytopathies. It involves the light microscopy (LM), immunofluorescence (IF) and electron microscopy (EM) study of renal parenchyma. Podocyte injury can produce various morphological patterns. There are four main renal biopsy-based morphological patterns, representing the four types of response to a podocyte injury. They are (a) podocyte injury without significant podocyte loss, as seen in MCD, (b) podocyte injury with podocytopenia as seen in FSGS, (c) podocyte injury with podocyte loss and reactive mild podocyte proliferation, as in diffuse mesangial sclerosis and (d) podocyte injury with podocyte loss and reactive prominent podocyte proliferation, as seen in CG. These classical examples of podocytopathies (MCD, DMS, FSGS and CG) are non-immune complex-mediated diseases with no evidence of immunoglobulin or complement deposition (indicating lack of antigen–antibody reaction and immune complex deposition and complement activation) in IF and EM studies. In the IF study, glomeruli may occasionally display some mild non-specific uptake IgM and C3 and represent macromolecule trapping. Other patterns of podocytopathies include diabetic nephropathy, membranous nephropathy, lupus podocytopathies, etc. These biopsy-based histologically defined entities are further detailed below.
The hallmark of MCD is that there is diffuse FPE, with none to minimal podocyte loss of no more than 20% [53]. It is a common cause of steroid-sensitive nephrotic syndrome in children and the elderly population. The LM shows normal-looking glomeruli and the IF study is negative for immunoglobulins IgA, IgG and complements C3 and C1q (Fig. 3A). The albumin IF stain typically shows evidence of proteinuria in the form of tubular resorption protein droplets (Fig. 3B). The abnormality in MCD is evident on EM study which depicts podocyte injury in the form of extensive to diffuse FPE without any significant podocyte loss or GBM denudation (Fig. 3C,D). Diagnosis of MCD is made based on the clinical history of nephrotic range proteinuria and the ultrastructural documentation of extensive to diffuse podocyte FPE on biopsy [57].
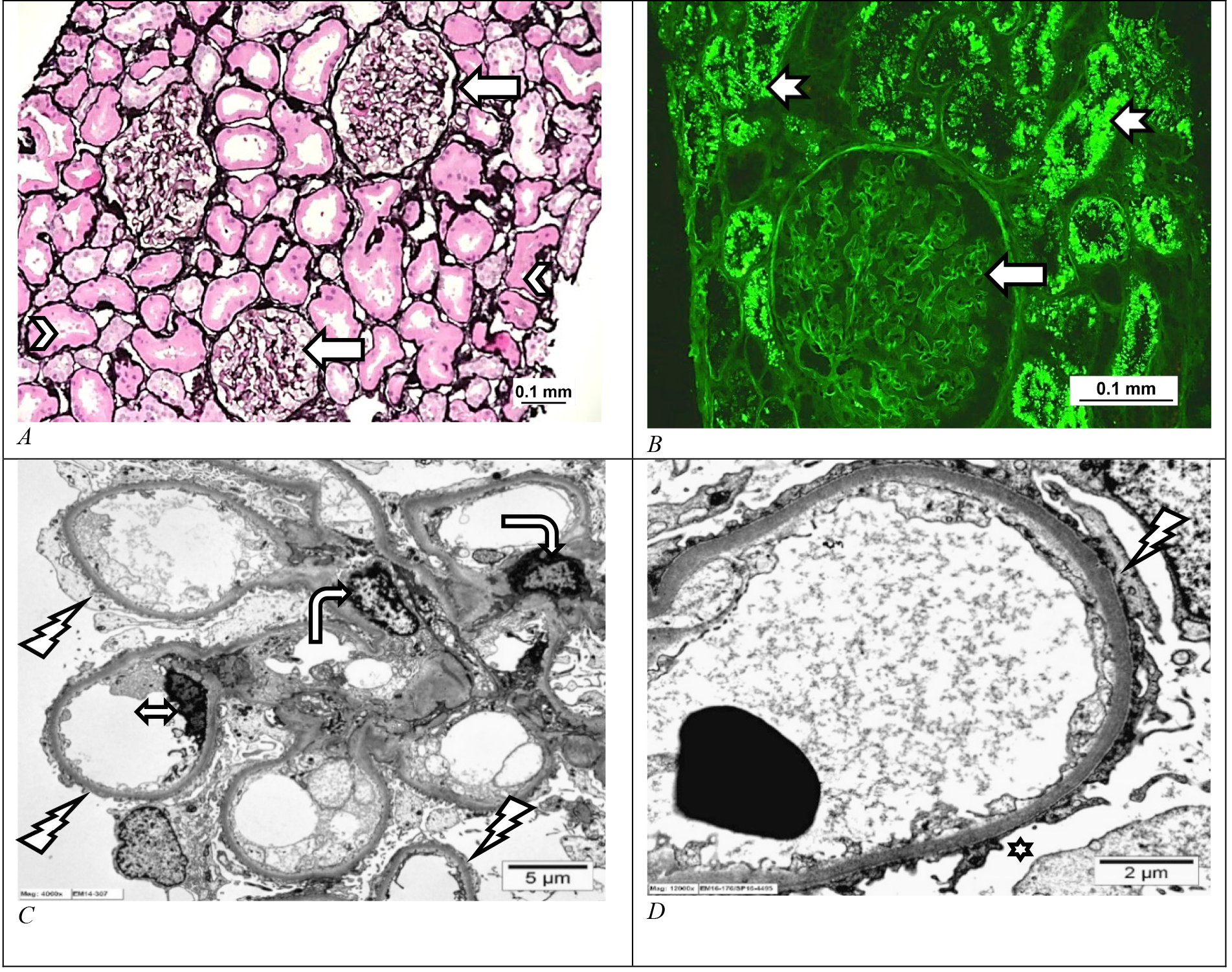 Fig. 3.
Fig. 3.Minimal change disease. (A) Light microscopic and electron microscopic photomicrograph of human kidney with minimal change disease showing a normal-looking kidney on histology with unremarkable glomeruli (arrows) and tubules (arrowheads), (Jones methenamine silver stain, Scale bar = 0.1 mm). (B) The albumin immunofluorescence stain shows an unremarkable-looking glomerulus (arrow) and immunofluorescent tubular resorption droplets in the proximal tubules (notched arrows) signifying proteinuria. (Immunofluorescent albumin stain, Scale bar = 0.1 mm). (C,D) Electron microscopic photomicrograph shows diffuse effacement of the podocyte foot processes (lightning arrow). Normal-looking mesangial (bent arrow) and endothelial cells (double-headed arrow) are also seen. (Uranyl acetate and lead citrate, C: Scale bar = 5 μm and D: Scale bar = 2 μm). Images from Pathology Department, King Khalid University Hospital-Medical City, King Saud University, Riyadh, Saudi Arabia.
DMS is a genetic disease commonly linked with WT-1 gene mutation, presenting as a severe nephrotic syndrome in children under 5 years of age, characterized by podocyte injury and loss. Since the podocyte regeneration capacity still exists until the age of 5 years, there is an attempt at regeneration and replacement of the lost cells by the podocytes. Also, as an initial response, the podocyte loss (of less than 20%) triggers the synthesis and deposition of ECM in the mesangium. The LM thus reveals diffuse mesangial expansion and solidification, along with hypertrophy and mild hyperplasia of the immature podocytes appearing as pseudocrescents in the urinary space [58]. IF is negative for immune deposits and ultrastructurally, there is patchy to extensive FPE and multilamination of the GBM.
Chronic podocyte injury and depletion (between 20% and 40%) and associated
denudation of the GBM surface, with no significant podocyte regeneration or
activation of PECs, all form the crux of FSGS [53]. FSGS is essentially a
response to chronic podocyte loss in which the rate of loss is greater than the
attempt by PECs to replace it. It is a chronic progressive condition
characterized histologically by segmental sclerosis of glomerular tufts,
involving some glomeruli (focal), often with adhesion between the affected tuft
and Bowman’s capsule [59]. The pathogenesis of FSGS begins with podocyte injury
followed by podocyte loss (podocytopenia), resulting in exposure of the
underlying GBM which subsequently adheres to the nearby Bowman’s capsule. The
podocyte loss upregulates the Notch gene, which in turn activates the PECs lining
the Bowman’s capsule to proliferate and migrate towards the glomerular tuft where
they produce and lay down ECM, resulting in sclerosis of that glomerular tuft
[52]. With time and as the disease advances, segmental glomerulosclerosis
progresses to become global glomerulosclerosis and brings about associated
tubular atrophy and interstitial fibrosis. The podocytopenia in FSGS can be
evidenced in urine as podocyturia and as urinary podocyte mRNA [60]. The podocyte
loss can be demonstrated in renal tissue, by immunostaining it with podocyte
marker WT-1, the expression of which is diminished in the affected glomeruli.
Immunostaining studies on the glomeruli of FSGS kidney show that as the disease
evolves and progresses, there is a significant reduction in podocyte marker WT-1
stain and concurrent increase in expressions of profibrogenic cytokine
TGF-
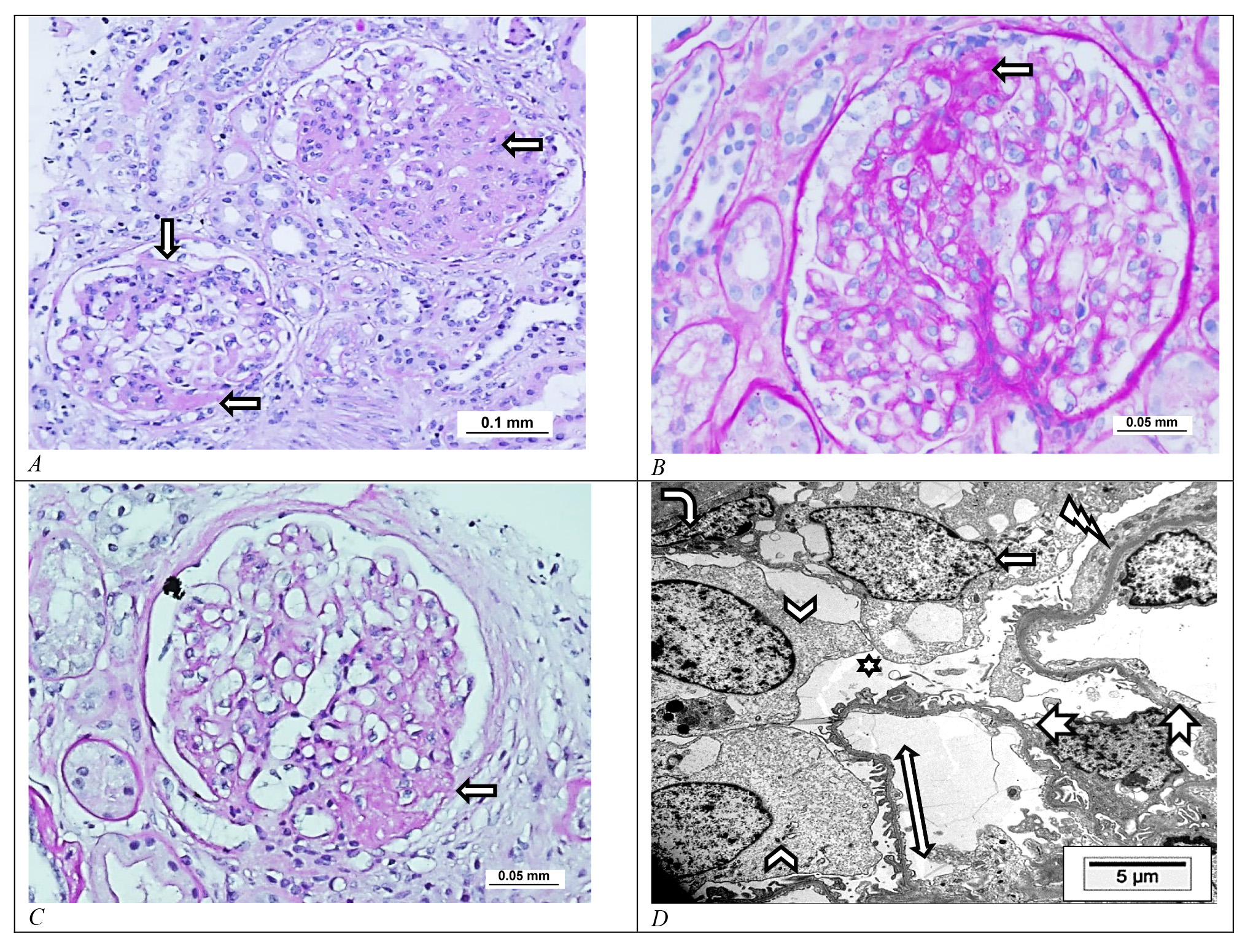 Fig. 4.
Fig. 4.Focal and segmental glomerulosclerosis. Light microscopic photomicrograph of focal and segmental glomerulosclerosis (FSGS) of human kidneys showing segmental glomerulosclerosis (arrows) with adhesion to the Bowman’s capsule of (A) NOS, (B) tip and (C) perihilar variants. (Periodic acid–Schiff stain, A: Scale bar = 0.1 mm; B: Scale bar = 0.05 mm; C: Scale bar = 0.05 mm). (D) Electron microscopic photomicrograph of the perihilar secondary type of FSGS shows the glomerular capillary loops (notched arrows) floating in the urinary space (star) with patchy focal effacement of the podocyte foot processes (lightning arrow), two normal-looking podocytes (arrowheads) corresponding to the normal looking foot processes (along the double-headed arrow) and one injured vacuolated podocyte (arrow) corresponding to the foot processes effacement site. A parietal epithelial cell (bent arrow) at the periphery is also identified. (Uranyl acetate and lead citrate, Scale bar = 5 μm). Images from Pathology Department, King Khalid University Hospital-Medical City, King Saud University, Riyadh, Saudi Arabia.
It is also referred to as the collapsing variant of FSGS. CG is characterized by the presence of at least one glomerulus with segmental capillary tuft collapse due to contraction and wrinkling of the GBM, with associated reactive and compensatory hyperplasia and hypertrophy of PECs in the urinary space, forming pseudo-crescents (Fig. 5A–C) [66, 67]. Any sudden massive podocyte loss (more than 40%) is the key trigger of CG and it sets into motion the implosive capillary loop collapse with aberrant proliferation of the PECs [53]. PECs attempt to regenerate and replace podocytes by proliferating but fail to completely differentiate into mature podocytes, and as a result, they accumulate in the urinary space as the characteristic pseudocrescents of CG. Patients typically present with severe steroid-resistant nephrotic syndrome with very high nephrotic range proteinuria. There is no evidence of deposits of IF or EM studies. EM typically shows diffuse FPE. It is associated with poor prognosis with relatively fast progression to ESRD. Based on etiology, CG can be idiopathic, genetic or reactive to insults like severe ischemia, autoimmune diseases, malignancies, certain drugs like interferon therapy and various infections including cytomegalovirus, parvovirus B19, human immunodeficiency virus (HIV) and also the new SARS-CoV2. HIVAN is a well-documented infection associated with podocytopathy and is typically reported in patients of African origin harboring the high-risk variant of the APOL1 gene. The virus directly infects the podocytes and triggers a florid proliferative response in podocytes via VEGF activation renal biopsy shows features of CG with pseudocrescent formation and tubuloreticular inclusions on EM (Fig. 5D) [13, 68].
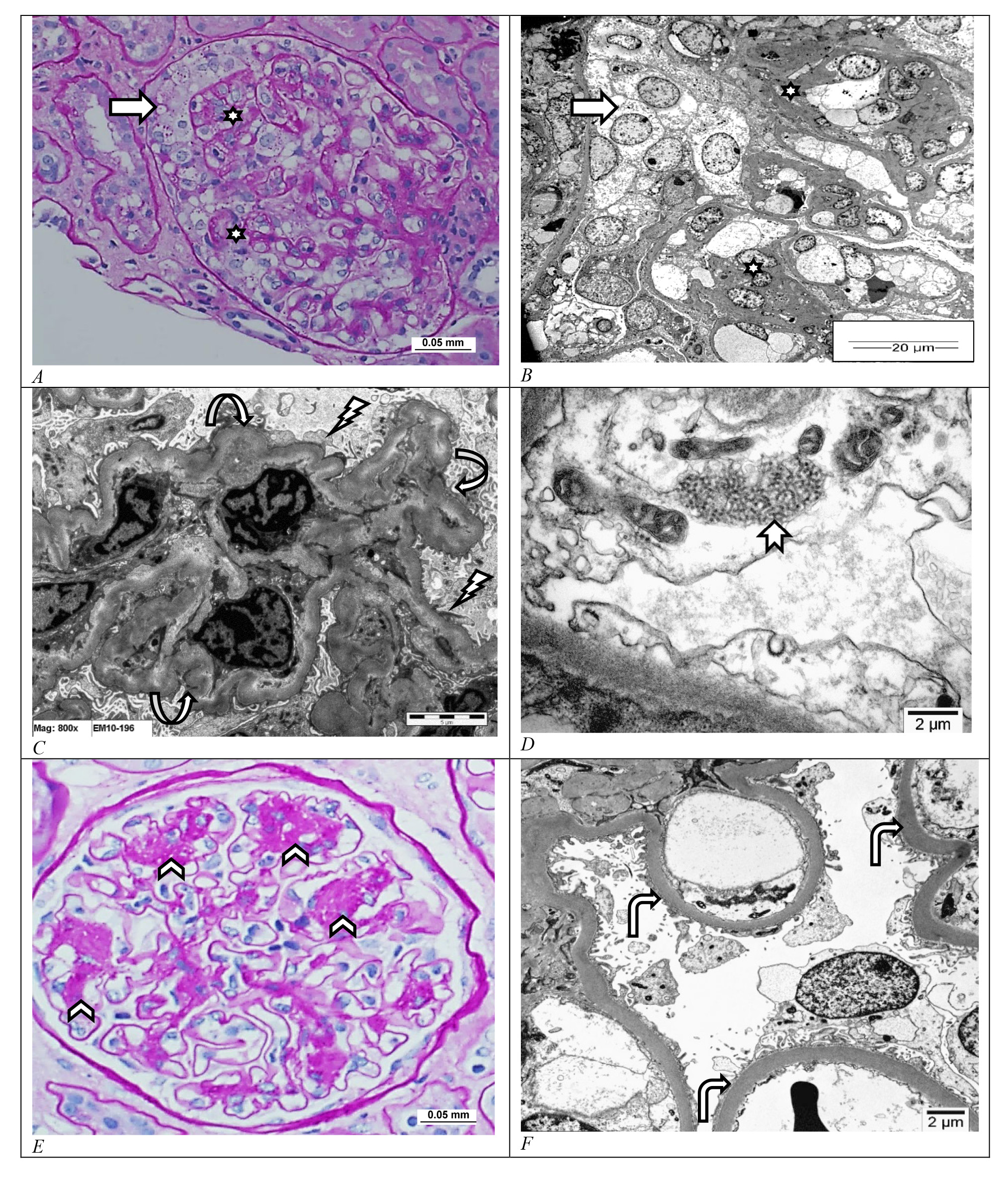 Fig. 5.
Fig. 5.Collapsing glomerulopathy and diabetic nephropathy. (A–D) Light microscopic and electron microscopic photomicrographs of human kidney with collapsing glomerulopathy show segmental glomerular capillary loop collapse and sclerosis (stars) with hypertrophy and hyperplasia of the overlying parietal epithelial cells (arrow). There is wrinkling of the glomerular basement membrane (circular arrows) with effacement of the overlying foot processes (lightning arrow). Patients with human immunodeficiency virus infection associated collapsing glomerulopathy as was the case in this kidney show tubuloreticular inclusions in the endothelial cell cytoplasm (notched arrow). ((A) Periodic acid–Schiff stain, Scale bar = 0.05 mm; (B–D) uranyl acetate and lead citrate, B: Scale bar = 20 μm, C: Scale bar = 5 μm and D: Scale bar = 2 μm). (E,F) Light microscopic and electron microscopic photomicrographs of human kidney with nodular form of diabetic nephropathy showing mesangial nodules with dilatation of the peripheral capillary loops (arrowheads) and diffuse thickening of the glomerular basement membrane (bents arrows). ((E) Periodic acid–Schiff stain, Scale bar = 0.05 mm and (F) uranyl acetate and lead citrate, Scale bar = 2 μm. Images from Pathology Department, King Khalid University Hospital-Medical City, King Saud University, Riyadh, Saudi Arabia.
Chronic long-standing poorly controlled diabetes mellitus can lead to diabetic nephropathy. It is a consequence of glomerular enlargement and podocyte loss [28]. Patients present with albuminuria, the severity of which depends on the extent of glycemic control and the duration of diabetes [29]. On LM, there is diffuse thickening of the glomerular capillary loops and expansion of the mesangial matrix. This mesangial expansion initially is mild and as the disease progresses, it eventually forms mesangial nodules (Kimmelsteil–Wilson nodules) with secondary microaneurysmal dilatation of the glomerular capillary loops around the nodules (Fig. 5E). The afferent and efferent arterioles usually show prominent hyalinosis in their walls. In later stages, segmental and global glomerulosclerosis ensue. On IF, although there is no evidence of immunoglobulin or complement deposition, the IgG and albumin stains show non-specific uptake (staining) along the GBM and the tubular basement membrane. Ultrastructurally, there is classical diffuse thickening of the GBM usually measuring more than 600 nm in average thickness, with variable patchy to extensive FPE and no electron-dense immune deposits (Fig. 5F).
MN is an immune complex-mediated renal disease in which the deposited sub-epithelial immune complexes directly injure the podocytes [69]. It is seen mainly in the adult population manifesting as nephrotic syndrome refractory to steroid therapy. About 80% of MNs are primary (idiopathic) autoimmune glomerular disease and majority (about 70%) are mediated by anti-phospholipase A2 receptor autoantibodies, and a small percentage (about 5%) are mediated by anti-thrombospondin domain-containing 7A autoantibodies. MN can also occur secondary to other conditions like various malignancies (carcinomas, leukemia and lymphomas), infections (e.g., hepatitis C or B viruses), autoimmune diseases (e.g., systemic lupus erythematosus with class V lupus nephritis) and certain therapeutic drugs (e.g., penicillamine, captopril, etc.). Classical renal biopsy of primary MN on histology shows diffuse thickening of the glomerular peripheral capillary loops, due to the sub-epithelial immune complex deposits (Fig. 6A). In the IF study, these immune complexes exhibit diffuse granular positivity along the peripheral capillary loops with immunoglobulin IgG, complement C3 and light chains kappa and lambda (Fig. 6B). EM shows diffuse FPE and sub-epithelial to intramembranous immune complex deposits (Fig. 6C,D). MN secondary to autoimmune conditions like systemic lupus erythematosus shows full house granular capillary wall staining with immunoglobulins IgA, IgG and IgM, complements C3 and C1q, and light chains kappa and lambda [70].
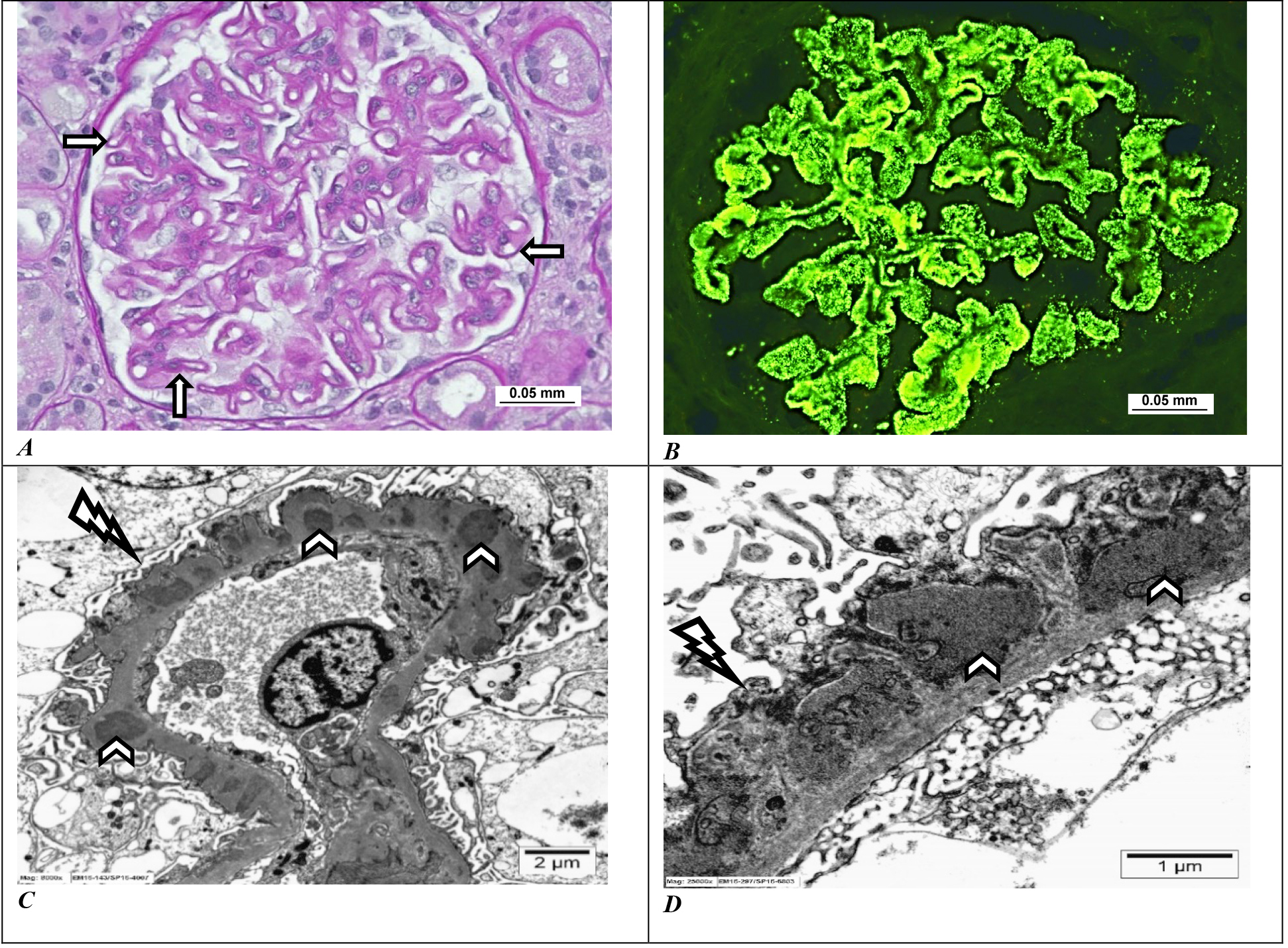 Fig. 6.
Fig. 6.Membranous nephropathy. (A) The light microscopy shows prominent thickening of the glomerular capillary loops (arrows) (periodic acid–Schiff stain, Scale bar = 0.05 mm). (B) Immunofluorescence with immunoglobulin IgG stain shows strong glomerular global peripheral capillary looping positivity (immunoglobulin IgG stain, Scale bar = 0.05 mm). (C,D) Ultrastructurally, there is diffuse effacement of podocyte foot processes (lightening arrows) along with multiple subepithelial electron-dense immune complex deposits (arrowheads) (uranyl acetate and lead citrate, C: Scale bar = 2 μm and D: Scale bar = 1 μm). Images from Pathology Department, King Khalid University Hospital-Medical City, King Saud University, Riyadh, Saudi Arabia.
It is a type of nephropathy seen in patients with active systemic lupus erythematosus. It is defined by nephrotic range proteinuria, severe to diffuse FPE with either no electron-dense immune complex deposits or only mesangial deposits [40, 71]. The biopsy shows features ranging from MCD to mesangial proliferation to FSGS with no endocapillary hypercellularity and no subendothelial or subepithelial immune deposits. The IF and EM studies are either negative for deposits or they may show evidence of full house mesangial staining with IgA, IgG, C3 and C1q and some mesangial deposits. The EM classically shows extensive to diffuse FPE with no subendothelial or subepithelial immune deposits. It contributes to about 1% of lupus nephritis biopsies [71]. The pathogenesis of lupus podocytopathy is not well understood and is most likely caused by a non-immune complex-mediated pathway in which there is podocyte injury secondary to abnormal cytokines, lymphokines and T-cells activity [72].
Alport syndrome (AS), a progressive renal disease due to mutations in COL4A3, COL4A4 and COL4A5 genes, causes deterioration of the GBM and podocyte damage. Patients present with nephrotic range proteinuria and some also develop sensorineural hearing loss and visual abnormalities. Histologically, initially, it appears as MCD and eventually progresses to FSGS. IF is negative for immunoglobulins and complements. The EM shows diffuse FPE and classical aberrations of the GBM with alternate thin and irregularly thickened segments, GBM splitting, multilamellation and fragmentation of the lamina densa, yielding a basket weave pattern with scattered small electron-dense granules [73]. Congenital nephrotic syndrome of the Finnish type which is present within the first few months of life with massive proteinuria, results from mutations of NPHS1 gene coding for SD structural protein nephrin, also shows histology ranging from MCD type glomeruli to mesangial proliferation to FSGS in a background of characteristic microcystic dilatation of renal tubules [74]. Typically, there is a loss of nephrin immunostain in the glomeruli and patients are positive for anti-nephrin antibodies [58]. IF is negative and EM shows extensive FPE. Conditions like C1q nephropathy and IgM nephropathy are the less common types of podocytopathies manifesting as steroid-resistant nephrotic range proteinuria [64]. Histologically, they too range from MCD to mesangial proliferation to FSGS-like appearance with dominant diffuse strong (2+ or more) mesangial staining with complement C1q or immunoglobulin IgM, respectively, on IF. Ultrastructurally, they show FPE and a few small mesangial/paramesangial deposits [58, 75]. COVID-19 caused by SARS-CoV2 can cause COVID-19-associated nephropathy (COVAN). The most common histologic type of COVAN is CG, especially in persons of African origin with high-risk APOL1 mutations, followed by MCD and non-collapsing FSGS [19, 20, 76]. Few cases of post-COVID-19 vaccination-associated podocytopathies have also been documented [77, 78]. Pregnancy-associated podocytopathies can also occur and they present as MCD, FSGS and even CG [18]. Nephrotoxic medications like interferon, pamidronate, adriamycin and lithium are chronicled in the development of MCD, FSGS or collapsing GN [23].
Correct diagnosis of the type of podocytopathy is essential for optimal management. A renal biopsy involving the LM, IF and EM studies is still the gold standard for obtaining a definitive diagnosis. However, podocytoapthies need to be further assessed beyond the realm of tissue biopsy by investigating the etiological cause as awareness of the etiology in individual cases may pave the path to precision medicine. Patients should be investigated for various infections, circulating factors, genetic testing, serum antinephrin antibodies, etc. Research in serum or urinary biomarkers and proteomic studies has suggested alpha-1 antitrypsin, transferrin, histatin-3, certain ribosomal proteins and calretinin as possible urinary markers in podocytopathies [79].
Current studies are exploring podocyte-tailored therapies. Podocytes, podocyte-specific proteins and pathways are being researched as targets of treatment. Notch signaling pathway inhibitors may be employed to prevent podocyte apoptosis and SOC and TRPC6 inhibitors may be used for maintaining calcium-dependent podocyte cytoskeletal integrity [30, 33, 80]. A mice study identified two endoplasmic reticulum calcium stabilizers (K201 and mesencephalic astrocyte-derived neurotrophic factor protein) that inhibit the leakage of calcium from the endoplasmic reticulum into the cytosol and this way, they maintain calcium homeostasis and protect against apoptosis [81]. Other possibilities are drugs that promote the progenitor PECs to regenerate podocytes. Drugs that induce upregulation of transcription factor WT-1 or those that suppress chemokine CXCL12, lead to activation of PECs to differentiate towards podocytes [82, 83]. Mitochondria-directed drugs that address oxidative podocyte injury and apoptosis are also being explored [84]. These are all potential therapeutic targets. Genetic testing identifies several types of familial podocytopathies and such patients can benefit from personalized treatments like focused genetically modified stem cell therapies and mRNA-based therapies [85]. Transgenic mice with APOL1 podocytopathy upon treatment with antisense oligonucleotides targeting APOL1 mRNA demonstrated a reduction in the expression of APOL1 and improvement in kidney functions [86]. Patients with genetic defects in biosynthesis coenzyme Q10 develop familial podocytopathy and they show improvement in renal function following oral supplementation of coenzyme Q10 [87]. The application of precision medicine promises to be the future for patients with proteinuric glomerular diseases.
Podocytes, the peripheral cells of the GFB, are central in the pathogenesis of various proteinuric diseases collectively called podocytopathies. While the diseases that fall under this umbrella have certain common overlapping etiologies and pathophysiologies, they also have several distinct, unique and characteristic features. The study of podocyte biology in the past decade has significantly enhanced our knowledge of these diseases and has clearly demonstrated the crucial pathogenic role of podocytes in disease evolution. All patients with podocytopathies should be investigated thoroughly for causative or triggering agents in order to provide a more personalized approach to management. It is still an evolving science and as our understanding of the role of podocytes in renal diseases improves, so will the therapeutic modalities.
The single author was responsible for the entire preparation of this manuscript.
According to the policy of the King Saud University Institutional Review Board (KSU IRB) and national regulations, the KSU IRB has determined that this study is exempt from further review.
Not applicable.
This research received no external funding.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.